

Summary
In glioblastoma cells the receptor tyrosine kinase c-Met is upregulated in response to Bevacizumab and plays an important role in promoting invasion and tumor recurrence. These data support novel links between VEGF-A and HGF and suggest that c-Met and its signaling effectors may be effective targets for anti-invasive therapies.
In this issue of Clinical Cancer Research Jahangiri et al make an important first step toward understanding mechanisms of resistance to the vascular endothelial growth factor-A (VEGF-A) neutralizing antibody Avastin/Bevacizumab (1). Tumor angiogenesis is regulated by a complex set of protein cues, but arguably the most important pro-angiogenic factor is VEGF-A (2). Unfortunately, various FDA-approved agents that inhibit components of the VEGF-A pathway have resulted in disappointing clinical outcomes. In particular, Avastin/Bevacizumab (3) has yielded minimal improvements in progression-free survival in many cancer patients due to VEGF-independent ‘neoangiogenesis’ and tumor recurrence (4). Additionally, in sub-sets of patients as well as some pre-clinical mouse models, anti-VEGF therapies unexpectedly lead to robust tumor cell invasion and metastases (5). While many VEGF-independent cues that promote angiogenesis have been identified (2), we understand significantly less about how anti-VEGF therapies lead to altered invasion and metastases. These responses may occur secondarily to unfavorable growth and survival conditions in the primary tumor microenvironment. Indeed, hypoxia-dependent gene regulatory pathways have been reported to induce tumor cell dispersal. In combination, circulating macrophages and various other stromal cell types within the tumor microenvironment may impact tumor cell dispersal via secreted pro-invasive cues. Alternatively, VEGF-A receptors including VEGFR-2 and Neuropilin-1 are expressed in many tumor cells. Thus, inhibition of VEGF signaling pathways could impact invasion via cell-intrinsic mechanisms.
The malignant brain cancer glioblastoma (GB) displays diffusely infiltrative growth patterns, with dispersive cells escaping surgical resection and invariably contributing to tumor recurrence. GBs are also highly vascularized, and are classified in part by the development of unique angiogenesis pathologies including blood vessels with glomeruloid-like tufts owing to aberrant microvascular cell proliferation and sprouting. GBs also develop edema and hemorrhage due to breakdown of the intratumoral blood-brain barrier. Bevacizumab results in improvements in GB progression-free survival owing to microvascular regression and reduced edema due in part to inhibition of VEGF-dependent endothelial cell survival and vascular permeability (6). However, there is no improvement in overall patient survival (5) because drug responses are transient, with recurrent tumors displaying resistance to continued therapy. During tumor progression and in most GBs that develop Bevacizumab resistance angiogenesis and invasion are tightly coupled pathologies. However, in approximately 30% of Bevacizumab-resistant tumors there is robust invasiveness with limited MRI contrast enhancement, suggesting an uncoupling of invasion and angiogenesis (7). Similar clinical findings have been reported for VEGF receptor antagonists (6). Therefore, it is important to identify pathways that drive invasion during GB progression as well as determine how invasion and angiogenesis may be uncoupled in response to anti-VEGF therapies. Characterizing pro-invasion pathways may also identify potential targets for intervention in GB, since there are currently few anti-invasive agents in the clinic.
Jahangiri et al. have identified multiple genes that are differentially expressed upon development of Bevacizumab resistance. They focus on the role of one select gene product, the receptor tyrosine kinase c-Met, which is upregulated in Bevacizumab-treated primary GB samples and U87 tumors selected for Bevacizumab resistance in mice. Met binds hepatocyte growth factor/scatter factor (HGF/SF) ligands and activates various intracellular pathways that promote cell growth and invasion (8). Jahangiri et al. report that autocrine HGF/SF-Met signaling promotes GB cell proliferation in a hypoxia-dependent manner. Furthermore, c-Met promotes tumor cell invasion, in part through Fak and Stat3 signaling effectors. RNAi-mediated silencing of MET gene expression or pharmacologic inhibition of c-Met kinase activities blocks tumor cell invasion and resistance to Bevacizumab. These results are consistent with a 2012 publication by Lu et al., showing that c-Met is upregulated in Bevacizumab-treated patient samples and in mosaic mouse models of GB genetically null for VEGF-A (9). Interestingly, in Bevacizumab-sensitive tumors c-Met and VEGFR-2 form heterodimeric complexes that suppress HGF-mediated tumor cell growth and invasion. Met/VEGFR-2 heterodimers associate with the cytoplasmic tyrosine phosphatase PTP1B, which dephosphorylates the c-Met kinase domain and inhibits interactions with pro-invasive signaling effectors. Collectively, the data from the Aghi and Bergers groups (both papers were published collaboratively) reveal cross talk between the HGF/SF and VEGF-A signaling pathways that negatively regulate tumor cell invasiveness during GB growth and progression. Met upregulation in response to Bevacizumab, probably in combination with other cell-intrinsic and cell-extrinsic events that promote resistance, alters this negative regulation and drives GB invasion (Figure 1).
Figure 1. Roles for c-Met in GB response and resistance to Bevacizumab.
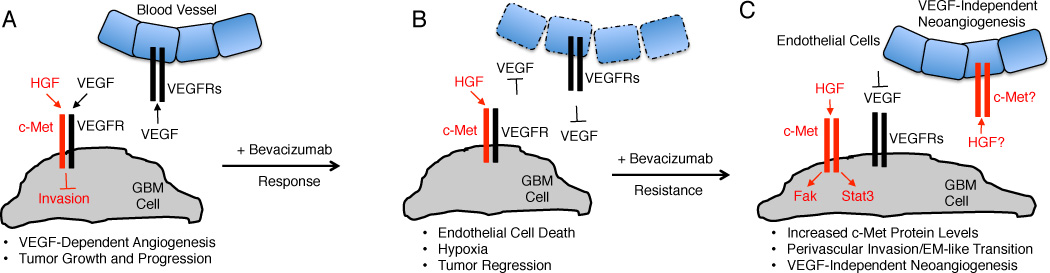
(A); Tumor cell proliferation and invasion are regulated via cross talk between the VEGF-A and HGF signaling pathways. During GB growth and progression c-Met and VEGFR-2 form heterodimeric complexes that suppress cell invasion via PTP1B-mediated Met tyrosine dephosphorylation. Meanwhile, VEGF-A produced by GB cells interacts with its receptors in endothelial cells and promotes angiogenesis and blood vessel permeability in the vascular niche. (B); Bevacizumab neutralizes VEGF-A signaling, leading to endothelial cell death, tumor regression, and transient improvements in overall and progression-free survival. (C); Responsiveness to Bevacizumab is followed by resistance, due to upregulation of c-Met gene expression in GB cells (acquired resistance) or selective survival of sub-populations of tumor cells that overexpress c-Met (intrinsic resistance). Met forms homodimers that bind HGF/SF and via an autocrine signaling loop promote Stat3 and Fak phosphorylation, promoting a mesenchymal-like phenotypes and perivascular tumor cell invasion. In Bevacizumab resistant tumors, paracrine HGF/SF signaling via Met in endothelial cells may also promote VEGF-independent neoangiogenesis.
These data raise several interesting questions. First, in Bevacizumab-resistant GBs what are the essential pathways that drive VEGF-independent angiogenesis? There are multiple pro-angiogenic factors; however, one obvious candidate is c-Met, which is expressed in endothelial cells and has pro-angiogenic functions via HGF/SF (8). Therefore, it is enticing to speculate that in addition to autocrine HGF/c-Met signaling events in invasive tumor cells, HGF/SF may also promote VEGF-independent neoangiogenesis via paracrine signaling between GB cells and blood vessels. Second, Jahangiri et al. have discovered multiple genes that are differentially expressed in GB samples upon development of Bevacizumab resistance. Are these genes differentially expressed in the majority of GB cells or in select sub-populations of cells? Stem-like GB cells overexpress VEGF-A and home to perivascular niches (10), suggesting that gene regulation in GB stem cells may contribute to acquired Bevacizumab resistance. Alternatively, GB cell death may lead to enrichment of sub-populations of intrinsically resistant tumor cells that express high endogenous levels of c-Met. The U87 xenograft selection models suggest acquired resistance since U87 cell lines have been cultured for decades and lack stem-like properties. However, Bevacizumab resistance in the U87 models and the primary GB cultures can be serially passaged, suggesting intrinsic stability. Delineating possible contributions by intrinsically resistant populations of GB cells in pre-clinical models and patient samples warrant further investigation. Third, can Bevacizumab-resistant tumors be classified based on their molecular genetic characteristics? Bevacizumab-sensitive GB cells are epithelial-like, whereas resistant cells upregulate some mesenchymal markers. However, the gene expression signatures of resistant tumors are distinct from mesenchymal GBs (11), suggesting that anti-vascular therapies may generate unique tumor sub-types that should be classified. Fourth, the exact Met-regulated intracellular signaling pathways that drive GB cell invasion remain to be determined, and identifying these effectors may lead to new therapies. Jahangiri report that Fak and Stat3 signaling effectors are hyperphosphorylated in invasive cells, and there are agents in the clinic that target these proteins. Another related question is whether the major c-Met signaling effector Gab-1 (8) is essential for pro-invasive signaling and if Met phosphorylation of Gab-1 is functionally linked to Fak and Stat3? Lastly, will combined inhibition of VEGF-A and HGF/SF pathways be effective anti-angiogenic and anti-invasive therapies? The dual-specific kinase inhibitor Cabozantinib, which targets Met and VEGFR-2, has shown encouraging results in pre-clinical models of pancreatic cancer (12) and has been approved for medullary thyroid cancers. However, it remains to be determined if clinical trials with this inhibitor or others will effectively impact GB recurrence and invasion.
Acknowledgements
I thank Dr. John de Groot (MDACC) for insightful comments on the manuscript.
Grant Support: R01NS059876 and R01NS078402 both from NIH/NINDS
Footnotes
The author declares no conflicts of interest.
References
- 1.Jahangiri A, De Lay M, Miller LM, Carbonell WS, Hu YL, Lu K, et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of anti-angiogenic therapy resistance. Clin Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-12-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sitohy B, Nagy JA, Dvorak HF. Anti-VEGF/VEGFR therapy for cancer: reassessing the target. Cancer Res. 2012;72:1909–1914. doi: 10.1158/0008-5472.CAN-11-3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 4.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nature reviews Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellis LM, Reardon DA. Cancer: The nuances of therapy. Nature. 2009;458:290–292. doi: 10.1038/458290a. [DOI] [PubMed] [Google Scholar]
- 6.Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610–622. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- 7.de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010;12:233–242. doi: 10.1093/neuonc/nop027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 9.Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. 2012;22:21–35. doi: 10.1016/j.ccr.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilbertson RJ, Rich JN. Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nature reviews Cancer. 2007;7:733–736. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- 11.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.You WK, Sennino B, Williamson CW, Falcon B, Hashizume H, Yao LC, et al. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. 2011;71:4758–68. doi: 10.1158/0008-5472.CAN-10-2527. [DOI] [PMC free article] [PubMed] [Google Scholar]