

Abstract
Objective
Familial aggregation of fibromyalgia has been increasingly recognized. The goal of the current study was to conduct a genome wide linkage scan to identify susceptibility loci for fibromyalgia.
Methods
We genotyped members of 116 families from the Fibromyalgia Family Study and performed a model-free genome-wide linkage analysis of fibromyalgia with 341 microsatellite markers, using the Haseman-Elston regression approach.
Results
The estimated sibling recurrence risk ratio (λs) for fibromyalgia was 13.6 (95% CI: 10.0–18.5), based on a reported population prevalence of 2%. Genome-wide suggestive evidence of linkage was found at marker D17S2196 (Empirical P =0.00030) and D17S1294 (Empirical P =0.00035) on chromosome 17p11.2-q11.2.
Conclusion
The estimated sibling recurrence risk ratio suggests a strong genetic component of fibromyalgia. This is the first study to report genome-wide suggestive linkage of fibromyalgia to the chromosome 17p11.2-q11.2 region. Further investigation of these multi-case families from the Fibromyalgia Family Study is warranted to identify potential causal risk variants for fibromyalgia.
Keywords: fibromyalgia, genome scan, linkage, sibling pairs, multi-case families
Fibromyalgia is a common, chronic pain disorder affecting an estimated 2% of the general population and defined by the American College of Rheumatology (ACR) as widespread pain of at least 3 months duration and pain on palpation in at least 11 of 18 tender point sites (1,2). Although the underlying pathophysiological mechanisms underlying fibromyalgia are not completely understood (3-5), Arnold et al. (6) reported that fibromyalgia strongly aggregates in families, confirming previous preliminary family studies (7-9), and supporting the possible role of genetic factors in the etiology of fibromyalgia (10).
Previous attempts to identify fibromyalgia susceptibility gene(s) have had limited success. Some candidate genes have been tested in population-based association studies, none of which has yet been confirmed as a susceptibility locus for fibromyalgia (11,12). More recently, Smith et al. (13), in the largest candidate gene association study of the fibromyalgia to date, observed significant differences in allele frequencies between cases and controls for several novel genes: GABRB3 (in the promoter region of the GABA-A β receptor gene), TAAR1 (trace amine-associated receptor 1), GBP1 (guanylate binding protein 1), RGS4 (regulator of G-protein signaling 4), CNR1 (CB-1 cannabinoid receptor gene), and GRIA4 (AMPA ionotropic glutamate receptor 4 subunit). Three of these genes, TAAR1, RGS4, and CNRI play roles in the modulation of analgesic pathways (13).
Genome wide association studies (GWAS) have revolutionized the dissection of genetic determinants of many complex traits. To date, GWAS has identified hundreds of robustly replicated loci for common traits (14). However, identified loci explain only a small portion of the heritable component associated with many complex traits. Clearly, additional loci that can explain a large proportion of the heritable component are still to be discovered. It has been suggested that one answer may reside in rare variants, which are not captured or poorly captured by current GWAS designs (15,16).
Family-based designs such as linkage studies have long been shown to have high power to detect loci with a large effect size. Genetic variants with a large effect size tend to be rare in the population, although those variants are likely more frequent in cohorts of multi-case families (17,18). Because the cost of re-sequencing and high-dimensional SNP chips has dropped significantly, there has been a resurgence of methods to detect rare variants that could cause complex traits. Recent developments of molecular genomic tools and statistical approaches enable investigators to capture almost exact genetic information about inheritance patterns and allele sharing and find rare variants of modest to large effect using family-based designs (19-21). Some investigators have speculated that rare variants obtained from re-sequencing of family-based data may show different properties than variants obtained from case-controls studies (22-24).
In an effort to determine if such designs can be implemented in investigations of fibromyalgia susceptibility, we conducted an autosomal genome wide linkage scan to map chromosome loci that may contain rare variants associated with fibromyalgia susceptibility using a cohort of multi-case families from the Fibromyalgia Family Study. To our knowledge, this is the first report of genome-wide linkage scan to focus on fibromyalgia in a family-based cohort.
PATIENTS AND METHODS
Study Population
Multi-case families with fibromyalgia were recruited and evaluated at four clinical sites: MetroHealth Medical Center/Case Western Reserve University (Cleveland), University of Texas Health Sciences Center (San Antonio), University of Illinois College of Medicine at Peoria (Peoria), and University of Cincinnati College of Medicine (Cincinnati, Ohio). The protocol was approved by the Institutional Review Board at each site. Probands (index cases) were recruited through the rheumatology clinics at each site, by referral from rheumatologists and other physicians, or self-referred through advertisements and patient support groups. A family was considered eligible if the proband (index case) met the 1990 American College of Rheumatology (ACR) criteria for primary fibromyalgia, and at least one first-degree relative of the proband met the ACR criteria for fibromyalgia (1). Families were excluded if the proband had a concomitant rheumatic disease, such as rheumatoid arthritis, or other medical explanation of their pain symptoms. If the proband’s affected first-degree relative was a parent or offspring, the family was included only if an unaffected female sibling of one of the affected individuals was available. (An unaffected male sibling was considered sufficient if one of the affected individuals in the family was male). If a family was eligible, available parents and female siblings (and male siblings if one of the affected individuals was male) of each affected individual were recruited. Only subjects who were age 12 or older were included.
Subject Evaluation
The investigators at each site (LMA, IJR, MBY, MAK) who are experienced with the evaluation of fibromyalgia conducted the diagnostic assessment of the subjects. The proband’s diagnosis of primary fibromyalgia was confirmed by physical examination, including a tender point examination, and review of medical records obtained after medical releases were signed by the subjects. All recruited family members were evaluated for the characteristic features of fibromyalgia, as defined by the 1990 ACR criteria. A subset of the family members agreed to provide a blood sample for the genetic analysis.
Sibling recurrence risk
To determine the extent of familial aggregation for fibromyalgia in the cohort of multi-case families, the sibling recurrence risk (Ks), defined as the proportion of affected siblings among all siblings of affected probands with fibromyalgia, was estimated by using a method described previously (25). The estimate of Ks was adjusted for sampling bias because families were recruited via an affected individual and because of the varying size of sibships. The single ascertainment strategy, i.e., when the probability of ascertaining a sibship is proportional to the number of affected individuals in that sibship, was used in our sample enrollment procedure. Sibling recurrence risk was calculated using Olson and Cordell’s formula for estimating sibling genetic risk parameters corrected for single ascertainment bias (25). In this formula, for ns(a) families with s offspring, a of whom are affected, which produces an unbiased estimate for single ascertainment strategies (25). The sibling recurrence risk ratio (λs) for fibromyalgia is calculated according to the formula (λs)= Ks/K. The population prevalence (K) of fibromyalgia is estimated at 2% based on a previous study (26).
Genome scan
We genotyped 341 markers on 22 autosomal chromosomes by using the Weber panel 8 marker set, which has an average marker spacing of 11 cM. After extracting DNA from the blood samples, we used a fluorescence-based genotyping method for the genome scan as previously described (27). In brief, after amplification reactions by using fluorescent-dyelabeled primers, multiplexed panels of markers were size separated on an ABI 3700 (Applied Biosystems) capillary gel by running the GeneScan 3.5 software for Windows NT. Rox 500 size standards were run in each lane. Besides the inlane standards, each 96-well plate contained four CEPH Centre d’Etude du Polymorphisme Humain (“Utah” residents with ancestry from northern and western Europe) controls (two samples each from individuals 1331-01 and 1331-02). Sixteen samples were placed in two different 96-well plates and one sample was placed in three different 96-well plates. The CEPH controls were used to standardize allele calling across plates, with the replicates being the safeguard for allele-calling reliability. The genotype data were collected and analyzed using the Genotyper 3.6 software for Windows NT. Initial marker placements were based on Marshfield map (28), although other placements were also investigated.
Error Checking
First, concordance between duplicates was assessed for consistency; inconsistent genotype data were changed to missing values. Second, the allelic data were checked for Mendelian inconsistencies, using the program MARKERINFO (S.A.G.E., version 6.1.0; http:// http://darwin.cwru.edu/sage/, provided by Case Western Reserve University, Cleveland, OH) (29). Mendelian inconsistent genotypes were set to missing for all members of a pedigree for the purpose of analysis.
Prior to performing the sibling pair-based linkage analysis, we reclassified the sibling pairs in each pedigree according to their likely true relationship, using the program RELTEST (S.A.G.E., version 6.1.0) (29). RELTEST is based on a Markov-process model of allele sharing along the chromosomes using genome scan data (30). The probability of misclassification depends on the total length of the genome scan and overall marker informativeness. If one or both members of a sibling pair have a high proportion of missing genotypes, their relationship may be misclassified. We reclassified 8 individuals in 4 full-sibships as half-siblings, and 2 individuals in 1 full-sibship as unrelated. Furthermore, 2 individuals in 1 full-sibship were reclassified as monozygotic twin; hence one sibling’s data was deleted.
Linkage Analysis
Maximum likelihood estimates of allele frequencies for each genetic marker were estimated using the program FREQ (S.A.G.E., version 6.1.0), which explicitly models pedigree structure to obtain unbiased estimates of allele frequencies (29). Multipoint identify-by-descent (IBD) sharing estimates were calculated for sibling pairs at the space of 2cM, using the program GENIBD (S.A.G.E., version 6.1.0) (29). Multipoint model-free linkage analyses were performed by using the Haseman-Elston regression approach (31), implemented in the program SIBPAL (S.A.G.E., version 6.1.0) (29). The Haseman-Elston linkage test was performed using multipoint IBD sharing estimates. The W4 weighting scheme (option W4 in SIBPAL) was used to adjust for nonindependence of sibling pairs within a sibship and of squared trait sums and differences as previously described (32). The reported P values were obtained assuming the usual asymptotic distribution of the likelihood-ratio test statistic. In addition to nominal P values, statistical significance was estimated via permutation testing, constructing an approximate empirical distribution of the test statistic. To verify all nominal P values < 0.00074, the Lander and Kruglyak’s criterion for suggestive linkage (33), we performed up to 100,000 permutations to obtain empirical P values.
RESULTS
Clinical Characteristics and Demographics
Members of 116 families in the Fibromyalgia Family Study were evaluated. Among the family members assessed for the presence of fibromyalgia, there were 342 siblings included in the assessment of sibling recurrence risk ratio (λs) (see below). A subset of family members contributed blood samples for the analysis (N=341 family members, including 257 siblings [203 sibling pairs]). The majority of individuals affected with fibromyalgia were women, consistent with the known preponderance of fibromyalgia in women (2). The mean age of fibromyalgia onset was 34.5 years. Characteristics of the family members, including the total group of family members genotyped and the subgroup of siblings who were included in the sibling-pair-based linkage analysis are presented in Table 1.
Table 1.
Characteristics of Fibromyalgia Family Study participants
| Characteristics | Total Genotyped Sample |
Siblings |
|---|---|---|
| Participants | 341 | 257 |
| Age (mean ± SD) | 49.6 ± 13.6 | 48.6 ± 11.5 |
| Sex (women, %) | 305 (89%) | 232 (90%) |
| Race | ||
| Caucasian | 305 (89%) | 231 (90%) |
| Hispanic | 22 (6.5%) | 15 (5.8%) |
| African American | 4 (1.2%) | 2 (0.8%) |
| Mixed | 8 (2.3%) | 8 (3.1%) |
| Unknown | 2 (0.6%) | 1 (0.4%) |
| Fibromyalgia1 | 264 (77%) | 198 (77%) |
| Age of fibromyalgia onset | 34.5 ± 13.5 | 34.5 ± 13.3 |
| (mean ± SD) |
Participants with Fibromyalgia
Sibling Recurrence Risk Ratio (λs)
Using the dichotomous definition of fibromyalgia affected (yes/no), we calculated the sibling recurrence risk ratio (λs) with correction for single ascertainment within the 116 families (25). We assumed a population prevalence of 2% (2, 36) and that birth order was interchangeable. The largest family consisted of 13 siblings. Among the 342 siblings (female and male) assessed for fibromyalgia, the observed sibling recurrence risk (Ks) for fibromyalgia was 27.2% (95% Confidence Interval (CI): 22.5% - 31.9%), which yielded a sibling recurrence risk ratio (λs) of 13.6 (95% CI: 10.0 – 18.5). The sibling recurrence risk (Ks ) for the 194 female siblings only was higher at 43.8% (95% CI: 36.7% - 50.8%). However, because the prevalence of fibromyalgia is higher in women than men in the general population (26), the corresponding sibling recurrence risk ratio (λs ) is 12.9 (95% CI: 9.4-17.6), which is similar to that observed for the entire set of data.
Genome Scan
Multipoint linkage analysis was performed on the 203 sibling pairs. The results of the genome scan multipoint linkage analysis are presented in Figure 1; detailed information for the most significant marker at each locus with a nominal significance of P ≤0.01 are shown in Table 2. Five of these loci with a nominal significance of P ≤0.01 were located on chromosomes 4q21.21-21.23, 8p21.2, 8q11.23, 9p24.3-24.1, and 17p11.2-q11.2. Markers D4S2361, D8S1048, D8S1110, D9S1779, and D17S2196 were the most significant markers at these loci (Table 2).
Figure 1.
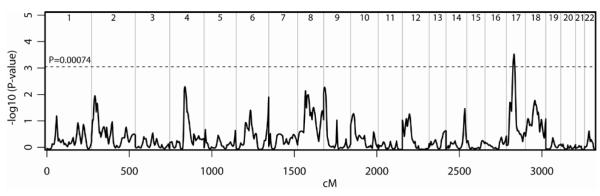
Results of genome-wide multipoint linkage scan for Fibromyalgia. For each chromosome (chr), genetic distance (cM) and −log10(P) are plotted on the X- and Y-axes, respectively. Horizontal line is drawn at P = 0.00074, the Lander and Kruglyak’s criterion for suggestive linkage.
Table 2.
Genetic locations and multipoint Haseman-Elston P values for markers demonstrating evidence of linkage (P ≤ 0.01) in the fibromyalgia genome scan
| Chromosome | Marker | Cytogenic location |
Position (cM) |
Position (hg19) |
Nominal P value |
|---|---|---|---|---|---|
| 4 | D4S3243 | 4q21.21 | 88 | ~80,932 kb | 0.0058 |
| 4 | D4S2361 | 4q21.23 | 93 | ~85,005 kb | 0.0045 |
| 8 | D8S1048 | 8p21.2 | 47.5 | ~26,811 kb | 0.0061 |
| 8 | D8S1110 | 8q11.23 | 60.5 | ~53,181 kb | 0.0092 |
| 9 | D9S1779 | 9p24.3 | 0 | ~516 kb | 0.0075 |
| 9 | D9S2169 | 9p24.1 | 14 | ~5,200 kb | 0.0094 |
| 17 | D17S2196 | 17p11.2 | 44 | ~17,264 kb | 0.00033* |
| 17 | D17S1294 | 17q11.2 | 50 | ~28,382 kb | 0.00048* |
P<0.00074, the Lander and Kruglyak’s criteria for genome-wide suggestive linkage.
We observed one chromosome region with a multipoint nominal P value < 0.00074, the Lander and Kruglyak’s criterion for genome-wide suggestive linkage (33), on chromosome 17p11.2-q11.2 (Table 3). Chromosome 17p11.2-q11.2 showed evidence of suggestive linkage to fibromyalgia in a 10-cM region spanning marker D17S2196 (nominal P=0.00033; 44 cM) to marker D17S1294 (nominal P=0.00048; 50 cM). The best signal on chromosome 17p11.2-q11.2 was at marker D17S2196 (nominal P=0.00033; empirical P=0.00030).
Table 3.
Genetic locations and multipoint P values (Haseman-Elston and empirical) for Chr17 locus demonstrating genome-wide suggestive linkage (P ≤ 0.00074) for fibromyalgia
| Chromosome | Marker | Cytogenic location |
Position (cM) |
Position (hg19) |
Nominal P value |
Empirical P value** |
|---|---|---|---|---|---|---|
| 17 | 40.0 | 0.00071* | 0.00044* | |||
| 17 | 42.0 | 0.00043* | 0.00036* | |||
| 17 | D17S2196 | 17p11.2 | 44.0 | ~17,264kb | 0.00033* | 0.00030* |
| 17 | 46.0 | 0.00025* | 0.00020* | |||
| 17 | 48.0 | 0.00029* | 0.00026* | |||
| 17 | D17S1294 | 17q11.2 | 50.0 | ~28,382kb | 0.00048* | 0.00035* |
P<0.00074, the Lander and Kruglyak’s criteria for genome-wide suggestive linkage.
Empirical P values were obtained through 100,000 permutations.
We converted the p-values to LOD (logarithm (to the base of 10) of odds) scores using Nyholt’s method (34). The P value for the best signal on chromosome 17p11.2-q11.2 at marker D17S2196 (nominal P=0.00033) is equivalent to a LOD score of 2.52, which is larger than a LOD score of 2.2, Lander and Kruglyak’s criterion for genome-wide suggestive linkage (33).
DISCUSSION
To our knowledge, the present study is the first genome wide linkage scan for fibromyalgia in a cohort of multi-case families. To explore whether there was excess risk among family members, we computed the sibling recurrence risk ratio (λs) in the studied cohort of multi-case families. The estimated sibling recurrence risk ratio (λs) of 13.6 suggests a strong genetic component of fibromyalgia, confirms previous reports that fibromyalgia aggregates in families (6,10), and is consistent with λs values reported for other complex disorders to which multiple genetic and environmental factors likely contribute (35).
By using model-free linkage methods, we detected one major locus for fibromyalgia on chromosome 17p11.2-q11.2 region, and several possible minor loci on other chromosomal regions. It is notable that chromosome 17p11.2-q11.2 region coincides with the map coordinate for two potential candidate genes for fibromyalgia: the serotonin transporter gene (SLC6A4) and the transient receptor potential (TRP) vanilloid 2 (TRPV2) genes.
Several converging lines of biological, pharmacological and genetic evidence suggest that the SLC6A4 gene is an attractive candidate gene for fibromyalgia susceptibility (5,6,36-43). The human serotonin transporter protein is encoded by a single gene (SLC6A4, LocusLink ID: 6532), mapped to chromosome 17q11.1-q12.12. A functional polymorphism (5-HTTLPR) in the 5′ regulatory region of the SLC6A4 gene involves two major alleles -- termed ‘S’ (Short) and ‘L’ (Long) allele-- that correspond to the presence of 14 or 16 repeat units of a 20-23 bp incomplete repeat (44). The short allele was found to reduce transcription efficiency for the SLC6A4 gene, resulting in decreased gene expression and serotonin uptake in lymphoblast cell lines (44). Association studies between this functional polymorphism and clinical pain syndromes such as fibromyalgia have generated conflicting results (36, 42, 43, 45). Further studies are needed to elucidate the role of SLC6A4 gene variants in etiology of fibromyalgia.
The 17p11.2-q11.2 chromosome region also coincides with map coordinates for the transient receptor potential (TRP) vanilloid 2 (TRPV2) gene. (46-48). There are compelling data for contribution to pain and specifically pathological pain associated with inflammatory and neuropathic states for other members of the TRP vanilloid subfamily, including TRPV1, TRPV4, and the ankyrin transmembrane protein TRPA1 (46). TRPV2 may play a role in mediating pain but more investigation is required to understand the role of TRPV2 in pain biology and chronic pain disorders like fibromyalgia.
However, the goal of a genome-wide linkage study is to explore all possible loci that may be involved in the causation of fibromyalgia, and any discussion about the role of SLC6A4 or TRPV2 genes in fibromyalgia is merely speculative. Furthermore, the region contains over one hundred other genes, and further molecular analyses including sequencing of the genes on chromosome 17p11.2-q11.2 region are warranted to identify additional variants for genetic testing.
Several loci (P <0.01) on 4q21.21-21.23, 8p21.2, 8q11.23, and 9p24.3-24.1 were identified through this genome scan. Whether mutations in these weaker loci are critical components in fibromyalgia etiology, or whether these loci act as modifiers, is a topic for further investigation. The majority of the loci identified in the genome scan do not coincide with map coordinates for previously postulated candidate genes for fibromyalgia, such as HTR2A (13q14-q21) (49), COMT (22q11.21) (50), DRD4 (11p15.5) (51), HLA region (52,53), or susceptibility genes identified in a recent candidate gene association study (13).
Whole exome sequencing (WES) is a comprehensive and unbiased survey of numerous types of genetic changes in the protein coding regions of the genome. Whole genome sequencing (WGS) is a similarly all-inclusive survey of virtually the entire coding and non-coding genome of an individual. One of the primary uses of next-generation sequence data is to improve our understanding of the correlation between phenotypes and genotypes (54,55). Genetic studies have shown a very high correspondence between the presence of rare, clinically meaningful mutations and acute disorder states, which can be identified through WES or WGS in both small and large families. For example, Bainbridge et al., (2011) (56) were able to perform WGS of a pair of dizygotic twins to identify a mutation in the sepiapterin reductase gene for dopa-responsive dystonia. Because we have 116 families enriched for affected pairs, we can similarly set up deep sequencing contrasts from targeted enrichment of the 17p11.2-q11.2 region to WES and WGS to find rare and common variants that may play a role in fibromyalgia etiology. The rapid advancement of these technologies will enable us to explore many hypotheses simultaneously.
Several limitations of this study should be considered. First, the findings should be considered preliminary based on the sample size of 203 affected sibling pairs. However, the identified loci contain two biological candidate genes including SLC6A4 and TRPV2, suggesting that these loci deserve further attention in future studies. Second, there is heterogeneity in the phenotypic expression of fibromyalgia. Future studies will examine the effect of phenotypic covariates in the analysis. Third, some genes are transcribed based on their parental origin, such that only one of the two alleles (either paternal or maternal) is expressed in a process called genomic imprinting (57). To dissect parent-of-origin effects, parental genotypes and preferably collection of extended families is required. In our current study, the family structures were not suitable for this analysis. However, future fibromyalgia models will need to consider parent-of-origin effects for the candidate genes.
In conclusion, we detected genome wide suggestive linkage to the chromosome 17p11.2-q11.2 region in a cohort of multi-case families from the Fibromyalgia Family Study. Further comprehensive sequencing analyses of the 17p11.2-q11.2 chromosome region in multi-case families are warranted to identify potential causal genetic risk variants for fibromyalgia.
Figure 2.

Detailed multipoint linkage results for Fibromyalgia on Chromosome 17. Horizontal line is drawn at P = 0.00074, the Lander and Kruglyak’s criterion for suggestive linkage.
Acknowledgments
The authors would like to thank the following members of the Scientific Advisory Committee who provided valuable advice during the trial: Robert Bennett, M.D., Harvey Moldofsky, M.D., Daniel J. Clauw, M.D., and Christopher Amos, Ph.D. We also thank the staff at the NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases for their support. We would like to acknowledge our research staff at each of the investigator sites. Dr. Jinbo Fan was supported in part by NIH/NCRR CTSA KL2RR024990. The program package S.A.G.E. is supported by a U.S. Public Health Service Resource Grant (RR03655) from the National Center for Research Resources. Finally, we thank the patients and their family members for their participation in this study.
Supported by NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases grant N01-AR-9-2235
REFERENCES
- 1.Wolfe F, Smythe HA, Yunus MB, Bennett RM, Bombardier C, Goldenberg DL, et al. The American College of Rheumatology 1990 criteria for the classification of fibromyalgia: report of the multicenter criteria committee. Arthritis Rheum. 1990;33:160–72. doi: 10.1002/art.1780330203. [DOI] [PubMed] [Google Scholar]
- 2.Wolfe F, Ross K, Anderson J, Russell IJ, Hebert L. The prevalence and characteristics of fibromyalgia in the general population. Arthritis Rheum. 1995;38:19–28. doi: 10.1002/art.1780380104. [DOI] [PubMed] [Google Scholar]
- 3.Russell IJ, Larson AA. Neurophysiopathogenesis of fibromyalgia syndrome: a unified hypothesis. Rheum Dis Clin Norht Am. 2009;35:421–35. doi: 10.1016/j.rdc.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Henriksson KG. Fibromyalgia--from syndrome to disease. Overview of pathogenetic mechanisms. J Rehabil Med. 2003;41(Suppl):89–94. doi: 10.1080/16501960310010215. [DOI] [PubMed] [Google Scholar]
- 5.Ablin J, Neumann L, Buskila D. Pathogenesis of fibromyalgia - a review. Joint Bone Spine. 2008 May;75:273–9. doi: 10.1016/j.jbspin.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Arnold LM, Hudson JI, Hess EV, Ware AE, Fritz DA, Auchenbach MB, et al. Family study of fibromyalgia. Arthritis Rheum. 2004;50:944–52. doi: 10.1002/art.20042. [DOI] [PubMed] [Google Scholar]
- 7.Pellegrino MJ, Waylonis GW, Sommer A. Familial occurrence of primary fibromyalgia. Arch Phys Med Rehabil. 1989;70:61–3. [PubMed] [Google Scholar]
- 8.Buskila D, Neumann L, Hazanov I, Carmi R. Familial aggregation in the fibromyalgia syndrome. Semin Arthritis Rheum. 1996;26:605–611. doi: 10.1016/s0049-0172(96)80011-4. [DOI] [PubMed] [Google Scholar]
- 9.Buskila D, Neumann L. Fibromyalgia syndrome (FM) and nonarticular tenderness in relatives of patients with FM. J Rheumatol. 1997;24:941–44. [PubMed] [Google Scholar]
- 10.Arnold LM, Hudson JI, Keck PE., Jr. Causes of familial aggregation of fibromyalgia, comment on the article by Arnold et al. [letter] Arthritis Rheum. 2004;50:3059–60. doi: 10.1002/art.20652. [DOI] [PubMed] [Google Scholar]
- 11.Ablin JN, Cohen H, Buskila D. Mechanisms of Disease: genetics of fibromyalgia. Nat Clin Pract Rheumatol. 2006;12:671–8. doi: 10.1038/ncprheum0349. [DOI] [PubMed] [Google Scholar]
- 12.Lee YH, Choi SJ, Ji JD, Song GG. Candidate gene studies of fibromyalgia: a systematic review and meta-analysis. Rheumatol Int. 2012;32:417–26. doi: 10.1007/s00296-010-1678-9. [DOI] [PubMed] [Google Scholar]
- 13.Smith SB, Maixner DW, Fillingim RB, Slade G, Gracely RH, Ambrose K, et al. Large candidate gene association study reveals genetic risk factors and therapeutic targets for fibromyalgia. Arthritis Rheum. 2012;64:584–93. doi: 10.1002/art.33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hindorff LA, Junkins HA, Manolio TA. [Accessed March 10, 2012];NHGRI Catalog of published genomewide association studies. at http://www.genome.gov/gwastudies.
- 15.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;7265:747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166–76. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- 17.Curtis D. Assessing the contribution family data can make to case-control studies of rare variants. Ann Hum Genet. 2011;75:630–8. doi: 10.1111/j.1469-1809.2011.00660.x. [DOI] [PubMed] [Google Scholar]
- 18.Shi G, Simino J, Rao DC. Enriching rare variants using family-specific linkage information. BMC Proc. 2011;5(Suppl 9):S82. doi: 10.1186/1753-6561-5-S9-S82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey-Wilson JE, Wilson AF. Linkage analysis in the next-generation sequencing era. Hum Hered. 2011;72:228–36. doi: 10.1159/000334381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gagnon F, Roslin NM, Lemire M. Successful identification of rare variants using oligogenic segregation analysis as a prioritizing tool for whole-exome sequencing studies. BMC Proc. 2011;5(Suppl 9):S11. doi: 10.1186/1753-6561-5-S9-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ott J, Kamatani Y, Lathrop M. Family-based designs for genome-wide association studies. Nat Rev Genet. 2011;12:465–74. doi: 10.1038/nrg2989. [DOI] [PubMed] [Google Scholar]
- 22.Zhu X, Feng T, Li Y, Lu Q, Elston RC. Detecting rare variants for complex traits using family and unrelated data. Genet Epidemiol. 2010;34:171–87. doi: 10.1002/gepi.20449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kazma R, Hoffmann TJ, Witte JS. Use of principal components to aggregate rare variants in case-control and family-based association studies in the presence of multiple covariates. BMC Proc. 2011;5(Suppl 9):S29. doi: 10.1186/1753-6561-5-S9-S29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin P, Hamm M, Hartz S, Zhang Z, Rice JP. Challenges and directions: an analysis of Genetic Analysis Workshop 17 data by collapsing rare variants within family data. BMC Proc. 2011;5(Suppl 9):S30. doi: 10.1186/1753-6561-5-S9-S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olson JM, Cordell HJ. Ascertainment bias in the estimation of sibling genetic risk parameters. Genet Epidemiol. 2000;18:217–35. doi: 10.1002/(SICI)1098-2272(200003)18:3<217::AID-GEPI3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 26.Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58:26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schick JH, Iyengar SK, Klein BE, Klein R, Reading K, Liptak R, et al. A whole-genome screen of a quantitative trait of age-related maculopathy in sibships from the Beaver Dam Eye Study. Am J Hum Genet. 2003;72:1412–24. doi: 10.1086/375500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broman KW, Murray JC, Sheffield VC, White RL, Weber JL. Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet. 1998;63:861–69. doi: 10.1086/302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.S.A.G.E. Statistical Analysis for Genetic Epidemiology. Version 6.1.0 A computer software package available through the Department of Epidemiology and Biostatistics at Case Western Reserve University; Cleveland, Ohio: 2010. [Google Scholar]
- 30.Olson JM. A general conditional-logistic model for affected-relative-pair linkage studies. Am J Hum Genet. 1999;65:1760–9. doi: 10.1086/302662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elston RC, Buxbaum S, Jacobs KB, Olson JM. Haseman and Elston revisited. Genet Epidemiol. 2000;19:1–17. doi: 10.1002/1098-2272(200007)19:1<1::AID-GEPI1>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 32.Shete S, Jacobs KB, Elston RC. Adding further power to the Haseman and Elston method for detecting linkage in larger sibships: weighting sums and differences. Hum Hered. 2003;55:79–85. doi: 10.1159/000072312. [DOI] [PubMed] [Google Scholar]
- 33.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–7. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 34.Nyholt DR. All LODs are not created equal. Am J Hum Genet. 2000;67:282–288. doi: 10.1086/303029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altmüller J, Palmer LJ, Fischer G, Scherb H, Wjst M. Genomewide scans of complex human diseases: True linkage is hard to find. Am J Hum Gene. 2001;69:936–950. doi: 10.1086/324069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Offenbaecher M, Bondy B, De Jonge S, Glatzeder K, Kruger M, Schoeps P, et al. Possible association of fibromyalgia with a polymorphism in the serotonin transporter gene regulatory region. Arthritis Rheum. 1989;42:2482–88. doi: 10.1002/1529-0131(199911)42:11<2482::AID-ANR27>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 37.Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- 38.Yunus MB, Dailey JW, Aldag JC, Masi AT, Jobe PC. Plasma tryptophan and other amino acids in primary fibromyalgia: a controlled study. J Rheumatol. 1992;19:90–4. [PubMed] [Google Scholar]
- 39.Wolfe F, Russell IJ, Vipraio G, Ross K, Anderson J. Serotonin levels, pain threshold, and fibromyalgia symptoms in the general population. J Rheumatol. 1997;24:555–9. [PubMed] [Google Scholar]
- 40.Russell IJ, Vaeroy H, Javors M, Nyberg F. Cerebrospinal fluid biogenic amine metabolites in fibromyalgia/fibrositis syndrome and rheumatoid arthritis. Arthritis Rheum. 1992;35:550–6. doi: 10.1002/art.1780350509. [DOI] [PubMed] [Google Scholar]
- 41.Russell IJ, Michalek JE, Vipraio GA, Fletcher EM, Javors MA, Bowden CA. Platelet 3H-imipramine uptake receptor density and serum serotonin levels in patients with fibromyalgia/fibrositis syndrome. J Rheumatol. 1992;19:104–9. [PubMed] [Google Scholar]
- 42.Ebstein RP, Buskila D, Cohen H, Neumann L. An association between FMS and the serotonin transporter promoter region (5-HTTLPR) polymorphism and relationship to anxiety-related personality traits. Am J Med Genet. 2001;105:627. doi: 10.1002/art.10103. [DOI] [PubMed] [Google Scholar]
- 43.Cohen H, Buskila D, Neumann L, Ebstein RP. Confirmation of an association between fibromyalgia and serotonin transporter promoter region (5-HTTLPR) polymorphism, and relationship to anxiety-related personality traits. Arthritis Rheum. 2002;46:845–7. doi: 10.1002/art.10103. [DOI] [PubMed] [Google Scholar]
- 44.Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–31. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]
- 45.Gursoy S. Absence of association of the serotonin transporter gene polymorphism with the mentally healthy subset of fibromyalgia patients. Clin Rheumatol. 2002;21:194–97. doi: 10.1007/s10067-002-8284-5. [DOI] [PubMed] [Google Scholar]
- 46.Levine JD, Alessandri-Haber N. TRP channels: targets for the relief of pain. Biochimica et Biophysica Acta. 2007;1772:989–1003. doi: 10.1016/j.bbadis.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 47.Mandadi S, Roufogalis BD. ThermaTRP channels in nociceptors: taking a lead from capsaicin receptor TRPV1. Curr Neuropharmacol. 2008;6:21–38. doi: 10.2174/157015908783769680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Broad LM, Mogg AJ, Beattie RE, Ogden AM, Blanco MJ, Bleakman D. TRP channels as emerging targets for pain therapeutics. Expert Opin Ther Targets. 2009;13:69–81. doi: 10.1517/14728220802616620. [DOI] [PubMed] [Google Scholar]
- 49.Bondy B, Speath M, Offenbacher M, Glatzeder K, Stratz T, Schwarz M, et al. The T102C polymorphism of the 5-HT2A-receptor gene in fibromyalgia. Neurobiol Dis. 1999;6:433–39. doi: 10.1006/nbdi.1999.0262. [DOI] [PubMed] [Google Scholar]
- 50.Martinez-Jauand M, Sitges C, Rodriguez V, Picornell A, Ramon M, Buskila D, et al. Pain sensitivity in fibromyalgia is associated with catechol-Omethyltransferase (COMT) gene. Eur J Pain. 2012 Apr 24; doi: 10.1002/j.1532-2149.2012.00153.x. doi: 10.1002/j.1532-2149.2012.00153.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 51.Buskila D, Cohen H, Neumann L, Ebstein RP. An association between fibromyalgia and the dopamine D4 receptor exon III repeat polymorphism and relationship to novelty seeking personality traits. Mol Psychiatry. 2004;9:730–1. doi: 10.1038/sj.mp.4001506. [DOI] [PubMed] [Google Scholar]
- 52.Burda CD, Cox FR, Osborne P. Histocompatibility antigens in the fibrositis (fibromyalgia) syndrome. Clin Exper Rheumatol. 1986;4:355–57. [PubMed] [Google Scholar]
- 53.Yunus MB, Khan MA, Rawlings KK, Green JR, Olson JM, Shah S. A genetic study of multicase families with fibromyalgia syndrome by HLA linkage analysis. J Rheumatol. 1999;26:408–411. [PubMed] [Google Scholar]
- 54.Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 55.Bick D, Dimmock D. Whole exome and whole genome sequencing. Curr Opin Pediatr. 2011;23:594–600. doi: 10.1097/MOP.0b013e32834b20ec. [DOI] [PubMed] [Google Scholar]
- 56.Bainbridge MN, Wiszniewski W, Murdock DR, Friedman J, Gonzaga-Jauregui C, Newsham I, et al. Whole-genome sequencing for optimized patient management. Sci Transl Med. 2011;3:87re3. doi: 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Falls JG, Pulford DJ, Wylie AA, Jirtle RL. Genomic imprinting: Implications for human disease. Am J Pathology. 1999;154:635–47. doi: 10.1016/S0002-9440(10)65309-6. [DOI] [PMC free article] [PubMed] [Google Scholar]