

Summary
Purpose
Epilepsy is a complex disease characterized by a predisposition toward seizures. There are numerous barriers to the successful treatment of epilepsy. For instance, current anti-epileptic drugs have adverse side effects and variable efficacies. Further, the pathophysiological basis of epilepsy remains largely elusive. Therefore, investigating novel genes and biological processes underlying epilepsy may provide valuable insight and enable the development of new therapeutic agents. We previously identified methylglyoxal (MG) as an endogenous GABAA receptor agonist. Here, we investigated the role of MG and its catabolic enzyme, glyoxalase 1 (GLO1), in seizures.
Methods
We pre-treated mice with MG before seizure induction with picrotoxin or pilocarpine and then assessed seizures behaviorally or by electroencephalography (EEG). We then investigated the role of GLO1 in seizures by treating mice with a pharmacological inhibitor of GLO1 before seizure induction with pilocarpine and measured subsequent seizure phenotypes. Next, we explored the genetic relationship between Glo1 expression and seizures. We analyzed seizure phenotypes among BXD recombinant inbred (RI) mice with differential Glo1 expression. Lastly, we investigated a causal role for Glo1 in seizures by administering pilocarpine to transgenic (Tg) mice that overexpress Glo1.
Key Findings
Pre-treatment with MG attenuated pharmacologically-induced seizures at both the behavioral and EEG levels. GLO1 inhibition, which increases MG concentration in vivo, also attenuated seizures. Among BXD RI mice, high Glo1 expression was correlated with increased seizure susceptibility. Tg mice overexpressing Glo1 displayed reduced MG concentration in the brain and increased seizure severity.
Significance
These data identify MG as an endogenous regulator of seizures. Similarly, inhibition of GLO1 attenuates seizures, suggesting that this may be a novel therapeutic approach for epilepsy. Furthermore, this system may represent an endogenous negative feedback loop whereby high metabolic activity increases inhibitory tone via local accumulation of MG. Finally, Glo1 may contribute to the genetic architecture of epilepsy, as Glo1 expression regulates both MG concentration and seizure severity.
Keywords: seizure, pilocarpine, picrotoxin, GABAA receptors, methylglyoxal, glyoxalase 1
Introduction
Epileptic seizures are transient interruptions of brain function due to abnormal (i.e. excessive or synchronous) neuronal activity (Fisher et al., 2005). Epilepsy is a neurological disorder characterized by a predisposition toward seizures (Fisher et al., 2005). It is not a single disease, but rather a group of disorders that share seizures as a common manifestation (Noe, 2011). Epilepsy is a complex trait with multiple genetic, non-genetic, and interacting causes (Mulley et al., 2005). This complexity poses challenges for identifying genes underlying epilepsy and for developing effective therapies. Current anti-epileptic drugs (AEDs) are not effective in all patients or types of epilepsy, and their efficacies are compromised by adverse effects in many patients (Brodie et al., 2011, Perucca & Tomson, 2011, Rossetti & Lowenstein, 2011). Identifying novel genes and biological pathways underlying epilepsy will provide valuable insight into its pathogenesis as well as therapeutic targets.
In the present study, we investigated methylglyoxal (MG) as a novel inhibitor of epileptic seizures. MG is an endogenous byproduct of glycolysis that is generated by the non-enzymatic fragmentation of dihydroxyacetone phosphate and glyceraldehyde-3-phosphate (Thornalley, 1993). Our recent work demonstrated that physiological concentrations of MG activate GABAA receptors (Distler et al., 2012; Distler and Palmer 2012). GABAA receptors are the primary regulators of fast inhibitory synaptic transmission in the central nervous system (Macdonald et al., 2010). Abundant clinical and experimental evidence has demonstrated that mutations in GABAA receptor-encoding genes can perturb GABAA receptor signaling and cause epileptic seizures (Briggs & Galanopoulou, 2011, Galanopoulou, 2010). Furthermore, several well-established AEDs activate or potentiate GABAA receptors, including benzodiazepines and barbiturates (Perucca & Tomson, 2011). Given the prominent role of GABAA receptors in epilepsy and MG’s action at GABAA receptors, we hypothesized that MG would protect against epileptic seizures. MG is metabolized by glyoxalase 1 (GLO1); therefore, we predicted that differences in Glo1 expression and activity would affect seizure susceptibility through regulating endogenous MG concentrations in the brain.
In the present study, we investigated whether direct administration of MG would inhibit epileptic seizures induced by the GABAA receptor antagonist, picrotoxin, and the muscarinic cholinergic agonist, pilocarpine. We also investigated whether changes in Glo1 expression or activity would affect seizure susceptibility and severity. Inhibition of GLO1 may potentiate an endogenous negative feedback loop, whereby high metabolic activity could increase inhibitory tone via GABAA receptors. As an anti-epileptic therapy, GLO1 inhibition might have a different, perhaps more favorable side effect profile than current AEDs.
Methods
Animals
All studies were approved by the IACUC at The University of Chicago or Emory University. Studies with MG and S-bromobenzylglutathione cyclopentyl diester (BrBzGCp2) were performed with C57BL/6J (B6) mice purchased from Jackson Laboratory (Bar Harbor, ME). Transgenic (Tg) mice were generated on an FVB/NJ (FVB) background as previously described (Distler et al., 2012) and were tested in parallel with their wild type (WT) littermates. Mice from two FVB Tg lines were tested and pooled into a common Tg group. All studies used male mice in order to reduce variability arising from the effects of the estrous cycle on seizure phenotypes (Foldvary-Schaefer et al., 2004, Scharfman & Gray, 2007, Veliskova, 2007).
Reagents
Picrotoxin (item P1675), pilocarpine hydrochloride (item P6503), and methylglyoxal (item M0252) were obtained from Sigma-Aldrich (St. Louis, MO). Atropine methylnitrate (item 417-102A) was obtained from Chem Service (West Chester, PA). BrBzGCp2 was synthesized at the Beckman Research Institute of the City of Hope, Duarte, CA as previously described (Distler et al., 2012).
Drug administration
For pre-treatment, MG (50 or 200 mg/kg at a volume of 10 ml/kg body weight) or vehicle (0.9% saline at a volume of 10 ml/kg body weight) was administered by intra-peritoneal (i.p.) injection 10 minutes before the seizure-inducing agent. For treatment after seizure onset, MG (200 mg/kg at a volume of 10 ml/g body weight) or vehicle (0.9% saline at a volume of 10 ml/kg body weight) was administered by i.p. injection 10 minutes after seizure onset. For GLO1 inhibition, BrBzGCp2 (50 mg/kg at a volume of 5 ml/kg body weight) or vehicle (8% DMSO and 18% Tween-80 at a volume of 5 ml/kg body weight) was administered by i.p. injection 2 hours before seizure induction.
Seizure induction for behavioral scoring
For behavioral analysis of picrotoxin-induced seizures, 5 mg/kg of picrotoxin in 0.9% saline was administered by i.p. injection at a volume of 10 ml/kg body weight. Seizures were scored for 1 hour after picrotoxin administration. For behavioral analysis of pilocarpine-induced seizures, mice were pre-treated with atropine (5 mg/kg in 0.9% saline at a volume of 10 ml/kg body weight) by i.p. injection in order to reduce the peripheral effects of pilocarpine. Thirty minutes after atropine administration, pilocarpine was administered by i.p. injection at a volume of 10 ml/kg body weight. B6 mice were treated with 250 mg/kg pilocarpine in 0.9% saline, and FVB mice (WT and Tg) were treated with 300 mg/kg pilocarpine in 0.9% saline. The B6 and FVB mice were given slightly different doses of pilocarpine (250 mg/kg and 300 mg/kg, respectively) due to strain differences in seizure susceptibility (Schauwecker, 2011, Schauwecker, 2012). Seizures were scored for 1.5 hours after pilocarpine administration.
Picrotoxin-induced seizures were scored as the presence of generalized convulsions as well as the latency to and duration of generalized convulsions.
Pilocarpine-induced seizures were scored as previously reported (Winawer et al., 2011):
Stage 1: Immobility/lying low.
Stage 2: Partial (limbic) seizures; non-continuous twitching/tremor/shaking of tail/head/body/limbs, forelimb and/or tail extension, rigid posture, repetitive movements, head bobbing.
Stage 3: Partial status epilepticus; continuous tremor/clonic seizures of body and tail while retaining posture.
Stage 4: Generalized seizures; rearing/hyperexcitability/running/falling, tonic extension/clonic seizures with loss of posture.
Stage 5: Generalized status epilepticus (continuous stage 4 seizures) resulting in death.
Seizure induction for EEG recording
Electroencephalographic (EEG) recording of mice treated with picrotoxin was performed at Emory University. Under isoflurane anesthesia, mice were surgically implanted sub-durally with four sterile stainless-steel screw electrodes for EEG recordings, and fine-wire electrodes were inserted into the neck muscle for electromyography (EMG), as previously described (Dutton et al., 2012, Martin et al., 2010, Martin et al., 2007, Papale et al., 2010). After a minimum of 10 days of recovery, each mouse was placed into a Plexiglas box (15 × 15 × 15 cm) and was attached to the EEG cable. Digital video/EEG/EMG recordings were amplified, filtered (0.3–35 Hz bandpass for the EEG and 10–70 Hz bandpass for the EMG), and digitized at a sampling rate of 200 Hz by Stellate Harmonie amplifier and software (Natus Medical, Inc.). The mice were injected i.p. with 7 mg/kg picrotoxin or vehicle (0.9% saline) at a volume of 10 ml/kg body weight. This dose of picrotoxin (7 mg/kg) was slightly higher than that used for behavioral scoring of seizures (5 mg/kg) at the University of Chicago, because it maximized seizure outcomes by EEG under the experimental conditions at Emory University. EEG and EMG data were collected for 1 hour. Seizures were characterized by the onset of sharp, highly synchronous spike discharges that increased in frequency and achieved amplitudes that were at least two times the background amplitude with detection in all cortical EEG channels and attenuation of the background rhythm. Simultaneous video recordings allowed the behavior of the mouse to be observed during the seizure. These data were used to assess latency to seizure onset, seizure duration, and number of seizures. Three mice failed to show seizures by EEG (2 pre-treated with vehicle and 1 pre-treated with MG) and were excluded from data analysis.
Gene network
Data on seizure susceptibility and Glo1 expression in BXD recombinant inbred (RI) lines were obtained from and analyzed using WebQTL at www.genenetwork.org (Wang et al., 2003). The record ID for hippocampal Glo1 expression was 1424109_a_at from the Hippocampus Consortium M430v2 (Jun06) PDNN database. The record ID for seizure susceptibility was 10388, representing data reported by McCall and Frierson (McCall & Frierson, 1981). Data were retrieved on January 28, 2012.
Measurement of MG concentration
MG concentration was measured in the brains of WT and Tg FVB mice by high-performance liquid chromatography (HPLC) as previously described (see supplemental data in Distler et al., 2012).
Statistical analyses
All statistical analyses were performed using StatView for Windows (SAS Institute, Inc.). All behavioral and EEG seizure outcomes were assessed using non-parametric tests, because they were not normally distributed. Two-group comparisons were made using Mann-Whitney U tests. Three-group comparisons were made using Kruskal-Wallis tests, and post-hoc comparisons were made using Mann-Whitney U tests. The relationship between Glo1 expression and seizure phenotypes in BXD recombinant inbred (RI) lines was assessed using Pearson correlations.
Results
MG treatment reduces the severity and duration of picrotoxin-induced seizures
First, we investigated whether MG could prevent or attenuate seizures by administering exogenous MG (50 and 200 mg/kg) or vehicle to mice before seizure induction. We previously demonstrated that this treatment dose-dependently increases MG concentration in the brain (Distler et al., 2012). Treatment with 50 mg/kg and 200 mg/kg MG is expected to increase the concentration of MG in the brain by approximately 16% and 43%, respectively, and does not cause cytotoxicty (Distler et al., 2012). After pre-treatment with MG, we induced seizures using picrotoxin. Picrotoxin is a GABAA receptor antagonist, which we selected based on its ability to induce seizures in mice (Fisher, 1989) and MG’s role as a GABAA receptor agonist (Distler et al., 2012). Pre-treatment with MG dose-dependently attenuated generalized convulsions induced by 5 mg/kg picrotoxin. Specifically, MG treatment delayed seizure onset (Figure 1a), reduced seizure duration (Figure 1b), and reduced the percentage of animals undergoing generalized convulsions (Figure 1c) at the behavioral level.
Figure 1. Exogenous MG attenuates picrotoxin-induced seizures at the behavioral level.
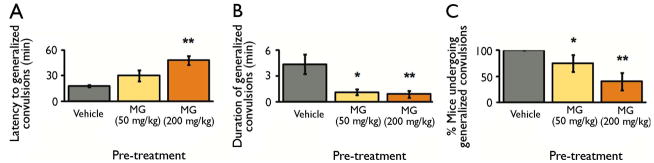
B6 mice were pre-treated with vehicle or MG (50 and 200 mg/kg) prior to seizure induction with picrotoxin (5 mg/kg). a, MG pre-treatment increased the latency to first generalized convulsion. P = 0.0002 by Kruskal-Wallis test. b, MG pre-treatment reduced the duration of generalized convulsions. P = 0.0025 by Kruskal-Wallis test. c, MG pre-treatment reduced the percentage of animals undergoing generalized convulsions. P = 0.0033 by Kruskal-Wallis test.
Data are mean ± SEM. n = 16 vehicle, 8 MG (50 mg/kg), 10 MG (200 mg/kg). * P < 0.05, ** P < 0.005 versus vehicle-treated group as determined by Mann-Whitney U tests.
In order to validate these behavioral data, we assessed seizure activity by EEG analysis of mice treated with 7 mg/kg picrotoxin. Again, pre-treatment with 200 mg/kg MG delayed seizure onset (Figure 2a), reduced seizure duration (Figure 2b), and reduced the number of seizures (Figure 2c) as measured by EEG. Representative EEG traces are shown in Figure 2d. Thus, MG attenuates seizures induced by GABAA receptor blockade at both the behavioral and EEG level.
Figure 2. Exogenous MG attenuates picrotoxin-induced seizures as assessed by EEG.

B6 mice were pre-treated with vehicle or MG (200 mg/kg) prior to seizure induction with picrotoxin (7 mg/kg). a, MG pre-treatment increased the latency to first EEG-confirmed seizure. P = 0.036 by Mann-Whitney U test. b, MG pre-treatment reduced the duration of EEG-confirmed seizures. P = 0.014 by Mann-Whitney U test. c, MG pre-treatment reduced the number of EEG-confirmed seizures. P = 0.028 by Mann-Whitney U test. d, Representative EEG recordings of a mouse during baseline period (prior to any injections), after acute saline and picrotoxin (7 mg/kg) injections, or after acute MG (200 mg/kg) and picrotoxin (7 mg/kg) injections. As shown in 2b, MG pre-treatment reduced the duration of EEG-confirmed generalized seizures (high amplitude and high frequency spike discharges located between arrows). Seizures are associated with high muscle activity, as indicated by the electromyography (EMG) trace. Note: animals from both groups (MG and saline) showed myoclonic jerks associated with high amplitude single spikes (▼). EEG traces correspond to four differential recordings from each of our four subdural electrodes (EEG1 and EEG2 (right cortical hemisphere), EEG3 and EEG4 (left cortical hemisphere)). EMG activity was recorded using two fine wires placed into the neck muscle. Calibration mark: 500 μV/mm and 1 second.
Data are mean ± SEM. n = 4 vehicle, 5 MG (200 mg/kg). * P < 0.05 versus vehicle-treated group as determined by Mann-Whitney U tests.
MG treatment reduces the severity and duration of pilocarpine-induced seizures
We next investigated MG’s anti-seizure effects in a mechanistically distinct seizure model. We induced seizures using pilocarpine, a muscarinic cholinergic agonist that induces severe, continuous limbic seizures after acute administration (Curia et al., 2008). We pre-treated mice with MG (50 mg/kg and 200 mg/kg) or vehicle and then induced seizures with 250 mg/kg pilocarpine. This dose of pilocarpine induced partial status epilepticus (stage 3 seizures), but not generalized status epilepticus, in vehicle- and MG-treated mice. Pre-treatment with MG (50 and 200 mg/kg) dose-dependently delayed acute seizure onset (Figure 3a), reduced seizure duration (Figure 3b), and reduced the highest seizure stage reached (Figure 3c) in response to pilocarpine.
Figure 3. Exogenous MG attenuates pilocarpine-induced seizures.

a–c, B6 mice were pre-treated with vehicle or MG (50 and 200 mg/kg) prior to seizure induction with pilocarpine (250 mg/kg). n = 19 vehicle, 12 MG (50 mg/kg), 8 MG (200 mg/kg). a, MG pre-treatment increased the latency to reach stage 3. P = 0.0033 by Kruskal-Wallis test. b, MG pre-treatment reduced the time spent in stage 3. P < 0.0001 by Kruskal-Wallis test. c, MG pre-treatment reduced the highest seizure stage reached. P = 0.0015 by Kruskal-Wallis test. d, Seizures were induced in B6 mice with pilocarpine (250 mg/kg); 10 minutes later, mice were treated with vehicle or MG (200 mg/kg). MG treatment reduced time spent in stage 3. n = 6 per group.
Data are mean ± SEM. * P < 0.05, ** P < 0.005 versus vehicle-treated group as determined by Mann-Whitney U tests.
We then investigated whether MG could stop or reduce the severity of ongoing seizures. We induced seizures with pilocarpine and then administered MG (200 mg/kg) 10 minutes after seizure induction. We selected this time point, because vehicle-treated mice first reach stage 3 seizures approximately 10 minutes after pilocarpine administration (Figure 3a). MG treatment reduced seizure duration in mice undergoing pilocarpine-induced seizures (Figures 3d). Neither vehicle- nor MG-treated mice returned to normal behavior during the observation period. Therefore, we conclude that this dose of MG reduced time spent in partial status epilepticus but did not eliminate seizure activity altogether. We did not explore higher doses of MG, because their potential cytotoxic effects could confound the interpretation of the results.
GLO1 inhibition reduces seizure severity
Next, we investigated the anti-seizure effects of BrBzGCp2 (Thornalley et al., 1996, Vince et al., 1971), a pharmacological inhibitor of GLO1. We previously demonstrated that administration of BrBzGCp2 increased MG concentration in the brain by approximately 20% (Distler et al., 2012). We pre-treated mice with BrBzGCp2 or vehicle two hours before administration of pilocarpine. Mice treated with BrBzGCp2 had shorter seizure durations than those treated with vehicle (Figure 4). However, GLO1 inhibition did not significantly affect seizure latency or highest seizure stage reached (data not shown). These data demonstrate that increasing endogenous levels of MG reduces seizure duration. Further, they indicate that GLO1 inhibition is a potential therapeutic intervention for seizures.
Figure 4. GLO1 inhibition attenuates pilocarpine-induced seizures.
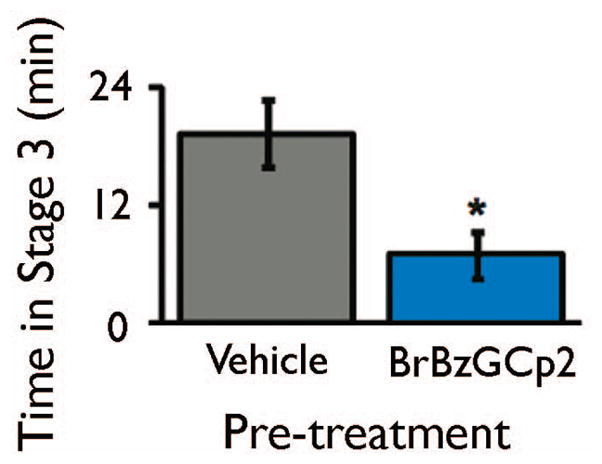
B6 mice were treated with vehicle or BrBzGCp2 (50 mg/kg). Two hours later, seizures were induced by pilocarpine (250 mg/kg). Pre-treatment with BrBzGCp2 reduced time spent in stage 3. Data are mean ± SEM. n = 12 vehicle, 8 BrBzGCp2. * P < 0.05 as determined by Mann-Whitney U test.
Differential Glo1 expression affects seizure susceptibility and severity
Finally, we explored whether Glo1 expression affects epileptic seizures. This may provide a link between the complex genetic architecture underlying epilepsy and MG, a novel mediator of seizures in mice. Here, we focused on Glo1, a gene that negatively regulates MG concentration in the brain (Distler et al., 2012). We hypothesized that mice with increased Glo1 expression would display increased seizure susceptibility and severity.
We utilized data from BXD RI lines (Peirce et al., 2004, Shifman et al., 2006, Williams et al., 2001), which are derived from intercrosses between B6 and DBA/2J (D2) inbred strains. The D2 strain carries a genomic duplication of Glo1 on chromosome 17, while the B6 strain does not; BXD lines that inherit the duplication show an approximately twofold increase in Glo1 expression (Williams et al., 2009). We used tools at Gene Network (www.genenetwork.org) to assess the correlation between Glo1 expression and published seizure phenotypes in BXD RI lines. A locus on chromosome 17 was significantly associated with seizure susceptibility at high atmospheric pressure among BXD RI lines (McCall & Frierson, 1981, Plomin et al., 1991). In this model, mice exposed to increasing pressure in a helium-oxygen atmosphere suffer from progressive convulsive seizures (Lever et al., 1971, Mansfield et al., 1980, McCall & Frierson, 1981). This model may be clinically relevant, because patients with epilepsy have an increased susceptibility to seizures at high atmospheric pressure (Doherty et al., 2007). We found that the locus for susceptibility to seizures at high atmospheric pressure co-localized with that of Glo1 expression (Figures 5a and 5b), which we previously attributed to the Glo1 duplication (Williams et al., 2009). Indeed, BXD RI lines with the Glo1 duplication displayed a significant reduction in seizure threshold compared to those without the duplication (Figure 5c). Further, there was a significant inverse correlation between Glo1 expression and seizure threshold (Figure 5d). This is consistent with our hypothesis that increased Glo1 expression increases seizure susceptibility. Thus, naturally occurring differences in Glo1 expression may regulate MG concentration and thus seizure sensitivity in mice.
Figure 5. Glo1 expression is associated with seizure susceptibility.

a, The position along the mouse genome is on the x-axis, and the logarithm of odds (LOD) score is on the y-axis. Pink and blue horizontal lines indicate genome-wide significance thresholds for Glo1 expression and seizure threshold, respectively. Hippocampal Glo1 mRNA expression was assessed by microarray (record ID 1424109_a_at). Seizure threshold data were obtained using record ID 10388 corresponding to data from McCall et al. (McCall & Frierson, 1981). The figure was generated by www.genenetwork.org. b, Data from (a) were replotted for chromosome 17 only. c, Seizure susceptibility in BXD RI lines with and without the Glo1 duplication based on their genotype at SNP rs3145545. d, Association between Glo1 expression in the hippocampus (record ID 1424109_a_at) and seizure threshold (record ID 10388). Pearson correlation: r2 = 0.59, P = 5.42 × 10−5.
Data were obtained from www.genenetwork.org on January 28, 2012 using the indicated record IDs. Glo1 expression was measured as previously described (Overall et al., 2009), and each unit represents a twofold difference in expression level. For seizure threshold, units represent the pressure at which animals seized. For d, black symbols represent BXD RI lines harboring the B6 Glo1 allele (single copy), and gray symbols represent BXD RI lines harboring the D2 Glo1 allele (duplicated) based on their genotype at SNP rs3145545.
BXD RI lines are a convenient population for investigating the effects of differential Glo1 expression on seizure susceptibility. However, there are numerous genetic differences between the lines, making it impossible to establish a causal relationship with a particular variant. To more directly test Glo1’s effect on seizures, we employed Tg mice that overexpress Glo1 (Distler et al., 2012). Tg mice displayed approximately 15% less MG in the brain than WT mice (Figure 6a). We administered pilocarpine (300 mg/kg) to WT and Tg mice and measured subsequent seizure activity. Tg mice displayed a non-significant trend toward reduced latency to first seizure (Figure 6b). Tg mice displayed significantly increased seizure duration and seizure severity compared to WT mice (Figures 6c–d). This suggests that differences in Glo1 expression or activity influence seizures. Thus, Glo1 polymorphisms may contribute to the genetic underpinnings of epilepsy.
Figure 6. Glo1 overexpression exacerbates seizure phenotypes.

a, MG concentration was measured by HPLC in the brains of FVB WT and Tg mice and is reported as relative concentration. n = 7 WT, 7 Tg. b–d, Seizures were induced in FVB WT and Tg mice with pilocarpine (300 mg/kg). n = 13 WT, 8 Tg. There was a non-significant trend for Tg mice to reach stage 4 seizures sooner than WT mice (b). Tg mice spent significantly more time in stage 4 (c) and had more severe seizures (d) than WT mice. Data are mean ± SEM. * P < 0.05 by Mann-Whitney U tests.
Discussion
In the present study, we demonstrated that pre-treatment with MG attenuated picrotoxin-and pilocarpine-induced seizures. MG’s efficacy in two seizure models demonstrates its broad anti-seizure effects across mechanistically distinct types of seizures. This is consistent with MG’s role in the CNS as an agonist at GABAA receptors, which are responsible for mediating neuronal inhibitory tone (Macdonald et al., 2010). At the behavioral level, MG affected three important measures of seizures: latency to first seizure, seizure duration, and seizure severity. For picrotoxin-induced seizures, pre-treatment with MG also reduced the percentage of mice exhibiting convulsive behavior. The behavioral data were corroborated by EEG recordings, demonstrating that MG pre-treatment attenuated EEG-confirmed picrotoxin-induced seizures. These data have important implications. First, they demonstrate that MG, an endogenous GABAA receptor agonist, protects against seizures. Second, they suggest that endogenous levels of MG may mediate seizure phenotypes in vivo. Third, they open the door for further investigation of MG’s therapeutic potential in the treatment of epilepsy.
We also used a GLO1 inhibitor, BrBzGCp2, to investigate the effects of increasing endogenous MG concentration on seizures. We found that pre-treatment with BrBzGCp2 decreased seizure duration. Therefore, our results highlight the potential for GLO1 inhibitors in the pharmacological treatment of epilepsy. This would represent a novel mechanism of action among AEDs. Future studies should explore whether GLO1 inhibition might also protect against brain damage associated with seizures. Current AEDs act mainly through modulating ion channels, including GABAA receptors (Brodie et al., 2011). GLO1 inhibition, on the other hand, would increase MG concentration in proportion with its endogenous production. This might avoid some of the adverse effects caused by traditional AEDs, a possibility that should be investigated in future studies.
MG levels increase under conditions of high metabolic load, positioning MG as an intermediate between metabolic state and neuronal inhibitory tone. Therefore, metabolic interventions that raise endogenous MG levels could be a promising therapeutic approach for epilepsy without the adverse side effects of AEDs (Brodie et al., 2011, Perucca & Tomson, 2011, Rossetti & Lowenstein, 2011). For instance, the ketogenic diet (KD) is a high-fat, low-carbohydrate diet that is administered to patients with epilepsy who do not respond to AEDs (Payne et al., 2011, Rossetti & Lowenstein, 2011). The mechanism by which KD controls seizures is unknown, but it has been hypothesized that the KD might increase MG levels (Beisswenger et al., 2005, Gasior et al., 2007, Hartman et al., 2007, Kalapos, 2007). In addition, other manipulations that increase MG could be investigated as novel methods for controlling seizures.
Finally, we investigated whether genetic polymorphisms regulating MG concentration underlie genetic susceptibility to seizures. Specifically, we examined the effect of Glo1 expression on seizures. We found that that differential expression of Glo1 in BXD RI lines was associated with susceptibility to seizures at high atmospheric pressure. We note that no other seizure phenotype from Gene Network (handling-induced convulsions, pentylenetetrazol -induced seizures, and audiogenic seizures) was significantly associated with Glo1 expression in BXD RI lines. We then used Tg mice overexpressing Glo1 to establish a causal role for Glo1 in increasing seizure susceptibility and severity. Tg mice had reduced MG concentration in the brain and increased susceptibility to pilocarpine-induced seizures (Figure 6). Therefore, we speculate that genetic variants in Glo1 may contribute to epilepsy.
Our previous work established a causal role for Glo1 in increasing anxiety-like behavior by reducing the concentration of MG in the brain (Distler et al., 2012; Distler and Palmer 2012). In those studies we did not observe hypolocomotion (sedation) or ataxia at the 50 mg/kg dose; however, 200 mg/kg produced sedation and a somewhat higher dose (300 mg/kg) produced ataxia. We have not observed sedation or ataxia following any dose of BrBzGCp2. Taken together these data suggest a therapeutic window in which anti-epileptic effects can be achieved without negative side effects like sedation and ataxia. More broadly, the results presented in this paper emphasize the importance of GLO1 and MG in the central nervous system. This pathway plays an important role in neuronal physiology through regulating neuronal inhibitory tone as well as associated pathophysiological conditions, including anxiety and epilepsy. Finally, our results may provide insight into the high comorbidity between epilepsy and anxiety disorders (Beyenburg et al., 2005, LaFrance et al., 2008, Schmidt, 2009, Titlic et al., 2009). There is a 25–50% prevalence of anxiety disorders among patients with epilepsy, which is twice the prevalence among the general population (LaFrance et al., 2008). The prevalence of comorbid anxiety disorders is especially high in epileptic patients who do not respond to AEDs (LaFrance et al., 2008, Schmidt, 2009). This could suggest a convergent pathologic mechanism that is not targeted by current AEDs. Polymorphisms in Glo1 and other genes that affect MG concentration may contribute to both anxiety and epilepsy, providing a common underlying pathway.
In conclusion, the present study demonstrated an important physiological role for MG in reducing seizure susceptibility and severity. Increasing endogenous MG by GLO1 inhibition had a similar effect and may be a useful therapeutic strategy. Finally, polymorphisms in Glo1 that regulate MG concentration may contribute to the genetic underpinnings of epilepsy and comorbid anxiety disorders.
Acknowledgments
This study was funded by the NIH grant R01MH079103 awarded to A.A.P. M.G.D. was supported by the NIH grant T32GM07281. M.R.W. was supported by the NIH grant R01NS061991. The authors thank Dr. P. Elyse Schauwecker for scientific advice and for histological processing of brains of mice treated with methylglyoxal. The authors also thank Mr. Austin Phillips for instruction and advice on the scoring of seizures.
Footnotes
Conflict of Interest: None of the authors has any conflict of interest to disclose
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- Beisswenger BG, Delucia EM, Lapoint N, Sanford RJ, Beisswenger PJ. Ketosis leads to increased methylglyoxal production on the Atkins diet. Ann N Y Acad Sci. 2005;1043:201–210. doi: 10.1196/annals.1333.025. [DOI] [PubMed] [Google Scholar]
- Beyenburg S, Mitchell AJ, Schmidt D, Elger CE, Reuber M. Anxiety in patients with epilepsy: systematic review and suggestions for clinical management. Epilepsy Behav. 2005;7:161–171. doi: 10.1016/j.yebeh.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Briggs SW, Galanopoulou AS. Altered GABA signaling in early life epilepsies. Neural Plast. 2011;2011:527605. doi: 10.1155/2011/527605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie MJ, Covanis A, Gil-Nagel A, Lerche H, Perucca E, Sills GJ, White HS. Antiepileptic drug therapy: does mechanism of action matter? Epilepsy Behav. 2011;21:331–341. doi: 10.1016/j.yebeh.2011.05.025. [DOI] [PubMed] [Google Scholar]
- Curia G, Longo D, Biagini G, Jones RS, Avoli M. The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods. 2008;172:143–157. doi: 10.1016/j.jneumeth.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distler MG, Plant LD, Sokoloff G, Hawk AJ, Aneas I, Wuenschell GE, Termini J, Meredith SC, Nobrega MA, Palmer AA. Glyoxalase 1 increases anxiety by reducing GABAA receptor agonist methylglyoxal. J Clin Invest. 2012;122:2306–2315. doi: 10.1172/JCI61319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distler MG, Palmer AA. Role of Glyoxalase 1 (Glo1) and methylglyoxal (MG) in behavior: recent advances and mechanistic insights. Front Gene. 2012;3:250. doi: 10.3389/fgene.2012.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty MJ, Youn C, Gwinn RP, Haltiner AM. Atmospheric pressure and seizure frequency in the epilepsy unit: preliminary observations. Epilepsia. 2007;48:1764–1767. doi: 10.1111/j.1528-1167.2007.01111.x. [DOI] [PubMed] [Google Scholar]
- Dutton SB, Makinson CD, Papale LA, Shankar A, Balakrishnan B, Nakazawa K, Escayg A. Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol Dis. 2012 doi: 10.1016/j.nbd.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RS. Animal models of the epilepsies. Brain Res Brain Res Rev. 1989;14:245–278. doi: 10.1016/0165-0173(89)90003-9. [DOI] [PubMed] [Google Scholar]
- Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, Engel J., Jr Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE) Epilepsia. 2005;46:470–472. doi: 10.1111/j.0013-9580.2005.66104.x. [DOI] [PubMed] [Google Scholar]
- Foldvary-Schaefer N, Harden C, Herzog A, Falcone T. Hormones and seizures. Cleve Clin J Med. 2004;71(Suppl 2):S11–18. doi: 10.3949/ccjm.71.suppl_2.s11. [DOI] [PubMed] [Google Scholar]
- Galanopoulou AS. Mutations affecting GABAergic signaling in seizures and epilepsy. Pflugers Arch. 2010;460:505–523. doi: 10.1007/s00424-010-0816-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasior M, French A, Joy MT, Tang RS, Hartman AL, Rogawski MA. The anticonvulsant activity of acetone, the major ketone body in the ketogenic diet, is not dependent on its metabolites acetol, 1,2-propanediol, methylglyoxal, or pyruvic acid. Epilepsia. 2007;48:793–800. doi: 10.1111/j.1528-1167.2007.01026.x. [DOI] [PubMed] [Google Scholar]
- Hartman AL, Gasior M, Vining EP, Rogawski MA. The neuropharmacology of the ketogenic diet. Pediatr Neurol. 2007;36:281–292. doi: 10.1016/j.pediatrneurol.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalapos MP. Possible mechanism for the effect of ketogenic diet in cases of uncontrolled seizures. The reconsideration of acetone theory. Med Hypotheses. 2007;68:1382–1388. doi: 10.1016/j.mehy.2006.10.041. [DOI] [PubMed] [Google Scholar]
- LaFrance WC, Jr, Kanner AM, Hermann B. Psychiatric comorbidities in epilepsy. Int Rev Neurobiol. 2008;83:347–383. doi: 10.1016/S0074-7742(08)00020-2. [DOI] [PubMed] [Google Scholar]
- Lever MJ, Miller KW, Paton WD, Smith EB. Pressure reversal of anaesthesia. Nature. 1971;231:368–371. doi: 10.1038/231368a0. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ, Gallagher MJ. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Physiol. 2010;588:1861–1869. doi: 10.1113/jphysiol.2010.186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield W, Brauer RW, Gillen HW, Nash K. Changes in CNS responses to high pressure during maturation of newborn mice. J Appl Physiol. 1980;49:390–397. doi: 10.1152/jappl.1980.49.3.390. [DOI] [PubMed] [Google Scholar]
- Martin MS, Dutt K, Papale LA, Dube CM, Dutton SB, de Haan G, Shankar A, Tufik S, Meisler MH, Baram TZ, Goldin AL, Escayg A. Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J Biol Chem. 2010;285:9823–9834. doi: 10.1074/jbc.M109.078568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet. 2007;16:2892–2899. doi: 10.1093/hmg/ddm248. [DOI] [PubMed] [Google Scholar]
- McCall RD, Frierson D., Jr Evidence that two loci predominantly determine the difference in susceptibility to the high pressure neurologic syndrome type I seizure in mice. Genetics. 1981;99:285–307. doi: 10.1093/genetics/99.2.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulley JC, Scheffer IE, Harkin LA, Berkovic SF, Dibbens LM. Susceptibility genes for complex epilepsy. Hum Mol Genet. 2005;14(Spec No 2):R243–249. doi: 10.1093/hmg/ddi355. [DOI] [PubMed] [Google Scholar]
- Noe KH. Seizures: diagnosis and management in the outpatient setting. Semin Neurol. 2011;31:54–64. doi: 10.1055/s-0031-1271310. [DOI] [PubMed] [Google Scholar]
- Overall RW, Kempermann G, Peirce J, Lu L, Goldowitz D, Gage FH, Goodwin S, Smit AB, Airey DC, Rosen GD, Schalkwyk LC, Sutter TR, Nowakowski RS, Whatley S, Williams RW. Genetics of the hippocampal transcriptome in mouse: a systematic survey and online neurogenomics resource. Front Neurosci. 2009;3:55. doi: 10.3389/neuro.15.003.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale LA, Paul KN, Sawyer NT, Manns JR, Tufik S, Escayg A. Dysfunction of the Scn8a voltage-gated sodium channel alters sleep architecture, reduces diurnal corticosterone levels, and enhances spatial memory. J Biol Chem. 2010;285:16553–16561. doi: 10.1074/jbc.M109.090084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne NE, Cross JH, Sander JW, Sisodiya SM. The ketogenic and related diets in adolescents and adults--a review. Epilepsia. 2011;52:1941–1948. doi: 10.1111/j.1528-1167.2011.03287.x. [DOI] [PubMed] [Google Scholar]
- Peirce JL, Lu L, Gu J, Silver LM, Williams RW. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet. 2004;5:7. doi: 10.1186/1471-2156-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perucca E, Tomson T. The pharmacological treatment of epilepsy in adults. Lancet Neurol. 2011;10:446–456. doi: 10.1016/S1474-4422(11)70047-3. [DOI] [PubMed] [Google Scholar]
- Plomin R, McClearn GE, Gora-Maslak G, Neiderhiser JM. Use of recombinant inbred strains to detect quantitative trait loci associated with behavior. Behav Genet. 1991;21:99–116. doi: 10.1007/BF01066330. [DOI] [PubMed] [Google Scholar]
- Rossetti AO, Lowenstein DH. Management of refractory status epilepticus in adults: still more questions than answers. Lancet Neurol. 2011;10:922–930. doi: 10.1016/S1474-4422(11)70187-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Gray WP. Relevance of seizure-induced neurogenesis in animal models of epilepsy to the etiology of temporal lobe epilepsy. Epilepsia. 2007;48(Suppl 2):33–41. doi: 10.1111/j.1528-1167.2007.01065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE. The relevance of individual genetic background and its role in animal models of epilepsy. Epilepsy Res. 2011;97:1–11. doi: 10.1016/j.eplepsyres.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE. Strain differences in seizure-induced cell death following pilocarpine-induced status epilepticus. Neurobiol Dis. 2012;45:297–304. doi: 10.1016/j.nbd.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D. Drug treatment of epilepsy: options and limitations. Epilepsy Behav. 2009;15:56–65. doi: 10.1016/j.yebeh.2009.02.030. [DOI] [PubMed] [Google Scholar]
- Shifman S, Bell JT, Copley RR, Taylor MS, Williams RW, Mott R, Flint J. A high-resolution single nucleotide polymorphism genetic map of the mouse genome. PLoS Biol. 2006;4:e395. doi: 10.1371/journal.pbio.0040395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornalley PJ. The glyoxalase system in health and disease. Mol Aspects Med. 1993;14:287–371. doi: 10.1016/0098-2997(93)90002-u. [DOI] [PubMed] [Google Scholar]
- Thornalley PJ, Ladan MJ, Ridgway SJ, Kang Y. Antitumor activity of S-(p-bromobenzyl)glutathione diesters in vitro: a structure-activity study. J Med Chem. 1996;39:3409–3411. doi: 10.1021/jm960129c. [DOI] [PubMed] [Google Scholar]
- Titlic M, Basic S, Hajnsek S, Lusic I. Comorbidity psychiatric disorders in epilepsy: a review of literature. Bratisl Lek Listy. 2009;110:105–109. [PubMed] [Google Scholar]
- Veliskova J. Estrogens and epilepsy: why are we so excited? Neuroscientist. 2007;13:77–88. doi: 10.1177/1073858406295827. [DOI] [PubMed] [Google Scholar]
- Vince R, Daluge S, Wadd WB. Studies on the inhibition of glyoxalase I by S-substituted glutathiones. J Med Chem. 1971;14:402–404. doi: 10.1021/jm00287a006. [DOI] [PubMed] [Google Scholar]
- Wang J, Williams RW, Manly KF. WebQTL: web-based complex trait analysis. Neuroinformatics. 2003;1:299–308. doi: 10.1385/NI:1:4:299. [DOI] [PubMed] [Google Scholar]
- Williams Rt, Lim JE, Harr B, Wing C, Walters R, Distler MG, Teschke M, Wu C, Wiltshire T, Su AI, Sokoloff G, Tarantino LM, Borevitz JO, Palmer AA. A common and unstable copy number variant is associated with differences in Glo1 expression and anxiety-like behavior. PLoS One. 2009;4:e4649. doi: 10.1371/journal.pone.0004649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RW, Gu J, Qi S, Lu L. The genetic structure of recombinant inbred mice: high-resolution consensus maps for complex trait analysis. Genome Biol. 2001;2:RESEARCH0046. doi: 10.1186/gb-2001-2-11-research0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winawer MR, Gildersleeve SS, Phillips AG, Rabinowitz D, Palmer AA. Mapping a mouse limbic seizure susceptibility locus on chromosome 10. Epilepsia. 2011;52:2076–2083. doi: 10.1111/j.1528-1167.2011.03256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]