

Abstract
Chromosomal replication machines contain coupled DNA polymerases that simultaneously replicate the leading and lagging strands1. However, coupled replication presents a largely unrecognized topological problem. Since DNA polymerase must travel a helical path during synthesis, the physical connection between leading and lagging strand polymerases causes the daughter strands to entwine, or produces extensive buildup of negative supercoils in the newly synthesized DNA2–4. How DNA polymerases maintain their connection during coupled replication despite these topological challenges is a mystery. Here, we examine the dynamics of the E. coli replisome, by ensemble and single-molecule methods that may solve this topological problem independent of topoisomerases. We find that the lagging strand polymerase frequently releases from an Okazaki fragment before completion, leaving single-strand gaps behind. Dissociation of the polymerase does not result in loss from the replisome due to its contact with the leading-strand polymerase. This behavior, referred to as “signal release”, had been thought to require a protein, possibly primase, to pry polymerase from incompletely extended DNA fragments5–7. However, we observe that signal release is independent of primase and does not appear to require a protein trigger at all. Instead, the lagging-strand polymerase is simply less processive in the context of a replisome. Interestingly, when the lagging-strand polymerase is supplied with primed DNA in trans, uncoupling it from the fork, high processivity is restored. Hence, we propose that coupled polymerases introduce topological changes, possibly by accumulation of superhelical tension in the newly synthesized DNA, that cause lower processivity and transient lagging-strand polymerase dissociation from DNA.
Coupled leading- and lagging-strand polymerases, as observed in phage and bacterial replisomes1,8, have dramatic implications for the topology of the daughter DNA products2–4,9. The topological problem is based on the a priori fact that polymerases generate a helical product, and thus either must travel a helical path or the DNA product must turn behind them. For example, a rotating leading-strand polymerase will take the attached lagging-strand polymerase and wind the DNA duplex 360° around the axis of the leading strand, forming precatenanes in the daughter helices (Figure 1a, left)2. At the rate of E. coli replication (650 bp/s) rotating polymerases quickly result in an impossible tangle2. Alternatively, if the DNA products rotate instead of the polymerases, negative supercoils accumulate (Figure 1a, right). Superhelical tension on the lagging strand may be relieved by rotation of single-strand DNA, but SSB forms large superstuctures, which likely constrain swivel motion10. The energy generated by only 3–4 supercoils11 is sufficient to disrupt protein-protein and protein-DNA interactions12. Thus topological tension could disrupt the replisome, unless the tension is periodically released. The supercoils produced by coupled polymerases behind the fork are negative supercoils, which can be resolved by Topo I and Topo III13. However, Topo III is non-essential, and the viability of Topo I mutants has been controversial14,15. Hence, topoisomerases may participate, but are insufficient to remove negative supercoils produced by coupled replisomes.
Figure 1. The topological problem caused by coupled leading- and lagging-strand polymerases.

The figure illustrates the topological problem only for the leading-strand polymerase. a The two Pols (yellow) are coupled through the clamp loader (green). Pols travel a helical path as they synthesize DNA. As the leading Pol spins, the lagging Pol is dragged around this path, twisting the lagging strand around the leading strand (left). Alternatively, the DNA turns instead (right), generating negative supercoils as indicated by the arrow. In either case, the stress can be released by b. transient dissociation of the leading Pol or, c. transient dissociation of the lagging Pol, which could re-bind the incomplete Okazaki fragment or a new RNA primer.
Interestingly, single-molecule studies demonstrate highly processive DNA synthesis (>100 kb) in the absence of topoisomerases, implying that replisomes possess an intrinsic solution to the problem of coupled replication16,17. Indeed, Cozzarelli originally proposed that the topological problem could be solved by transient release of one polymerase of a coupled replisome from DNA (Figure 1b), enabling the negative supercoils on both strands to relax2. If the leading polymerase detaches from DNA, it will rebind the same primer terminus for continued extension. If the lagging polymerase dissociates it will reattach to the same Okazaki fragment and complete it provided a new primed site is not yet formed. During extension of longer Okazaki fragments a new priming event is more likely to occur; thus a dissociated lagging polymerase may rebind the at the new RNA primer, leaving the original Okazaki fragment incomplete (Figure 1b). In fact incomplete Okazaki fragments have been observed in bacterial and phage systems5–7. Premature polymerase dissociation is referred to as “signal release” because it has been presumed that the lagging-strand polymerase is “signaled” by a replisome component to dissociate and alleviate supercoil tension5–7.
To gain insight into the topological challenge of coupled DNA replication, we developed assays to study signal release. The E. coli replisome is assembled on a 5′ biotinylated rolling circle substrate and then attached to streptavidin beads (Fig. 2a). The substrate has only three nucleotides on either strand allowing either leading or lagging strand to be labeled depending on which radioactive nucleotide is used. Replication is initiated after wash steps to remove unbound proteins, and ssDNA gaps left by signal release are detected by treating the (α-32P)-dTTP labeled lagging-strand template with S1 endonuclease to digest the gaps (Supplementary Fig. 1). If all Okazaki fragments are completely extended the product will be S1-resistant. Conversely, 100% signal release will leave gaps between every Okazaki fragment, and S1 digestion will yield products of similar size to Okazaki fragments (Supplementary Fig. 2). An intermediate length of DNA will result if signal release is <100%.
Figure 2. Signal release does not require primase and correlates with increasing Okazaki fragment length.
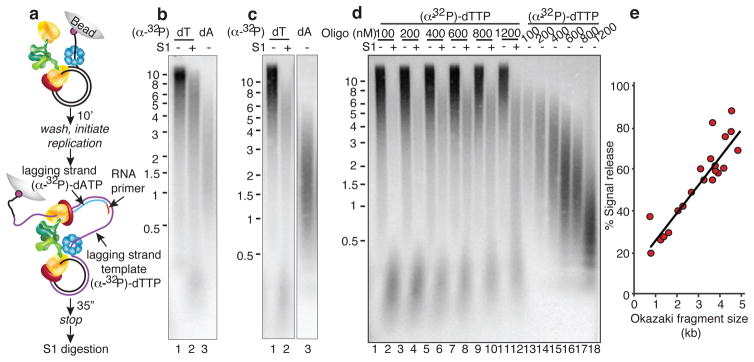
a, The replisome, consisting of DNA helicase (blue), Pol III* (one clamp loader (green) that binds 3 Pols (yellow); only two Pols are shown for clarity) and β-clamps (red) is assembled on a 5′ biotinylated rolling circle DNA, then attached to beads. After unbound proteins are removed, replication is initiated by adding primase, β, SSB, ATP and (α-32P)-dNTPs. Reactions are quenched and treated with S1 to cleave gaps left by signal release, prior to analysis on alkaline gels. The newly synthesized leading strand (purple) is the template for lagging-strand synthesis (blue), initiated by RNA primers (red). b, Replication reactions using (α-32P)-dTTP either before (lane 1) or after (lane 2) S1 analysis, or using (α-32P)-dATP (lane 3). c, Reactions in the absence of primase, using 800 nM DNA 20-mers to prime the lagging strand. d, Titration of DNA 20-mers into reactions. e, Plot of Okazaki fragment length versus % signal release.
The S1 analysis shows an intermediate-sized (α-32P)-dTTP labeled DNA (Fig. 2b, lane 2) that migrates between the length of undigested DNA (lane 1) and the size of Okazaki fragments (lane 3). Size analysis of S1-treated products reveals that about one-third of all Okazaki fragments are incomplete (Supplementary Fig. 3, Full Methods). To validate the assay, several controls were performed (Supplementary Fig. 4): 1) S1 digestion eliminates ssDNA produced in a reaction without primase (i.e. no lagging-strand synthesis), 2) S1 does not cleave double-strand Okazaki fragments, 3) ssDNA gaps produced by signal release can be filled in by addition of Pol II, 4) the presence of complete Okazaki fragments is confirmed using ligase.
Earlier studies implied that primase triggers polymerase signal release5,6. To test this hypothesis, we performed the reaction in the complete absence of primase, using exogenously added DNA primers to initiate Okazaki fragments. However, the result is unchanged (Fig. 2c). Studies in the T4 system suggest that a loaded clamp triggers polymerase release7. Although clamps cannot be omitted without negating lagging-strand synthesis altogether, titrations of the clamp (+/− clamp loader) do not influence signal release (Supplementary Fig. 5). Importantly, the assays of Fig. 2 remove excess (unbound) polymerase-clamp loader (i.e. Pol III*) and DnaB helicase, and show that primase, soluble polymerase, clamp loader and helicase are not required to trigger polymerase dissociation.
We find that increasing Okazaki fragment size by lowering DNA primer concentration results in more frequent signal release (Fig 2d, e and Supplementary Fig. 6). Okazaki fragments of 1 kb give only 20% signal release while 5 kb Okazaki fragments give 80% signal release. Hence, small Okazaki fragments are more likely to be completed than long Okazaki fragments, implying that the lagging-strand polymerase is not very processive. In contrast, studies on model lagging-strand templates show the polymerase has high intrinsic processive and extends a primed 5.4 kb φX174 ssDNA without dissociating18,19. To exclude that this result is an artifact of using DNA primers, we altered Okazaki fragment size by changing the concentration of primase20, but the results were upheld (Supplementary Fig. 7). That signal release correlates with long Okazaki fragments was confirmed using Pol II to fill in ssDNA gaps, demonstrating that ssDNA gaps are associated with long Okazaki fragments and yield an average gap size of 800 nt (Supplementary Fig. 8).
To further test for a protein circuit that lowers processivity by triggering polymerase release, we designed an assay to directly measure lagging-strand polymerase processivity. Since priming at a replication fork occurs randomly, producing heterogeneous sized products, we attached a 5.4 kb primed φX174 circular ssDNA to the lagging-strand polymerase in trans before initiating replication (Fig. 3a). Considering that all replisomal proteins are present, protein-induced signal release should still occur, and the length of extension from the defined primed site will define the processivity of the lagging-strand polymerase.
Figure 3. Lagging-strand polymerase processivity is restored using a separate DNA molecule.

a, The replisome is assembled on DNA as in the legend to Fig. 2a. Unbound proteins are washed away prior to addition of primed φX174 5.4 kb ssDNA. b, Reactions are initiated in the presence (lanes 7–12 and 19–24) or absence (lanes 1–6 and 13–18) of primase. Supernatants containing φX174 replication products (lanes 1–12) are separated from bead-bound rolling circle products (lanes 13–24).
A time-course of replication shows a most surprising result (Fig. 3b). The 5.4 kb φX174 DNA is fully replicated by the lagging polymerase (Fig. 3b, lanes 1–6 and 13–18), in striking contrast to the low processivity during Okazaki fragment synthesis (i.e. 80% dissociation by 5 kb, Fig. 2d, e). We obtained similar results in the presence or absence of primase, confirming that neither primase nor primed sites trigger signal release (Fig. 3b, lanes 7–12 and 19–24). The major difference in reactions with or without the primed φX174 DNA in trans is the connection of the two polymerases through the DNA, indicating that the connection of two polymerases to the same DNA molecule causes a decrease in lagging-strand polymerase processivity. One possible non-protein source is the topological strain produced in DNA due to coupled replication (Fig. 1). Lagging-strand loops are an unlikely source, as bending of large DNA stretches requires very little energy21. To examine the dynamics of lagging-strand polymerase action we turned to single-molecule studies to determine if signal release is characteristic of all replisomes, and whether it takes place during long replication times. Replication is performed in a flow cell in which the replisome-DNA complex is attached to a lipid bilayer. Replicating DNA is held in place at a lipid diffusion barrier (Fig. 4a, and Full Methods). Upon initiating replication, product DNA is observed using TIRF microscopy and a fluorescent dsDNA intercalator (see Fig. 4b).
Figure 4. Single-molecule TIRF microcopy.

a, The replisome is assembled on the rolling circle DNA and attached to a lipid bilayer. Replication is initiated upon flowing replication buffer at 10 μl/min. b, DNA curtain produced at 10 μl/min. c, Normalized line plot intensity of a representative DNA strand. Diagrams at the top illustrate dsDNA and ssDNA regions. d, Analysis as in c, but with Pol III* in the buffer flow. e. Quantification of line plot analysis at indicated flow rates. n = 9 ± SD for 10 μl/min or n = 13 ± SD for 100 μl/min. f. DNA curtain at 100 μl/min. g. Line plot analysis as in c, at 100 μl/min. h. Histogram relating pixel intensity to dsDNA at 10 μl/min (purple) and 100 μl/min (bright blue). i, Scheme at 100 μl/min.
We initially performed experiments at 10 μl/min, which translates into an applied force of 0.23 pN on the DNA, far below that needed to disrupt most protein-protein and protein-DNA interactions12. After replication is complete, we increased the flow to 100 μl/min (1.45 pN) to stretch DNA, enabling examination of the product for ssDNA gaps, which appear as regions with decreased fluorescence intensity16 (Fig. 4b, c). The resolution of the CCD camera is approximately 0.27μm per pixel, the length of about 880 bp of dsDNA. Hence ssDNA gaps should result in an uneven pixel density over the length of the molecule and enable a quantitative evaluation of the ssDNA/dsDNA ratio per pixel (Full Methods). All the molecules examined contained pixel intensity differences along their entire length (Fig. 4c, and Supplementary Fig. 9). If the pixel intensity differences are indeed ssDNA they should be eliminated upon adding Pol III, which fills gaps, in the flow buffer, and this was the case (Fig. 4d). Summation of the pixel intensities over 298.4 μm of DNA within nine different DNA molecules indicates 13% ssDNA and 87% dsDNA (assuming SSB-bound ssDNA equals dsDNA length5 (Fig. 4e)). This result is within 2-fold that observed in ensemble studies (i.e. Okazaki fragments average 2.3 kb, and give 65% signal release at 10 nM primase; thus an 800 bp average gap size yields a value of 22.3% ssDNA and 77.7% dsDNA). Overall, these results demonstrate that signal release occurs over the length of long DNA products and is a characteristic of all replisomes.
We also performed replication at a 10-fold higher flow rate (100 μl/min; 1.45 pN) (Fig. 4f, g). Unexpectedly, the amount of ssDNA relative to dsDNA increased nearly two-fold relative to the 10 μl/min flow rate (Fig. 4h), from 13% at 10 μl/min to 22% at 100 μl/min (Fig. 4e). An explanation for this difference is that when the lagging-strand polymerase undergoes a transient release cycle (signal release), the increased velocity of the flow carries away the “old” 3′ terminus, making the polymerase more likely to associate with the next RNA primer synthesized at the fork (illustration in Fig. 4i), resulting in more ssDNA gaps.
Notably, previous studies of signal release that suggested a protein trigger are also consistent with polymerase release triggered by torsional constraints due to coupled replication5–7. For example, in T4, increasing the concentration of clamps may accelerate clamp assembly on new RNA primers and increase the probability that a released polymerase binds the new RNA primed site7. In the T7 system additional primase may enhance priming frequency providing more opportunity for a dissociated polymerase to bind a new site5.
Negative supercoiling is required for a variety of DNA metabolic actions, including replication, recombination and transcription22,23. Negative superhelicity is a property of genomic DNA in all organisms and appears constant over the entire genome24. Hence it is conceivable that coupled leading-/lagging-strand replication provides a method by which negative supercoils can be placed into DNA across the entire genome without topoisomerase action.
Our data suggest that signal release is caused by DNA topology and thus may provide a topoisomerase-independent solution to the topological problem incurred by coupled DNA polymerases during replication. The results demonstrate that: (a) signal release does not require primase, (b) the release process is stochastic implying an inherent low processivity of the lagging polymerase, (c) signal release requires connection of both polymerases to the same DNA and (d) addition of DNA in trans eliminates signal release in the presence of all replisomal proteins. Despite the correlation of premature signal release to the topological problem inherent in coupled polymerases, the problem of coupled polymerase action is not definitively solved in this report and will require further studies.
FULL METHODS
Reagents and proteins
Proteins were purified as described:α, ε, γ, τ, δ,δ′, χ, ψ, θ, β26, SSB27, Pol III* ((αεθ)3τ3δδ′χψ) andγ complex (γ3δδ′χψ) were reconstituted as described26,28,29. Solid support bead-based assays were performed using Pol III* containing an ε mutant (D12A and E14A) that eliminates the 3′-5′ exonuclease activity to prevent removal of the 3′ dideoxyterminal nucleotide of the Trap DNA. Replication buffer is 20 mM Tris-HC1 (pH 7.5), 4% glycerol, 0.1 mM EDTA, 40 μg/ml BSA, 5 mM DTT, 10 mM MgOAc2. S1 buffer is 40 mM NaOAc (pH 4.5), 300 mM NaCl and 2 mM ZnSO4.
Rolling circle replication reactions
Twenty μl streptavidin-coupled magnetic beads (Invitrogen) were washed and equilibrated in replication buffer. Replisomes were assembled in 20 μl replication buffer RB (20 mM Tris-HC1 (pH 7.5), 4% glycerol, 0.1 mM EDTA, 40 μg/ml BSA, 5 mM DTT, 10 mM MgOAc2) containing 100 fmol synthetic 5′-biotinylated 100-mer rolling circle DNA (prepared as in 25), 60 μM each dATP and dGTP, 50 μM ATP-γ-S, 4 pmol DnaB6, 2.5 pmol β2, and 0.5 pmol Pol III*. Reactions were incubated 5 min at 37°C prior to immobilization on beads for 10 min at 25°C. Beads were washed three times in 500 μl replication buffer containing 30 μM dATP and dGTP, 12 pmol β2 and 50 μM ATP-γ-S prior to resuspending in 10 μl replication buffer containing 60 μM dCTP, 60 μM dGTP and 2.5 pmol β2. Replication was initiated by addition of 10 μl 0.5 mM ATP, 200 nM primase (unless indicated otherwise), 480 nM SSB4, 50 μM NTPs and either: 60 μM dATP, 10 μM TTP, 3 μCi (α–32P)-TTP to label the lagging-strand template (i.e. leading-strand product is the lagging-strand template in a rolling circle reaction), or 60 μM dTTP, 10 μM dATP and 3μCi (α–32P)-dATP to label lagging-strand Okazaki fragments. After 15 sec 5 μl replication buffer containing 20 nM Trap DNA was added to sequester any polymerase that dissociates during replication, thereby preventing it from filling gaps produced by signal release. Indicated concentrations reflect final reaction conditions in 25 μl. Replication was allowed to proceed for a total of 35 sec. The Trap DNA consists of M13mp18 ssDNA primed with a 3′ dideoxyterminated DNA 30-mer (5′-GTTAAAGGCCGCTTTTGCGGGATCGTCACddC-3′) onto which the β clamp was pre-assembled in a 2-min reaction at 37°C containing 4.8 pmol SSB4, 2.5 pmol β2, 1 pmol γ-complex. Reactions were quenched by adding 25 μl of 40 mM EDTA and 1% SDS. Where indicated, primase was substituted with 20-mer DNA oligonucleotides (5′-GCAAAAGCAGGCACAGCAAC-3′). Beads were washed twice in Buffer E (20 mM Tris-HC1 (pH 7.5), 4% glycerol, 0.1 mM EDTA, 40 μg/ml BSA, 5 mM DTT) and once in S1 buffer (200 mM sodium acetate (pH 4.5), 1.5 M NaCl and 10 mM ZnSO4).
S1 endonuclease digestion
Beads were resuspended in 20 μl S1 buffer and 5 μl of the reaction was withdrawn and incubated with 1 U S1 nuclease (Fermentas) for 7.5 min at 37°C in 15 μl total reaction volume. Reactions were quenched by adding 15 μl of 40 mM HEPES (pH 7.5), 40 mM EDTA and 6% SDS. DNA was released from beads by incubating at 95°C for 3 min and resolved on 0.8 % alkaline agarose gels.
Control reactions for S1 assay
Where indicated, S1 treatment was performed on either: 0.2 μg supercoiled pIK31 plasmid DNA, pIK31 linearized with PstI (pIK31-PstI), pIK31 nicked on one strand with Nb.BvCI (pIK31-BvCI), or 0.4 μg circular M13mp18 ssDNA. Rolling circle replication reactions were performed as described above. Reactions that were treated with Pol II were first replicated and quenched as described above, washed three times in replication buffer before resuspending in 25 μl buffer containing 20 nM Pol II, 60 μM dCTP and dGTP, 2.5 pmol β2, 1 pmol γ-complex and 0.5 mM ATP and incubated for 5 min at 37°C. Replication was initiated by adding 60 μM dTTP, dATP and incubated for 10 min at 37°C. Replication for fill-in reactions with Pol II to label gaps were performed similarly and initiated with dATP, dTTP and (α–32P)-dATP, where indicated.
Ligation of Okazaki fragments
Replication reactions that were treated with T4 DNA ligase (New England Biolabs) were first replicated in the presence of (α–32P)-dATP and 800 nM 5′-phosphorylated oligonucleotide (5′-P-GCAAAAGCAGGCACAGCAAC-3′) for lagging-strand synthesis and quenched as described. Beads were washed three times in 20 mM Tris-HC1 (pH 7.5), 4% glycerol, 0.1 mM EDTA, 40 μg/ml BSA, 5 mM DTT. Beads were resuspended in T4 quick ligase buffer (NEB) and 1 μl of T4 DNA quick ligase for 15 min at 25°C. Reactions were quenched by adding 15 μl of 40 mM HEPES (pH 7.5), 40 mM EDTA and 6% SDS. DNA was released from the beads by incubating at 95°C for 3 min and resolved in 0.8 % alkaline agarose gels.
Analysis of lagging-strand polymerase processivity on primed φX174 ssDNA in trans
Replisomes were assembled on the biotinylated rolling circle substrate as described above. After the wash steps, beads were resuspended in 20 μl replication buffer containing 8 nM φX174 ssDNA (5.4 kb) primed with a DNA oligonucleotide (5′-CAAGCAGTAGTAATTCCTGCTTTATCAAG-3′), 2.4 pmol SSB4, 1 pmol β2, 0.08 pmol γ-complex, 60 μM dCTP and 60 μM dGTP for 2 min at 37°C. Beads were immobilized, washed and replication was initiated as described above.
Data analysis
Lanes in alkaline gels were scanned using a phosphorimager with ImageQuant Software. The intensity of radioactivity at each pixel was normalized to the corresponding molecular weight in order to correct for the fact that longer products incorporate more radiolabel. Size markers were used as a reference in the gel. Percent signal release was calculated from the size difference of S1 treated leading strand and lagging strand products (fit with a single Gaussian distribution as described in the legend to Supplementary Fig. 3) using the calculation:
SR = Signal release; Size (lg) = Size of lagging strand product; Size (ld + S1) = Size of S1 digested leading strand product (i.e. lagging strand template).
Single-molecule replication assays
Replisomes were assembled onto 530 fmol (10.5 nM) 5′ biotinylated 100-mer rolling circle DNA by first incubating with 18 pmol DnaB helicase (365 nM) in 50 μl Buffer A (20 mM Tris- HCl, pH 7.5, 5 mM DTT, 40 μg/ml BSA, 4% glycerol) containing 8 mM MgOAc2 and 0.25 mM ATP, followed by incubation for 30 sec at 37°C. Then a 25 μl solution of Buffer A containing Pol III* (675 fmol (αεθ)τ3 δδ′ψχ, 27 nM as complex), β2 (1.85 pmol, 74 nM as dimer), 60 μM each dCTP and dGTP and 8 mM MgOAc2 was added followed by a further 1 min at 37°C. To immobilize the replisome-DNA complex in the flow cell, 1 μl of the above reaction was diluted 1000-fold into 1 ml of Buffer B (8 mM MgOAc2, 60 μM each of dCTP and dGTP, and 50 nM Yo-Pro1 in Buffer A), and then the diluted reaction was passed through the flow cell at 500 μl/min for 30 sec, and a further 2 min at 10 μl/min. DNA replication was initiated upon flowing Buffer A containing 60 μM of each dNTP, 250 μM each of CTP, TTP, and UTP, 1 mM ATP, 462 nM SSB4, 100 nM primase, 50 nM β2, 50 mM potassium glutamate, 50 nM Yo-Pro1, 0.8% glucose, 0.01% β-mercaptoethanol, 0.57U glucose oxidase, and 2.1U catalase. The flow rate was either 10 μl/min or 100 μl/min, as indicated. After 10 min of replication, the flow rate was adjusted to 100 μl/min to analyze replication products.
Intensity Analysis of the DNA products
In order to correct for flow disturbances in our analysis, we averaged between 30–40 frames for each DNA strand (over two adjacent pixels); the same operation was performed for an adjacent empty region to serve as background correction that takes into consideration the signal variation of non-uniform intensities displayed by the bilayer. In addition to this background subtraction, we performed a signal normalization. As the DNA strand grows longer and extends further away from the Biotin-Streptavidin anchoring point in the lipid bilayer, there is an additional “slow-drift” in intensity that is due to higher strand mobility into the flow. Therefore we normalized the slow-drift intensity signal to 100% dsDNA by applying a rank-order filter, a method best suited for removing shot noise. This method finds the specified percentile of data points in the data window around each point in the data set (here the corrected line plot intensity) and replaces that point with the percentile. The analysis was performed using Matlab software.
Supplementary Material
Acknowledgments
We thank Drs. L. Langston, A. Libchaber and B. Michel for helpful suggestions on the manuscript. We are also grateful to NIH (GM 38839) for supporting this work.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions I.K. and M.O.D. conceived the project, I.K. and R.G. performed experiments, I.K., R.G. and M.O.D. designed the experiments, analyzed data and wrote the paper.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no conflict of interest.
References
- 1.Kornberg A, Baker TA. DNA replication. 2. W.H. Freeman; 1992. [Google Scholar]
- 2.Ullsperger CJ, Volgodskii AV, Cozzarelli NR. Unlinking of DNA by Topoisomerases During DNA Replication. 9. Springer-Verlag; 1995. [Google Scholar]
- 3.Hingorani MM, O’Donnell M. Sliding clamps: a (tail)ored fit. Curr Biol. 2000;10:R25–29. doi: 10.1016/s0960-9822(99)00252-3. [DOI] [PubMed] [Google Scholar]
- 4.Wyman C, Botchan M. DNA replication. A familiar ring to DNA polymerase processivity. Curr Biol. 1995;5:334–337. doi: 10.1016/s0960-9822(95)00065-0. [DOI] [PubMed] [Google Scholar]
- 5.Hamdan SM, Loparo JJ, Takahashi M, Richardson CC, van Oijen AM. Dynamics of DNA replication loops reveal temporal control of lagging-strand synthesis. Nature. 2009;457:336–339. doi: 10.1038/nature07512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Marians KJ. Two distinct triggers for cycling of the lagging strand polymerase at the replication fork. J Biol Chem. 2000;275:34757–34765. doi: 10.1074/jbc.M006556200. [DOI] [PubMed] [Google Scholar]
- 7.Yang J, Nelson SW, Benkovic SJ. The control mechanism for lagging strand polymerase recycling during bacteriophage T4 DNA replication. Mol Cell. 2006;21:153–164. doi: 10.1016/j.molcel.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 8.Johnson A, O’Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- 9.Postow L, Peter BJ, Cozzarelli NR. Knot what we thought before: the twisted story of replication. Bioessays. 1999;21:805–808. doi: 10.1002/(SICI)1521-1878(199910)21:10<805::AID-BIES1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 10.Chastain PD, 2nd, Makhov AM, Nossal NG, Griffith J. Architecture of the replication complex and DNA loops at the fork generated by the bacteriophage t4 proteins. J Biol Chem. 2003;278:21276–21285. doi: 10.1074/jbc.M301573200. [DOI] [PubMed] [Google Scholar]
- 11.Vologodskii AV, Lukashin AV, Anshelevich VV, Frank-Kamenetskii MD. Fluctuations in superhelical DNA. Nucleic Acids Res. 1979;6:967–982. doi: 10.1093/nar/6.3.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leu FP, Georgescu R, O’Donnell M. Mechanism of the E. coli tau processivity switch during lagging-strand synthesis. Mol Cell. 2003;11:315–327. doi: 10.1016/s1097-2765(03)00042-x. [DOI] [PubMed] [Google Scholar]
- 13.Espeli O, Marians KJ. Untangling intracellular DNA topology. Mol Microbiol. 2004;52:925–931. doi: 10.1111/j.1365-2958.2004.04047.x. [DOI] [PubMed] [Google Scholar]
- 14.Stockum A, Lloyd RG, Rudolph CJ. On the viability of Escherichia coli cells lacking DNA topoisomerase I. BMC Microbiol. 2012;12:26. doi: 10.1186/1471-2180-12-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stupina VA, Wang JC. Viability of Escherichia coli topA mutants lacking DNA topoisomerase I. J Biol Chem. 2005;280:355–360. doi: 10.1074/jbc.M411924200. [DOI] [PubMed] [Google Scholar]
- 16.Yao NY, Georgescu RE, Finkelstein J, O’Donnell ME. Single-molecule analysis reveals that the lagging strand increases replisome processivity but slows replication fork progression. Proc Natl Acad Sci U S A. 2009;106:13236–13241. doi: 10.1073/pnas.0906157106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanner NA, et al. Real-time single-molecule observation of rolling-circle DNA replication. Nucleic Acids Res. 2009;37:e27. doi: 10.1093/nar/gkp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Donnell ME, Kornberg A. Complete replication of templates by Escherichia coli DNA polymerase III holoenzyme. J Biol Chem. 1985;260:12884–12889. [PubMed] [Google Scholar]
- 19.McHenry CS. DNA replicases from a bacterial perspective. Annu Rev Biochem. 2011;80:403–436. doi: 10.1146/annurev-biochem-061208-091655. [DOI] [PubMed] [Google Scholar]
- 20.Wu CA, Zechner EL, Reems JA, McHenry CS, Marians KJ. Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. V. Primase action regulates the cycle of Okazaki fragment synthesis. J Biol Chem. 1992;267:4074–4083. [PubMed] [Google Scholar]
- 21.Marko JF, Siggia ED. Stretching DNA. Macromolecules. 1995;28:8759–8770. [Google Scholar]
- 22.Dorman CJ. DNA supercoiling and bacterial gene expression. Sci Prog. 2006;89:151–166. doi: 10.3184/003685006783238317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Travers A, Muskhelishvili G. A common topology for bacterial and eukaryotic transcription initiation? EMBO Rep. 2007;8:147–151. doi: 10.1038/sj.embor.7400898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benyajati C, Worcel A. Isolation, characterization, and structure of the folded interphase genome of Drosophila melanogaster. Cell. 1976;9:393–407. doi: 10.1016/0092-8674(76)90084-2. [DOI] [PubMed] [Google Scholar]
- 25.McInerney P, O’Donnell M. Functional uncoupling of twin polymerases: mechanism of polymerase dissociation from a lagging-strand block. J Biol Chem. 2004;279:21543–21551. doi: 10.1074/jbc.M401649200. [DOI] [PubMed] [Google Scholar]
- 26.Onrust R, Finkelstein J, Turner J, Naktinis V, O’Donnell M. Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. III. Interface between two polymerases and the clamp loader. J Biol Chem. 1995;270:13366–13377. doi: 10.1074/jbc.270.22.13366. [DOI] [PubMed] [Google Scholar]
- 27.Yao N, Hurwitz J, O’Donnell M. Dynamics of beta and proliferating cell nuclear antigen sliding clamps in traversing DNA secondary structure. J Biol Chem. 2000;275:1421–1432. doi: 10.1074/jbc.275.2.1421. [DOI] [PubMed] [Google Scholar]
- 28.Bailey S, Wing RA, Steitz TA. The structure of T. aquaticus DNA polymerase III is distinct from eukaryotic replicative DNA polymerases. Cell. 2006;126:893–904. doi: 10.1016/j.cell.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 29.Georgescu RE, et al. Mechanism of polymerase collision release from sliding clamps on the lagging strand. Embo J. 2009;28:2981–2991. doi: 10.1038/emboj.2009.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.