

Abstract
Purpose
RNA interference has the potential to specifically knock down the expression of target genes, and thereby transform cancer therapy. However, lack of effective delivery of small inhibitory RNA (siRNA) has dramatically limited its in vivo applications. We have developed a multistage vector (MSV) system, composed of discoidal porous silicon particles loaded with nanotherapeutics, that directs effective delivery and sustained release of siRNA in tumor tissues. In this study, we evaluated therapeutic efficacy of MSV-loaded EphA2 siRNA (MSV/EphA2) with murine orthotopic models of metastatic ovarian cancers as a first step towards development of a new class of nanotherapeutics for the treatment of ovarian cancer.
Experimental design
Tumor accumulation of MSV/EphA2 and sustained release of siRNA from MSV were analyzed after i.v. administration of MSV/siRNA. Nude mice with metastatic SKOV3ip2 tumors were treated with MSV/EphA2 and paclitaxel, and therapeutic efficacy was assessed. Mice with chemotherapy-resistant HeyA8 ovarian tumors were treated with a combination of MSV/EphA2 and docetaxel, and enhanced therapeutic efficacy was evaluated.
Results
Treatment of SKOV3ip2 tumor mice with MSV/EphA2 biweekly for 6 weeks resulted in dose-dependent (5, 10 and 15 μg/mice) reduction of tumor weight (36%, 64%, and 83%) and number of tumor nodules compared with the control groups. In addition, tumor growth was completely inhibited when mice were treated with MSV/EphA2 in combination with paclitaxel. Furthermore, combination treatment with MSV/EphA2 and docetaxel inhibited growth of HeyA8-MDR tumors, which were otherwise resistant to docetaxel treatment.
Conclusion
These findings indicate that MSV/EphA2 merits further development as a novel therapeutic agent for ovarian cancer.
Keywords: ovarian cancer, siRNA, nanoliposome, porous silicon, multistage vector, sustained release
Introduction
The EphA2 gene encoding the epithelial cell receptor protein-tyrosine kinase is overexpressed in multiple cancer types (1–3). Its expression level is associated with aggressive features of tumor growth (4), and is a predictive marker for tumor recurrence and patient survival (5, 6). We have previously shown that EphA2 expression was associated with angiogenesis in ovarian tumors (7), and that knockdown of EphA2 expression with gene-specific siRNA oligos delivered in dioleoyl phosphatidylcholine (DOPC) neutral liposomes led to inhibition of tumor growth in orthotopic mouse models of ovarian cancer (3). In an effort to develop a tumor-specific delivery system with sustained release of siRNA oligos, we loaded liposomal siRNA into the 40–65 nm size pores inside the 1.6 μm hemispherical porous silicon particles to create a nanoparticle-in-microparticle multistage vector (MSV) delivery system (8). Once delivered in vivo via intravenous administration, the silicon particles travel in circulation, settle at the tumor vasculature where the liposomal siRNA gets released when the porous silicon degrades (9, 10). In a proof-of principle study, we have shown that knockdown of EphA2 expression lasted for as long as 3 weeks from a single administration of this new formulation, which resulted in reduced tumor cell proliferation and tumor angiogenesis, and eventually tumor growth (8). Our results also indicated that the MSV delivery system did not cause significant toxicity to major organs such as liver and kidney (8).
The micrometer-size, nanoporous silicon particles were designed to accumulate in tumor endothelial and perivascular depots (11). Unlike most nanocarriers for drug delivery that rely on the leaky vasculature to accumulate in tumor tissues, these porous silicon particles do not solely exploit the enhanced permeability and retention effect of the tumor vasculature. Instead, they travel hematogenously and interact with endothelial cells taking advantage of the optimal hydrodynamic force and interfacial interaction within the tumor vasculature. Our recent studies have revealed that size, shape, and surface chemical property of the porous silicon particles are major determinants of tissue distribution (12–14). More discoidal particles accumulates in tumor tissues than the spherical or cylindrical particles in mouse model of MDA-MB-231 breast cancer (12), or the hemispherical particles in a mouse model of melanoma (13). Thus, the discoidal particles represent the best option as a vector for delivery of nanotherapeutics.
In the current study, we loaded EphA2 siRNA DOPC liposomes into the 1000 × 400 nm discoidal MSV particles to assemble the final therapeutic agent discoidal MSV/EphA2-siRNA-DOPC liposome (MSV/EphA2), and characterized siRNA release profile in vitro and biodistribution in vivo. We tested the efficacy of MSV/EphA2 and the taxanes in chemotherapy-sensitive and –resistant models. The results from these studies support the development of MSV/EphA2 as an effective therapeutic agent for the treatment of metastatic ovarian cancer.
Materials and Methods
Fabrication and characterization of discoidal porous silicon microparticles
Discoidal porous silicon microparticles were fabricated by electrochemical etching of silicon wafer as previously described (15). Their physical dimension and pore size were verified by high-resolution scanning electron microscope. The porosity was verified by nitrogen absorption analysis as previously described (13). The porous silicon particles were modified with 2% APTES to obtain positively charged particles for loading of neutral to slight negatively charged nanoliposomes (16). Stability of the modified particles was measured by incubating 0.5 billion particles with 1 ml 10% fetal bovine serum (FBS), pH5.7. Particles degradation was monitored with a Multisizer 4 (Beckman Coulter) to measure the diameter of individual particles, and a JEOL 6500F scanning electron microscope.
Preparation of siRNA DOPC nanoliposomes and assembly into MSV
siRNA oligos were incorporated into 1,2-dioleoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids, INC, Alabaster, AL) by lyophilization. Briefly, 15 μg siRNA and DOPC were mixed in the presence of excess t-butanol at a ratio of 1:10 (w/w) as described previously (3, 7, 8). After Tween-20 was added, the mixture was frozen in an acetone-dry ice bath and lyophilized. Nanoliposomes were reconstituted by adding 40 μl water into the vial followed by a brief mix. The nanoliposome suspension was then mixed with 6×108 discoidal porous silicon particles, and sonicated for 5 minutes.
Tumor cells, culture condition, and in vitro release of MSV/siRNA
The human ovarian cancer cells SKOV3ip2 and HeyA8-MDR have been described previously (17). Cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) in 5% CO2/95% air at 37°C.
To measure siRNA release from MSV in cancer cells, SKOV3ip2 and HeyA8 cells were seeded in 4-chamber tissue culture slides and MSV/Alexa555-labeled siRNA particles were added into cell culture 24 hours later at a ration of 1/50 (cell/MSV). Cells were washed the next day to get rid of free particles, and fluorescent intensity from Alexa-555 siRNA was visualized with a confocal microscope 1, 2, and 7 days after particle addition. Cells were also stained with DAPI for visualization of the nuclei. To measure sustained knockdown of EphA2 expression in tumor cells in vitro, SKOV3 cells were incubated with MSV/Scramble siRNA or MSV/EphA2 siRNA. Samples were collected on days 3, 5, 7, and 9, and processed for Western blot analysis. EphA2 expression level was detected with an anti-EphA2 antibody from Invitrogen. Time-dependent cell viability was also measured with the Cell Counting Kit-8 from Dojindo.
Orthotopic tumor implantation
Female athymic nude mice (NCr-nu, 8 to 12 weeks old) were purchased from Taconic (Hudson, NY, USA). To generate tumors, SKOV3ip2 (1×106 cells/0.2 mL HBSS) or HeyA8-MDR (5×105 cells/0.2 mL HBSS) cells were injected into the peritoneal cavity of nude mice. For efficacy evaluation, mice were treated with therapeutics 2 weeks after tumor implantation.
In vivo distribution of siRNA
Porous silicon particles were covalently conjugated to the Dylight-488 fluorescent dye, and Cy3-labeled siRNA oligos were used to prepare nanoliposomes. Nude mice bearing SKOV3ip2 tumors were treated i.v. with 15 μg siRNA in Dylight-MSV/Cy3-siRNA. Animals were sacrificed 12 hr, 24 hr, 72 hr, 120 hr, or 168 hr later. Tumor nodules and major organs including kidney, spleen, liver, lung and heart were collected, and tissues with equal size and shape were deposited into a 96-well plate for measurement of fluorescent intensities. Images were captured with a Xenogen IVIS Spectrum imaging system (Caliper Life Sciences, Hoptkinton, MA) with a 535 nm excitation filter and 580 nm emission filter for Cy3, and 605 nm excitation filter and 660 nm emission filter for Dylight.
Therapeutic activity of MSV/EphA2
Paclitaxel (PTX) and docetaxel (DTX) were purchased from M D Anderson Cancer Center pharmacy and diluted in PBS to 2mg/kg and 3mg/kg before use. To assess the therapeutic activity of MSV/EphA2 alone or in combination with PTX, nude mice bearing SKOV3ip2 tumors were randomly divided into 8 groups (10 mice per group) and treated with: 1) empty MSV, 2) MSV/scramble siRNA (MSV/Scr, 15 μg siRNA), 3) MSV/EphA2 (5 μg siRNA), 4) MSV/EphA2 (10 μg siRNA), 5) MSV/EphA2 (15 μg siRNA), 6) PTX (75 μg), 7) MSV/Scr plus PTX 8) MSV/EphA2 (15 μg) plus PTX. PTX was dosed i.p. weekly, and empty MSV and MSV/siRNA were dosed i.v. biweekly. Animals were sacrificed after 6 weeks of treatment, and tumor samples were removed and processed for immunohistochemical (IHC) analyses. To assess gene expression by Western blot analysis, the tumor mice were treated with the indicated therapeutics once, and sacrificed 7 days later. Tumor nodules were collected, and processed directly with sample buffers.
To evaluate the therapeutic activity of MSV/EphA2 alone or in combination with DTX, HeyA8-MDR cells were inoculated i.p into nude mice. Fourteen days later, mice were randomly assigned to five treatment groups (10 mice per group) and treated with: 1) DTX (50 μg), 2) MSV/Scr, 3) MSV/EphA2, 4) MSV/Scr plus DTX, 5) MSV/EphA2 plus DTX. MSV/siRNA (15 μg siRNA/injection) was dosed i.v. biweekly and DTX was dosed i.p. weekly.
IHC analysis of tumor samples
EphA2 expression was analyzed by IHC analysis using paraffin-embedded tumors as described previously (3, 8). IHC analysis for CD31 (microvessel density) was done on freshly cut frozen tissue with the anti-CD31 antibody (platelet/endothelial cell adhesion molecule-1, rat IgG; BD Pharmingen, San Diego, CA). To quantify microvessel density (MVD), 5 random fields at 20× final magnification were examined for each tumor, and the number of microvessels per field was counted.
Assessment of cell apoptosis by terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) assay was carried out by IHC analysis using freshly cut frozen tissue as described previously (3, 7, 8). To quantify TUNEL positive cells, the number of positive cells was counted in 5 random fields at 20× magnification.
Statistical analysis
For in vivo experiments, differences in continuous variables (tumor weight, MVD, and TUNEL staining) were analyzed using the Student’s t test for comparing two groups and by ANOVA for multiple group comparisons with p < 0.05 considered statistically significant and p < 0.01 very significant.
Results
Characterization of porous silicon microparticles
The 1,000 × 400 nm discoidal porous silicon particles were fabricated by a combination of electrochemical etching and photolithography (15). The unmodified particles had a negative surface charge (−30 mV) since there was a thin-layer of oxidized silicon on the surface as a result of the fabrication process, and were vulnerable to degradation in aqueous solution. In serum, most particles were dissolved after several hours of incubation (15). We modified the silicon particles by chemically conjugating APTES onto the surface (16). APTES modification not only stabilized the particles, but also converted the otherwise negatively charged surface to slightly positive (zeta potential +3 to +7 mV), which facilitated loading of the slightly negative to neutral liposomes (30–35 nm in diameter) into the nanopores (average diameter 60 nm).
Stability of the APTES-modified porous silicon particles were tested in PBS containing 10% FBS. SEM images showed that the newly fabricated discoidal silicon particles consisted of a thin, high-density top layer and a thick, high-porosity bottom layer (Fig. 1A). The nanoliposomes would be loaded into the high-porosity layer. The uniformly looking particles had clear edges and well-defined nanoporous structure. After 24-hour incubation in FBS, the particles had lost part of the porous structure already. After 3 days in FBS, most particles had lost a substantial proportion of the high-porosity layer. Only the high-density layer was left with some particles. However, some particles still retained part of the porous structure after 10 days of incubation. Particle degradation was also confirmed by size measurement with a Multisizer based on particle diameter (Fig. 1B, upper panel) and volume (Fig. 1B, bottom panel). While the median sizes of particles were close between the pre-incubation and day 1 samples, a 32% drop on median particle size (from 0.13 to 0.088 μm3) was noticed after 3 days of incubation. By day 10, most particles were much smaller than the pre-inoculation samples with a 46% drop on median particle size.
Figure 1. Stability of silicon particles in serum.

A. Discoidal porous silicon particles (1,000 ×400 nm) were incubated in FBS, pH 5.7. Aliquots of particle suspension were samples after 1, 3, and 10 days. Particle structure was analyzed using a scanning electron microscope. B. Particle size was measured with a Multisizer 4 instrument, and displayed based on diameter (upper panel) and volume (bottom panel). The dashed lines indicate the size of median volume (0.13, 0.125, 0.088 and 0.07 μm3 on days 0, 1, 3 and 10).
Characterization of MSV/siRNA
To prepare MSV/siRNA, DOPC liposomes incorporated with siRNA oligos were loaded into the 1,000 × 400 nm discoidal porous silicon particles. Six hundred million porous silicon particles were needed in order to load 15 μg siRNA oligos. We have previously shown that 15 μg siRNA/injection/mouse is needed to ensure sufficient knockdown of EphA2 expression in tumor samples (8). To assess siRNA release inside cells, we prepared DOPC liposomes with the fluorescent Alexa555 siRNA oligos and loaded them into porous silicon. MSV/Alexa555 siRNA was added into culture of human SKOV3ip2 and HeyA8 cancer cells, and fluorescent intensity inside the cells was monitored. Multiple silicon particles could be found within each cell one day after incubation (Fig. 2A). We have previously demonstrated that particles are internalized by phagocytosis and/or macro-pinocytosis (18). All particles were localized in the cytosol where the liposomal siRNA got released. Bright fluorescent intensity could still be detected on day 2 and day 7, indicating that the particles were stable inside the cell and that sustained release of siRNA could be achieved. It is interesting to note that the cells still carried MSV/Alexa555 siRNA by day 7, when the original cells should have gone through several cycles of cell division (Fig. 2A). The result implies that the MSV particles were partitioned evenly during cell division.
Figure 2. Sustained release of liposomal siRNA in tumor cells.

A. Alexa555-labeled siRNA oligos were packaged in DOPC nanoliposomes, and loaded into MSV (MSV/Alexa555 siRNA). Human ovarian tumor cells SKOV3ip2 and HeyA8 were incubated with MSV/Alexa555 siRNA, and release of Alexas555 siRNA from MSV was monitored by confocal microscopy over the next 7 days. Nuclei were stained in blue with DAPI, and the fluorescent Alexa555 siRNA was in red. B. Western blot analysis of EphA2 expression in SKOV3 cells incubated with MSV/EphA2 siRNA. C. CCK-8 assay to measure cell viability after treatment with MSV/EphA2.
We incubated SK-OV-3 cells with MSV/Scramble siRNA or MSV/EphA2 siRNA, and monitored knockdown of EphA2 expression by Western blot analysis. EphA2 knockdown was apparent on day 3, and maximum knockdown was reached on day 7 (Fig. 2B). Knockdown of EphA2 expression reduced cell viability by 25% on day 5 and 40% by day 9 (Fig. 2C).
Biodistribution of MSV/siRNA
To determine siRNA distribution in vivo, we packaged Cy3-siRNA into DOPC liposomes, loaded them into Dylight-conjugated MSV particles, and administrated MSV/siRNA i.v. into the SKOV3ip2 orthotopic tumor mice. Mice were sacrificed at different time points, major organs and tumor tissues were collected, and fluorescence from Cy3 and Dylight was measured with a Xenogen IVIS Spectrum imaging system. Liver and tumor accumulation of MSV particles was apparent 12 hours post administration based on fluorescent intensity from Dylight (Fig. 3A). Dylight accumulation in the kidney was also observed at the 12-hour time point, suggesting a substantial amount of the fluorescent dye had already been cleaved from the particle. A 5-fold increase of Cy3 fluorescent intensity was observed in tumor tissues from mice in the treatment group comparing to the untreated mice at the 12-hour time point (Fig. 3B). Fluorescent intensity from Cy3-siRNA in tumor tissues was maintained at 2 fold above the background level over the next 4 days (24 hour to 120 hour time points), and started to drop on day 7 (Fig. 3B). This result indicates that a relatively high level of siRNA in tumor tissues can be maintained by sustained release of siRNA from MSV. Interestingly, Cy3 intensity was comparable in the major organs including the liver between control and treated mice, although more liver accumulation of MSV particles could be detected (Fig. 3A). It is possible that liver served as a depot of MSV/siRNA. As soon as liposomal siRNA was released from MSV, they entered the circulation and accumulated in tumor tissues as a result of the enhanced permeability and retention (EPR) effect of the tumor (19).
Figure 3. Biodistribution of MSV/siRNA.

SKOV3ip2 tumor mice were administrated i.v. with Dylight-MSV/Cy3-siRNA, and sacrificed 12, 24, 72, 120, and 168 hours later (n=3). Major organs and tumor tissues were collected, and fluorescent intensities from individual samples were measured. A, Dylight fluorescent intensity. B, Cy3 fluorescent intensity.
Dose-dependent inhibition of tumor growth by MSV/EphA2 siRNA
Mice with metastatic SKOV3ip2 tumors were treated biweekly with MSV/EphA2 siRNA at 5, 10, or 15 μg siRNA /mouse/treatment for 6 weeks. In the control groups, animals were treated either with empty MSV or MSV/Scr. At the end of the treatment, all animals were sacrificed, and the number of tumor nodules and total weight of tumor tissues per mouse were measured. Overall, treatment with MSV/EphA2 resulted in significantly reduced tumor growth (Fig. 4). Dose-dependent inhibition was apparent among the EphA2 siRNA groups. Treatments with 5, 10, or 15 μg siRNA resulted in 36% (p<0.05), 64% (p<0.05), and 83% (p<0.01) reduction of total tumor weight respectively comparing to the MSV/Scr group (Fig. 4). Dose-dependent inhibition was mostly reflected in the tumor weight, as the average number of tumor nodules per mouse was close in the three treatment groups.
Figure 4. In vivo therapeutic efficacy of sustained EphA2-siRNA-DOPC delivery by discoidal MP in combination with paclitaxel.
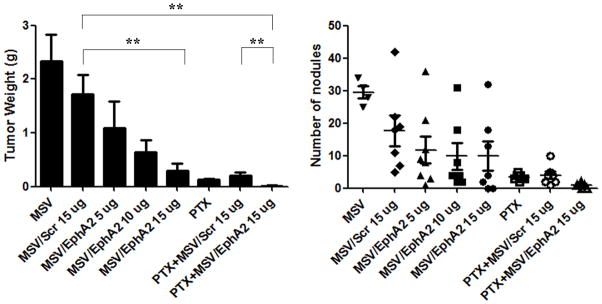
Nude mice were inoculated i.p. with SKOV3ip2 cells and randomly divided into eight treatment groups (n = 10): 1) MSV, 2) MSV loaded with non-silencing scramble siRNA-DOPC (MSV/Control, 15 μg siRNA), 3) MSV loaded with 5 μg EphA2-siRNA-DOPC (MSV/EphA2, 5 μg), 4) MSV loaded with 10 μg EphA2-siRNA-DOPC (MSV/EphA2, 10 μg), 5) MSV loaded with 15 μg EphA2-siRNA-DOPC (MSV/EphA2, 15 μg), 6) paclitaxel (PAX), 7) PTX and MSV/Control combination, and 8) PTX and MSV/EphA2 combination. Mice were dosed i.v. biweekly for 6 weeks. They were sacrificed at the end of the treatment, and total tumor weight and tumor nodules were measured. **: p<0.01.
Pathological analysis of tumor samples revealed that treatment with MSV/5 μg EphA2 siRNA only partially inhibited gene expression (Fig. 5A). Increasing siRNA to 10 and 15 μg/treatment dramatically reduced EphA2 expression. To confirm knockdown of EphA2 expression, we carried out another experiment by treating tumor mice once instead of 3 times so that there would be enough tumor nodules left for Western blot analysis (Fig. 5B). While there was still EphA2 expression in the 5 μg EphA2 siRNA treatment group, no EphA2 expression was detected in tumor samples treated with 10 or 15 μg EphA2 siRNA.
Figure 5. Analysis on knockdown of EphA2 expression, tumor angiogenesis, and tumor cell apoptosis.




A. EphA2 expression in tumor tissues. Paraffin tissue blocks were processed, and EphA2 expression was analyzed by immunohistochemistry. B. Western blot analysis of EphA2 expression in tumor samples 7 days post treatment. Two tumor samples per MSV/EphA2 treatment group were included. C. Tumor microvessel density analysis. Tumor microvessels were determined after immunohistochemical staining for CD31. D. TUNEL assay for cell apoptosis. Tumor sections from each group were stained for TUNEL. Representative slides from each group are shown. E. Quantitation of tumor microvessels and apoptotic cells. The number of microvessels and apoptotic cells was counted. Five fields per slide and at least 5 slides per group (all from different animals) were counted. *: p<0.05, **: p<0.01.
Knockdown of EphA2 expression resulted in reduction of total number of microvessels in tumor samples (Fig. 5C). Multiple apoptotic cells could be identified in MSV/EphA2 treated tumors, but not in samples from the control groups (Fig. 5D). Dose-dependent cell apoptosis was apparent in the MSV/EphA2 treatment groups (Fig. 5E). These results indicate that MSV/EphA2 siRNA is an effective agent to treat metastatic ovarian tumor.
Taxanes are widely used for the treatment of ovarian cancer in clinic. We treated SKOV3ip2 tumor-bearing mice with PTX, and compared therapeutic efficacy between monotherapy and combination therapy with MSV/EphA2 siRNA. As expected, PTX treatment alone significantly inhibited tumor growth, which was reflected by the 90% reduction in tumor weight and dramatic reduction in the number of tumor nodules (Fig. 4A). Interestingly, PTX treatment also caused slight reduction of EphA2 expression in the residual tumor nodules (Fig. 5A). This probably reflects the fact that the majority of tumor cells had been killed after 6-week treatment with this chemotherapy drug, since a single treatment with PTX did not alter EphA2 expression (Fig. 5B). The greatest therapeutic benefit was from combination treatment with PTX and MSV/EphA2. Almost all tumor nodules were eliminated, so that there was no tumor sample available from this treatment group for pathological analysis at the end of the 6-week treatment. This result provides support for an effective therapy strategy in clinic to target the EphA2 gene in combination with standard of care chemotherapy drugs.
Enhanced therapeutic efficacy in taxane-resistant tumor
While taxanes are effective drugs to treat ovarian cancers in clinic, most cancers eventually develop therapy resistance. We generated HeyA8-MDR mice to explore the possibility of fighting taxane-resistant tumor by knocking down EphA2 expression. The HeyA8-MDR tumor cells overexpress drug efflux transporters (20), and have cross-resistance to cisplatin, adriamycin, and many other chemotherapy drugs (17). As expected, no therapeutic efficacy was observed in mice treated with 50 μg DTX biweekly for 6 weeks (Fig. 6A). As with the SKOV3ip2 tumor mice, there was an 80% reduction of tumor weight in the HeyA8-MDR mice treated with MSV/15 μg EphA2 siRNA compared to those in the control groups (DTX and MSV/Scr). DTX and MSV/EphA2 combination treatment further reduced tumor weight, but not the number of tumor nodules (Fig. 6A).
Figure 6. Therapeutic efficacy of MSV/EphA2 in combination with docetaxel on HeyA8-MDR tumors.
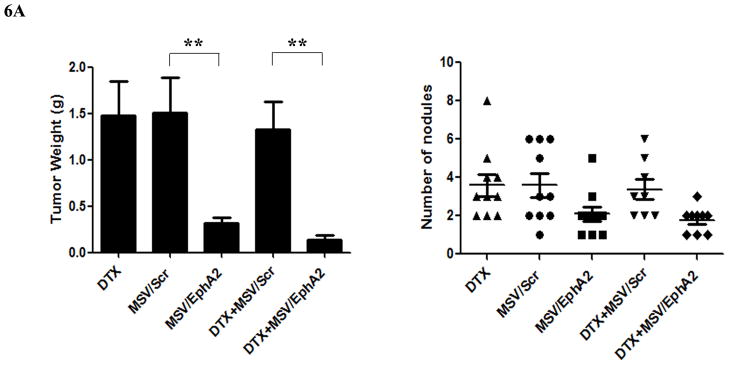
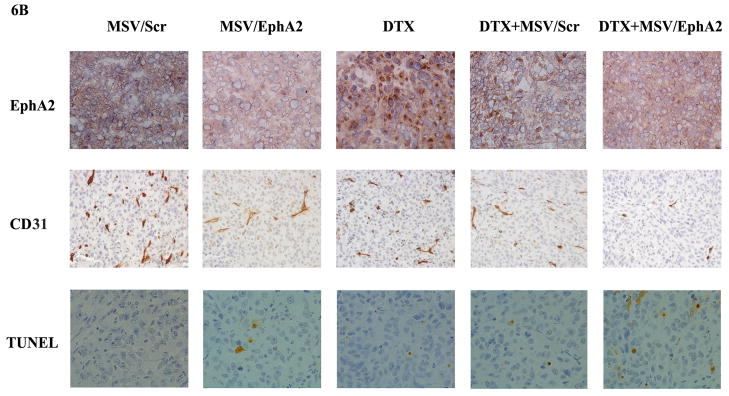
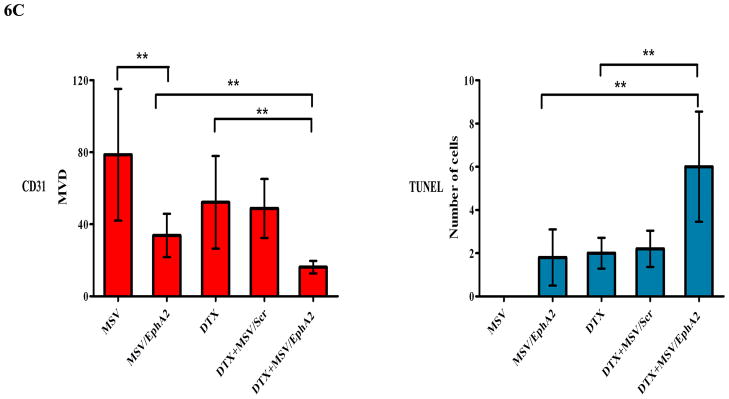
Nude mice were inoculated i.p. with HeyA8-MDR cells, and randomly allocated to one of five treatment groups (n = 10): 1) docetaxel (DTX), 2) MSV loaded with non-silencing scramble siRNA-DOPC (MSV/Control, 15 μg), 3) MSV loaded with 15 μg EphA2-siRNA-DOPC (MSV/EphA2, 15 μg), 4) DTX and MSV/Control combination (DTX + MSV/Control), 5) DTX and MSV/EphA2 combination (DTX + MSV/EphA2). Mice were treated biweekly for 6 weeks. At the end of the treatment, all mice were sacrificed, and total tumor weight and the number of tumor nodules were measured. A. Distribution of tumor weight and tumor nodules among the treatment groups. B. Immunohistochemical staining for EphA2 expression, tumor microvessels by CD31, and cell apoptosis by TUNEL assay. C. Quantitation of tumor microvessels and apoptotic cells. Five fields per slide and at least 5 slides per group (all from different animals) were counted. *: p<0.05, **: p<0.01.
As expected, MSV/EphA2 treatment resulted in knockdown of EphA2 expression, and consequently reduced number of microvessels and increased cell apoptosis in HeyA8-MDR tumor (Fig. 6B and 6C). In contrast to the SKOV3ip2 tumors, treatment with the chemotherapy drug did not significantly alter EphA2 expression level (Fig. 6B). DTX treatment reduced the number of microvessels (Fig. 6B and 6C); however, the result did not have any significant impact on overall tumor weight or the number of tumor nodules (Fig. 6A). Combination treatment reduced microvessel by 80% and dramatically increased the number of apoptotic cells in tumor tissues (Fig. 6B and 6C).
Discussion
We have demonstrated here efficient multistage delivery of siRNA to tumor tissues. The delivery system allowed for sustained release of nanoliposomal siRNA from the primary uptake sites of MSV and subsequent siRNA accumulation into tumors. Treatment with MSV/EphA2 siRNA sensitized tumors to chemotherapy and overcame drug resistance in mice bearing tumors overexpressing multidrug resistant genes.
Most newly diagnosed patients with ovarian cancer are treated with surgery and chemotherapy (21–23). Taxanes and cisplatin analogues are the front-line chemotherapeutic agents of choice for advanced ovarian cancer (22). Initial responsiveness to drug treatment is high; however, the majority of patients will relapse with resistant diseases. The poor patient survival is directly attributable to advanced and metastatic disease and the lack of effective treatment strategies. Drug resistance is the leading cause of therapy failure in the treatment of ovarian cancer (23). There are many potential mechanisms for resistance to chemotherapy treatment. Among them is P-glycoprotein-mediated efflux to reduce cellular accumulation of the drug (24). It has been suggested that the current therapy regimens only eliminate vulnerable cells, with the remaining cells eventually developing a drug-resistant phenotype, promoting tumor growth and metastasis. So there is an urgency in developing novel treatment strategies that will lead to better outcomes in patients with ovarian cancer. In the current study, we have demonstrated effective siRNA delivery to tumor tissues with the multistage system and synergy between MSV/EphA2 siRNA and chemotherapy drugs in the treatment of ovarian cancer with both taxane-sensitive and –resistant tumor mouse models.
The use of siRNA is an attractive strategy for cancer treatment by targeting essential genes for cancer cell survival. This strategy can be applied to the vast majority of cancer targets that are either considered non-druggable by conventional drug development approaches or their small molecule inhibitors are yet to be developed (25). However, application of siRNA therapeutics in clinic has proven to be challenging so far due to the lack of effective delivery vectors to overcome the multiple biological barriers (9, 10). Our multistage vector delivery system serves as a bridge from bench to clinic. In comparison to the hemispherical silicon particles used in our previous study (8), the discoidal silicon particles used in this study have optimal hemodynamic properties in circulation and larger surface interaction area with the tumor vasculature, and consequently, improved tumor accumulation (12, 13). In a recent study, we have shown that tumor accumulation of discoidal particles was dramatically improved over spherical particles of similar sizes (15). Interestingly, the 1000 × 400 nm discoidal particles had the best tumor accumulation comparing to bigger (1,800 × 600 nm) or smaller (600 × 200 nm) particles, and exhibited the best tumor-to-liver ratio, as the smaller particles were more extensively uptaken in the liver and spleen (13). Toxicity evaluation that discoidal MSV loaded with siRNA oligos did not trigger acute innate immune responses judged by changes in levels of 32 serum cytokines, chemokines, and colony-stimulating factors (26). Repetitive treatments with escalating dosages of MSV/siRNA did not cause sub-acute toxicities either including body weight changes, morphological changes of major organs, blood chemistry on liver and kidney functions, and hematology (26). These studies have demonstrated that discoidal silicon particles loaded with siRNA oligos in nanoliposomes are safe to use as cancer therapeutics. We have also developed a protocol for large-scale fabrication of discoidal silicon particles. Instead of fabricating particles one layer at the time, we can now manufacture them in stacks of 10 to 20 layers at a time (unpublished data), with enough flexibility to fabricate silicon particles with any size and porosity. This protocol can be directly translated to large-scale cGMP production of MSV/siRNA. Thus, MSV/EphA2 is a novel approach to solid tumor treatment and it warrants further drug development.
Translational Relevance.
This study describes experimental therapy of ovarian cancer by suppressing EphA2 expression. Although RNA interference has been widely used to demonstrate gene functions in vitro, in vivo application for cancer therapy has remained to be challenging due to lack of effective delivery vehicles for the gene-silencing agents. Here we have used the multistage vector (MSV) system to achieve efficient delivery of liposomal EphA2 siRNA oligos to metastatic ovarian tumor tissues. Sustained release of EphA2 siRNA from MSV resulted in effective knockdown of gene expression in tumor tissues. Treatment of murine model of SKOV3ip2 tumor with MSV/EphA2 siRNA in combination with paclitaxel completely inhibited tumor growth. In addition, combination treatment with MSV/EphA2 siRNA and docetaxel inhibited growth of HeyA8-MDR tumors that were other wise resistant to taxanes. Our study provides a strong rational for the development of EphA2 siRNA therapeutics for the treatment of ovarian cancer in clinic.
Acknowledgments
The authors acknowledge financial support from the following sources: Department of Defense grant W81XWH-09-1-0212, National Institute of Health grants U54CA143837, U54CA151668, RC2GM092599, and P50 CA083639, the CPRIT grant RP121071 from the state of Texas, the RGK Foundation and the Gilder Foundation, and the Ernest Cockrell Jr. Distinguished Endowed Chair. M. Ferrari is the founding scientist and a member of the Board of Directors of Leonardo Biosystems, a member of Board of Directors of Arrow Head Research Corporation, and hereby discloses potential financial interests in the companies. The other authors disclosed no potential conflicts of interest.
References
- 1.Walker-Daniels J, Coffman K, Azimi M, Rhim JS, Bostwick DG, Snyder P, et al. Overexpression of the EphA2 tyrosine kinase in prostate cancer. Prostate. 1999;41(4):275–80. doi: 10.1002/(sici)1097-0045(19991201)41:4<275::aid-pros8>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 2.Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001;61(5):2301–6. [PubMed] [Google Scholar]
- 3.Landen CN, Jr, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65(15):6910–8. doi: 10.1158/0008-5472.CAN-05-0530. [DOI] [PubMed] [Google Scholar]
- 4.Thaker PH, Deavers M, Celestino J, Thornton A, Fletcher MS, Landen CN, et al. EphA2 expression is associated with aggressive features in ovarian carcinoma. Clin Cancer Res. 2004;10(15):5145–50. doi: 10.1158/1078-0432.CCR-03-0589. [DOI] [PubMed] [Google Scholar]
- 5.Kinch MS, Moore MB, Harpole DH., Jr Predictive value of the EphA2 receptor tyrosine kinase in lung cancer recurrence and survival. Clin Cancer Res. 2003;9(2):613–8. [PubMed] [Google Scholar]
- 6.Merritt WM, Kamat AA, Hwang JY, Bottsford-Miller J, Lu C, Lin YG, et al. Clinical and biological impact of EphA2 overexpression and angiogenesis in endometrial cancer. Cancer Biol Ther. 2011;10(12):1306–14. doi: 10.4161/cbt.10.12.13582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin YG, Han LY, Kamat AA, Merritt WM, Landen CN, Deavers MT, et al. EphA2 overexpression is associated with angiogenesis in ovarian cancer. Cancer. 2007;109(2):332–40. doi: 10.1002/cncr.22415. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka T, Mangala LS, Vivas-Mejia PE, Nieves-Alicea R, Mann AP, Mora E, et al. Sustained small interfering RNA delivery by mesoporous silicon particles. Cancer Res. 2010;70(9):3687–96. doi: 10.1158/0008-5472.CAN-09-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrari M. Frontiers in cancer nanomedicine: directing mass transport through biological barriers. Trends Biotechnol. 2010;28(4):181–8. doi: 10.1016/j.tibtech.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen H, Sun T, Ferrari M. Nanovector delivery of siRNA for cancer therapy. Cancer Gene Ther. 2012 doi: 10.1038/cgt.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrari M. Vectoring siRNA therapeutics into the clinic. Nat Rev Clin Oncol. 2010;7(9):485–6. doi: 10.1038/nrclinonc.2010.131. [DOI] [PubMed] [Google Scholar]
- 12.Decuzzi P, Godin B, Tanaka T, Lee SY, Chiappini C, Liu X, et al. Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release. 2010;141(3):320–7. doi: 10.1016/j.jconrel.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 13.van de Ven AL, Kim P, Haley O, Fakhoury JR, Adriani G, Schmulen J, et al. Rapid tumoritropic accumulation of systemically injected plateloid particles and their biodistribution. J Control Release. 2011 doi: 10.1016/j.jconrel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Godin B, Tasciotti E, Liu X, Serda RE, Ferrari M. Multistage nanovectors: from concept to novel imaging contrast agents and therapeutics. Acc Chem Res. 2011;44(10):979–89. doi: 10.1021/ar200077p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Godin B, Chiappini C, Srinivasan S, Alexander JF, Yokoi K, Ferrara M, et al. Discoidal porous silicon particles: fabrication and biodistribution in breast cancer bearing mice. Adv Funct Mater. 2012 doi: 10.1002/adfm.201200869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen H, You J, Zhang G, Ziemys A, Li Q, Bai L, et al. Cooperative, nanoparticle-enabled thermal therapy of breast cancer. Adv Healthcare Mater. 2012;1(1):84–9. doi: 10.1002/adhm.201100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halder J, Landen CN, Jr, Lutgendorf SK, Li Y, Jennings NB, Fan D, et al. Focal adhesion kinase silencing augments docetaxel-mediated apoptosis in ovarian cancer cells. Clin Cancer Res. 2005;11(24 Pt 1):8829–36. doi: 10.1158/1078-0432.CCR-05-1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serda RE, Mack A, van de Ven AL, Ferrati S, Dunner K, Jr, Godin B, et al. Logic-embedded vectors for intracellular partitioning, endosomal escape, and exocytosis of nanoparticles. Small. 2010;6(23):2691–700. doi: 10.1002/smll.201000727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 20.De Souza R, Zahedi P, Badame RM, Allen C, Piquette-Miller M. Chemotherapy dosing schedule influences drug resistance development in ovarian cancer. Mol Cancer Ther. 2011;10(7):1289–99. doi: 10.1158/1535-7163.MCT-11-0058. [DOI] [PubMed] [Google Scholar]
- 21.Kyrgiou M, Salanti G, Pavlidis N, Paraskevaidis E, Ioannidis JP. Survival benefits with diverse chemotherapy regimens for ovarian cancer: meta-analysis of multiple treatments. J Natl Cancer Inst. 2006;98(22):1655–63. doi: 10.1093/jnci/djj443. [DOI] [PubMed] [Google Scholar]
- 22.Parmar MK, Ledermann JA, Colombo N, du Bois A, Delaloye JF, Kristensen GB, et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet. 2003;361(9375):2099–106. doi: 10.1016/s0140-6736(03)13718-x. [DOI] [PubMed] [Google Scholar]
- 23.Yap TA, Carden CP, Kaye SB. Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer. 2009;9(3):167–81. doi: 10.1038/nrc2583. [DOI] [PubMed] [Google Scholar]
- 24.Parekh H, Wiesen K, Simpkins H. Acquisition of taxol resistance via P-glycoprotein- and non-P-glycoprotein-mediated mechanisms in human ovarian carcinoma cells. Biochem Pharmacol. 1997;53(4):461–70. doi: 10.1016/s0006-2952(97)83383-7. [DOI] [PubMed] [Google Scholar]
- 25.Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432(7014):173–8. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 26.Xu R, Huang Y, Guo X, Mai J, Koay EJ, Li Q, et al. Toxicity evaluation of multistage vectored ATM siRNA targeting DNA damage response pathways for breast cancer therapy. Small. 2012 (in press) [Google Scholar]