

Abstract
In the present study, the efficacy of indole-3-carbinol (I3C), a key bioactive component of cruciferous vegetables, for prevention of cancer in offspring exposed in utero to the environmental carcinogen dibenzo[def,p]chrysene (DBC) was evaluated using an estrogen receptor beta (ERβ) knockout mouse model. I3C was provided either through the maternal diet coincident with carcinogen exposure during pregnancy or directly to offspring post initiation with DBC. I3C was effective at reducing T-cell acute lymphoblastic lymphoma/leukemia (T-ALL)-related mortality in offspring only if provided via the maternal diet, although a gender difference in the role of ERβ in mediating this response was evident. In female offspring, chemoprevention of T-ALL by maternal dietary I3C required expression of ERβ; survival in Esr2 wild-type and heterozygous female offspring was >90% compared to 66% in Esr2 null females. Alternatively, ERβ status did not significantly impact the transplacental chemoprevention by I3C in males. The possible role of ERβ in mediating lung carcinogenesis or chemoprevention by I3C was similarly complicated. Lung tumor incidence was unaltered by either dietary intervention, whereas lung tumor multiplicity was substantially reduced in Esr2 null females on the control diet and marginally lower in Esr2 null males exposed to I3C via the maternal diet compared to their wild-type and heterozygous counterparts. These findings suggest that I3C may act via ERβ to prevent or suppress DBC-initiated transplacental carcinogenesis, but that the involvement of this receptor appears to differ depending on the cancer type and gender of the offspring.
Keywords: Cancer prevention, indole-3-carbinol, estrogen receptor beta, T-cell acute lymphoblastic leukemia, lung carcinogenesis, polycyclic aromatic hydrocarbon
Introduction
Indoles are degradation products of glucosinolates, which occur naturally in cruciferous vegetables, and have been widely studied for their chemopreventive properties (reviewed in 1). One such bioactive food chemical, indole-3-carbinol (I3C), has been shown to prevent or reduce the risk of cancer in a number of animal models of carcinogenesis including, in part, cancer of the colon, mammary gland, skin, lung and endometrium (see reviews 2, 3). Alternatively, dietary I3C can enhance tumorigenesis, particularly hepatocellular carcinogenesis in rat and trout (4, 5). The chemopreventive activity of I3C may be mediated by one or more of its acid concentration products, such as 3,3′-diindolylmethane (DIM). The anticancer effects of I3C (or its derivatives) are facilitated by interaction with several nuclear transcription factors, including the aryl hydrocarbon receptor (AhR), the estrogen receptor (ER), Sp1 or nuclear factor κB (NFκB) to alter processes associated with carcinogen detoxification, cell cycle progression, apoptosis and DNA repair (reviewed in 6).
The fetus is particularly sensitive to the toxic, teratogenic and carcinogenic effects of environmental chemicals due to underdeveloped detoxification and elimination pathways and unique sensitivities to chemicals. Therefore, there is great concern that perinatal exposure to environmental chemicals could be linked to childhood and adult cancers. Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous in the environment and are derived from incomplete combustion of biomass and fossil fuels. The most potent PAH tested to date is dibenzo[def,p]chrysene (DBC), also called dibenzo[a,l]pyrene, a component of PAH mixtures in the environment (7). DBC is a multi-organ carcinogen that initiates cancers of the skin, endometrium, ovary, lung, and liver as well as lymphoma (8–11). A recent study by members of our research group showed that in utero exposure to DBC (15 mg/kg orally on gestation day 17) in mice results in a high incidence of mortality in young adults due to aggressive T-cell acute lymphoblastic lymphoma/leukemia (T-ALL) (12), a disease that is also observed in human adolescents (13). All surviving offspring exposed to DBC in utero had lung tumors, and more than half of the males had liver tumors. Moreover, Yu et al., showed that dietary I3C consumed by the mother during gestation and lactation significantly reduced offspring mortality due to DBC-dependent T-ALL and reduced the number of lung tumors in offspring surviving to middle age (14). This study examined the influence of a responsive versus non-responsive AhR phenotype in mediating the protective effect of I3C against these cancers, although neither maternal nor fetal AhR phenotype influenced the efficacy of I3C as an anticancer agent. While I3C is an effective chemoprotective compound transplacentally, the mechanism by which it blocks or suppresses tumorigenesis in this cancer model is currently not known. Many of the molecular actions of I3C or its derivatives involve direct interaction with the ER, modulation of ER-dependent gene expression and/or alteration of estrogen metabolism pathways (15–19). Thus, it is possible that dietary I3C may act through the estrogen receptor to exert its anticancer effects in vivo.
The main objective of this study was to evaluate the role of ERβ, which is highly expressed in the fetal thymus and lung (20, 21), in modulating the chemopreventive properties of dietary I3C in an animal model of transplacental carcinogenesis using the ERβ knockout mouse model. Our working hypothesis was that protection by dietary I3C against DBC-initiated transplacental cancer required expression of ERβ in the offspring. Secondarily, we also examined the timing of dietary I3C exposure, either administered via the maternal diet or fed directly to offspring after weaning. Herein, we show for the first time that transplacental cancer prevention by I3C administered to the dam during gestation/lactation requires at least one gene copy of ERβ.
Methods
Chemicals
DBC (CAS # 191-30-0, formerly called dibenzo[a,l]pyrene) was obtained from the National Cancer Institute Chemical Reference Standard Repository and determined by HPLC analysis to be 98% pure DBC (12). I3C was obtained from Sigma-Aldrich (St. Louis, MO). All other chemicals used were of reagent grade and purchased from general laboratory suppliers.
Animals and design of transplacental cancer study
The Oregon State University Institutional Animal Care and Use Committee approved all procedures for the handling and treatment of mice in this study (protocol #3468). Mice were maintained in a pathogen free vivarium at 20 ± 1°C and 50 ± 10% humidity on a 12:12 hour dark:light cycle and housed in microisolator cages (Life Products, Inc., Seaford, DE) with Care FRESH bedding. Breeding pairs of male B6.129P2-Esr2tm1Unc/J (βERKO mouse; Esr2−/−) and female 129S1/SvImJ mice were obtained from Jackson Laboratories (Bar Harbor, ME) and acclimated to the vivarium for one week prior to breeding. The B6129SF1/J mouse strain was previously shown to be sensitive to DBC as a transplacental carcinogen (12). Because the βERKO mouse had been backcrossed to C57BL6 mice for eight generations at Jackson Laboratories, we bred Esr2−/− males with 129S1/SvImJ females (Esr2+/+) to generate heterozygous hybrids, B6129SF1/J-Esr2+/− for carcinogen exposure (Supplementary Fig. S1). Esr2 hybrid male and female mice were randomized to six experimental groups and paired for breeding. Females were monitored daily for detection of a vaginal plug, which was designated gestation day 0 (GD 0). On GD 17, dams in groups 1, 3 and 5 were dosed with an oral gavage of corn oil (5 ml/kg), whereas dams in groups 2, 4 and 6 were dosed with 15 mg/kg DBC in corn oil. The numbers of dams and offspring and the average litter sizes are presented in Supplementary Table S1.
AIN93G diet (Research Diets, Inc., New Brunswick, NJ) was provided to dams and then offspring until 3 months of age, at which point AIN93M diet was used for the remainder of the study. Pregnant mice and offspring in groups 1 and 2 were fed AIN93G/M diet without modification (control diet) until 10 months of age. Dams in experimental groups 3 and 4 were fed AIN93G diet supplemented with 2000 ppm I3C (I3C:Maternal, or I3C:M diet) from GD 9 until weaning at 3 weeks of age; offspring were then fed unmodified AIN93G/M diet until 10 months of age. Dams in groups 5 and 6 were fed unmodified AIN93G throughout pregnancy and lactation; after weaning at 3 weeks, offspring were fed AIN93G/M diet supplemented with 2000 ppm I3C (I3C:Offspring, or I3C:O diet) until 24 weeks of age followed by unmodified AIN93M diet until 10 months. Preparation, storage and quality control analysis of I3C-supplemented AIN diet has been described previously (22). All diets were provided ad lib., and I3C-supplemented diets were replaced daily.
All mice were observed daily for any signs of distress or discomfort. Any mice exhibiting signs of morbidity, pain or distress were humanely euthanized with an overdose of CO2 followed by cervical dislocation, and then necropsied. At necropsy, presence of any tumor was noted (thymic lymphoma, lung, liver, ovarian, skin, etc.) and the number and size (diameter measured by digital caliper) of lung and liver tumors were recorded.
Genotyping protocol
All offspring were genotyped by PCR for presence of the Esr2tm1Unc allele following the standard protocol provided by Jackson Laboratories. Briefly, genomic DNA was obtained from ear tissue samples (Qiagen DNeasy Blood and Tissue Kit) and then subject to standard PCR (Amplitaq Gold Master Mix, Life Technologies, Carlsbad, CA) using primers for the wild-type (oIMR3145, 5′-GTTGTGCCAGCCCTGTTACT), heterozygous (oIMR3146, 5′-TCACAGGACCAGACACCGTA) and mutant (oIMR3147, 5′-GCAGCCTCTGTTCCACATACAC) alleles. PCR products were separated by gel electrophoresis on a 2% agarose gel. Mice were classified as wild-type when a single PCR product of 106 bp was detected, as heterozygous when two products of 106 and 160 bp were detected, and mutant when only the 160 bp product was observed. The ratios of Esr2 genotypes for offspring in each treatment group are presented in Supplementary Table S1.
Histopathology
In the present study, all mice surviving to 10 months were euthanized and necropsied as described above. Multiple tissues (thymus, lung, liver, kidney, spleen, testes, ovaries, colon, skin and lymph nodes) were first examined by gross necropsy for abnormalities and then preserved in 10% formalin. Fixed tissues were processed to paraffin blocks and then subjected to hematoxylin and eosin staining for examination by a board-certified pathologist at the Utah Veterinary Diagnostics Laboratory. As has been observed repeatedly in prior studies (12, 14, 23, 24), between 3 and 6 months of age, approximately half of the offspring initiated with DBC in utero succumbed to lymphoma that was characterized as T-cell in origin (CD3+, B220−) with various antigen phenotypes (CD4−CD8+; CD4+CD8+), and thus, classified as T-ALL. Additionally, lung carcinogenesis (hyperplasia, adenoma, adenoma with progression and/or carcinoma) was evident in nearly all offspring surviving to 10 months of age, and some male offspring developed liver neoplasms. Histopathological analysis of samples from the present study performed at the Utah Veterinary Diagnostics Laboratory (in consultation with the Oregon State University Veterinary Diagnostics Laboratory) confirmed the same disease profile (lymphoma, lung adenomatous hyperplasia and adenoma, liver neoplasia) as was observed in prior studies using DBC as a transplacental carcinogen (12, 14, 23, 24). Representative histopathology images are shown in Supplementary Figure S2.
Statistical analyses
Following the example previously published (24), the general strategy for analyses of individual offspring responses was 1) to account for cluster (litter) effects in offspring data, 2) to assess main effects of the categorical predictors carcinogen, diet, Esr2 genotype and gender and 3) to assess Esr2 genotype and gender effects within each diet group in DBC-initiated offspring. First, simple univariate analyses were performed to determine the association between the categorical predictors carcinogen, diet, genotype and gender and offspring survival (SAS 9.3, lifetest procedure); all factors were considered potential significant contributors to the final model (Log rank test of equality over strata P values < 0.25; Supplemental Figure S3). All potential interactions between these experimental factors were similarly examined, although none were considered to be significant (P > 0.25). As the primary outcome of interest in this study was survival in carcinogen-exposed offspring, analyses of survival curves were limited to data for offspring initiated in utero with DBC using the SAS lifetest procedure (Kaplan-Meir method with Šidák correction for multiple comparisons). Initial tests examined main effects of the categorical variables diet, Esr2 genotype and gender, followed by pairwise tests to examine the effects of either Esr2 genotype within each DBC/diet group. These pairwise analyses were performed for all offspring, and then separately for male and female offspring. Similar analyses were performed using proportional hazards (Cox) regression analysis (SAS phreg procedure) with the robust sandwich score test to account for grouping of offspring by litter, which yielded comparable results (not shown). Graphical analysis of pairs of survival curves reasonably satisfied the proportional hazards assumption.
Analyses of lung tumor incidences were performed using a quasi-likelihood logistic regression (SAS genmod procedure) where the apparent variation between litters was used to account for over-dispersion in the grouped binomial data. Main effects of diet, gender and Esr2 genotype for DBC-initiated offspring were calculated, followed by pairwise analyses to determine effects of either Esr2 genotype or gender within each DBC/diet treatment group. Similar results were obtained using logistic regression ignoring litters (SAS logistic procedure; results not shown). Mixed models analyses (SAS mixed procedure) of tumor multiplicities (log transformed) or tumor sizes were performed with data for DBC-exposed offspring that survived to 10 months of age. Analyses were performed in a stepwise fashion as described above with the parameter litters included as a random factor to account for data over-dispersion. Again, these analyses were performed first for all offspring, and then separately for male and female offspring. Because of the relatively few liver tumors observed in this study, there was insufficient statistical power to reliably perform a robust analyses of the data on liver tumor outcome.
Results
T-cell acute lymphoblastic/lymphoma-dependent survival
Although a few offspring died prior to weaning (anemia related to DBC exposure), there was not a significant effect of carcinogen or diet group on litter size or body weight at weaning (Supplementary Table S1). Offspring born to dams initiated in utero on gestation day 17 with 15 mg/kg DBC exhibited a high rate of mortality between three to six months of age due to aggressive T-ALL (Fig. 1A; Table 1), whereas deaths after six months of age were primarily attributed to lung cancer morbidity. Survival analyses were performed using mortality data for death due to T-ALL (Fig. 1) or death due to any cause (results in Supplementary Table S2, figure not shown). When considering offspring survival data stratified according to diet group, survival in male offspring exposed to I3C via the maternal diet (91%) was significantly greater (P = 0.003) compared to offspring exposed to the control diet (61%) (Fig. 1B). However, survival in offspring fed I3C directly (66%) was not significantly different from the control group (P = 0.9896). On the other hand, an overall significant effect of diet was not evident in females, where survival was 78% in control animals, 89% in I3C:M-treated offspring and 75% in I3C:O-treated offspring (Fig. 1C).
Figure 1.
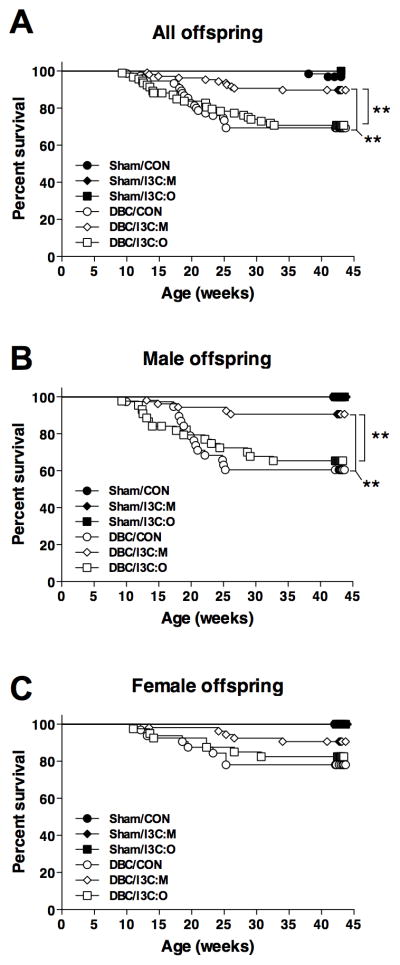
Impact of experimental diets and Esr2 genotype on T-ALL dependent mortality in offspring initiated in utero with DBC on gestation day 17. A–C, Survival curves for all offspring (A), male offspring (B) and female offspring (C) according to carcinogen/diet groups: sham/control (●), sham/I3C:M (◆), Sham/I3C:O (■), DBC/control (○), DBC:I3C/M (◇) and DBC/I3C:O (□). ** P < 0.01, significant difference in survival for the indicated pairwise comparisons as determined by Lifetables (Kaplan Meir method) analysis. Complete statistical results for these analyses (as well as results of similar analyses for mortality due to any cause, such as anemia or other cancers) are provided in Supplementary Table S1.
Table 1.
Effects of experimental diet, gender and Esr2 genotype on offspring survival to 10 months of age.
| All offspring
|
Male offspring
|
Female offspring
|
||||
|---|---|---|---|---|---|---|
| Any cause | T-ALL* | Any cause | T-ALL* | Any cause | T-ALL* | |
| Offspring stratified by diet group | ||||||
| Sham-initiated | ||||||
| Control | 3.1% (2/65) | 0.0% (0/65) | 0.0% (0/26) | 0.0% (0/26) | 0.0% (0/39) | 0.0% (0/39) |
| I3C:M | 0.0% (0/65) | 0.0% (0/65) | 0.0% (0/34) | 0.0% (0/34) | 0.0% (0/31) | 0.0% (0/31) |
| I3C:O | 1.6% (1/64) | 0.0% (0/64) | 0.0% (0/29) | 0.0% (0/29) | 2.9% (1/35) | 0.0% (0/34) |
| DBC-initiated | ||||||
| Control | 56.7% (68/120)a | 30.7% (23/75)a | 60.3% (35/58)a | 39.5% (15/38)a | 53.2% (33/62)a | 21.6% (8/37)a |
| I3C:M | 20.0% (24/120)b | 10.3% (11/107)b | 19.0% (11/58)b | 9.4% (5/53)b | 21.3% (13/61)b | 11.1% (6/54)a |
| I3C:O | 45.5% (55/121)a | 29.0% (27/93)a | 53.2% (33/62)a | 34.1% (15/44)a | 37.3% (22/59)ab | 24.5% (12/49)a |
| DBC-exposed offspring stratified by gender | ||||||
| Male | 44.1% (79/179)a | 25.9% (35/135)a | ||||
| Female | 37.4% (68/182)a | 18.3% (26/142)a | ||||
| DBC-exposed offspring stratified by Esr2 genotype | ||||||
| Wild-type | 28.2% (24/85)a | 17.6% (13/74)a | 30.2% (13/43)a | 21.1% (8/38)a | 26.2% (11/42)a | 13.9% (5/36)a |
| Heterozygous | 32.9% (56/170)a | 18.0% (25/139)a | 38.6% (34/88)a | 23.9% (17/71)a | 26.8% (22/82)a | 11.8% (8/68)a |
| Null | 50.8% (30/59)ab | 29.3% (12/41)a | 54.8% (17/31)a | 30.0% (6/20)a | 46.4% (13/28)a | 28.6% (6/21)a |
| DBC-exposed offspring stratified by diet, then genotype | ||||||
| CON diet group | ||||||
| Wild-type | 38.7% (12/31)a | 29.6% (8/27)a | 37.5% (6/16)a | 33.3% (5/15)a | 40.0% (6/15)a | 25.0% (3/12)a |
| Heterozygous | 57.1% (32/56)a | 31.4% (11/35)a | 62.1% (18/29)a | 42.1% (8/19)a | 51.9% (14/27)a | 18.8% (3/16)a |
| Null | 66.7% (10/15)a | 28.6% (2/7)a | 66.7% (4/6)a | 33.3% (1/3)a | 66.7% (6/9)a | 4.0% (1/4)a |
| I3C:M diet group | ||||||
| Wild-type | 4.0% (1/25)a | 4.0% (1/25)a | 7.7% (1/13)a | 7.7% (1/13)a | 0.0% (0/12)a | 0.0% (0/12)a |
| Heterozygous | 10.8% (7/65)a | 4.9% (3/61)a | 23.1% (3/31)a | 6.7% (2/30)a | 11.8% (4/34)a | 3.2% (1/31)a |
| Null | 41.7% (10/24)b | 30.0% (6/20)b | 38.5% (5/13)a | 20.0% (2/10)a | 45.5% (5/11)b | 40.0% (4/10)b |
| DBCI3C:O diet group | ||||||
| Wild-type | 37.9% (11/29)a | 18.2% (4/22)a | 42.9% (6/14)a | 20.0% (2/10)a | 33.3% (5/15)a | 16.7% (2/12)a |
| Heterozygous | 34.7% (17/49)a | 25.6% (11/43)a | 46.4% (13/28)a | 31.8% (7/22)a | 19.0% (4/21)a | 19.0% (4/21)a |
| Null | 50.0% (10/20)a | 28.6% (4/14)a | 66.7% (8/12)a | 42.9% (3/7)a | 25.0% (2/8)a | 14.3% (1/7)a |
Note: Different superscripts indicate a significant difference within each comparison group (as indicated by row headings). Life tables analyses (Kaplan-Meir method) (SAS Lifetest procedure) of survival in DBC-initiated offspring were performed in a stepwise manner, wherein death due to any cause or to T-ALL were considered separately. Effects of diet, gender or genotype in sham-initiated animals were not considered as so few deaths occurred in these groups. Initial analyses included all data within the DBC exposure group stratified by gender, diet group or genotype. Then, pair-wise analyses for Esr2 genotype within each DBC/diet group were performed. Analyses were run irrespective of different genders, as well as separately for male and female offspring. Log-rank, Šidák-adjusted P values for pair-wise comparisons between indicated data groups are available in Supplementary Table S2. Incidences of mortality or morbidity are reported as percentages (deaths/total).
Animals that died or were euthanized due to a cause unrelated to lymphoma (anemia, other tumors [lung, liver, skin or ovarian] or cause unknown) during the course of the study were excluded from this tally. Thus, these numbers represent mortality specific to T-cell acute lymphoblastic lymphoma/leukemia (T-ALL).
Although only male offspring appeared to be responsive to the protective effects of maternal dietary I3C, beneficial effects of this intervention were observed in females when diet group and Esr2 genotype were considered as nested factors in the statistical analysis (Supplementary Table S2). Figure 2 shows the impact of Esr2 genotype on survival of all offspring (Fig. 2A–C), males only (Fig. 2D–F) or females only (Fig. 2G–I) stratified by experimental diet. In the absence of any diet modification for dams or offspring, Esr2 genotype did not significantly impact male or female offspring survival in the control diet group (Fig. 2D,G). However, in offspring exposed to I3C via the maternal diet, lymphoma-dependent survival in females expressing one (100%) or both (97%) wild-type Esr2 alleles was significantly greater (P = 0.0051 and P = 0.0058, respectively) than survival in Esr2 null females (60%) (Fig. 2H). Alternatively, Esr2 genotype did not significantly alter male offspring survival in the I3C:M diet group (Fig. 2E). Moreover, Esr2 status in offspring fed I3C directly post carcinogen exposure did not influence survival for either sex (Fig. 2F,I). Similar results were observed when deaths due to any cause were included in the statistical model (Supplementary Table S2).
Figure 2.

Impact of Esr2 genotype on T-ALL dependent mortality in offspring initiated with DBC in utero within each experimental diet group. A–C, Survival curves for all offspring (A–C), male offspring (D–F) or female offspring (G–I) within each diet group: control (A, D, G), I3C:M (B,E,H) or I3C:O (C,F,I). Data are stratified according to Esr2 genotype: wild-type (○), heterozygous (△) and null (■). * P <0.05; ** P < 0.01; *** P < 0.001, significant difference in survival for the indicated pairwise comparisons as determined by Lifetables (Kaplan Meir method) analysis. Complete statistical results for these analyses (as well as results of similar analyses for mortality due to any cause, such as anemia or other cancers) are provided in Supplementary Table S1.
Lung carcinogenesis in 10 month-old survivors
Morbidity due to lung tumorigenesis (adenoma, adenocarcinoma) was generally greater than 80% in 10 month-old surviving male and female offspring initiated in utero with DBC compared to a spontaneous incidence of <12% and <3% in sham-initiated male and female offspring, respectively (Table 2). Dietary I3C did not significantly reduce lung tumor incidence whether administered via the maternal diet or fed directly to the offspring in either males or females (Supplementary Table S3). A modest, but significant, decrease in lung tumor incidence was observed in Esr2 wild-type males in the control diet group compared to their heterozygous counterparts (Table 2).
Table 2.
Effects of experimental diet, gender and Esr2 genotype on lung tumorigenesis in 10-month old surviving offspring
| All offspring
|
Male offspring
|
Female offspring
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Incidence | Multiplicity | Size | Incidence | Multiplicity | Size | Incidence | Multiplicity | Size | |
| Offspring stratified by diet group | |||||||||
| Sham-initiated | |||||||||
| CON | 1.6% (1/63) | 1.0±0.0 | 3.5±0.0 | 0.0% (0/26) | 2.6% (1/39) | 1.0±0.0 | 3.5±0.0 | ||
| I3C:M | 6.2% (4/65) | 1.5±0.3 | 1.6±0.3 | 11.8% (4/34) | 1.5±0.3 | 1.6±0.3 | 0.0% (0/31) | ||
| I3C:O | 4.8% (3/63) | 1.0±0.0 | 1.3±0.3 | 6.9% (2/29) | 1.0±0.0 | 1.5±0.5 | 2.9% (1/34) | 1.0±0.0 | 1.1±0.0 |
| DBC-initiated | |||||||||
| CON | 90.6% (48/53)a | 13.1±1.4a | 1.7±0.1ab | 87.5% (21/24)a | 11.9±1.6ab | 1.9±0.2a | 93.1% (27/29)a | 14.0±2.1a | 1.6±0.2a |
| I3C:M | 91.7% (88/96)a | 9.2±0.8b | 1.9±0.1a | 91.7% (44/48)a | 9.8±1.2a | 2.0±0.1a | 91.7% (44/48)a | 8.8±1.1a | 1.7±0.2a |
| I3C:O | 87.7% (57/65)a | 13.7±1.2a | 1.5±0.1b | 92.9% (26/28)a | 16.8±1.7b | 1.5±0.1b | 83.8% (31/37)a | 11.0±1.4a | 1.4±0.1a |
| DBC-exposed offspring stratified by gender | |||||||||
| Male | 91.0% (91/100)a | 12.2±0.9a | 1.8±0.1a | ||||||
| Female | 89.5% (102/114)a | 10.9±0.9a | 1.6±0.1a | ||||||
| DBC-exposed offspring stratified by Esr2 genotype | |||||||||
| Wild-type | 86.9% (53/61)a | 11.6±1.1a | 1.7±0.1a | 80.0% (24/30)a | 13.3±1.8a | 1.9±0.2a | 93.5% (29/31)a | 10.2±1.4a | 1.5±0.1a |
| Heterozygous | 90.4% (103/114)a | 12.4±0.9a | 1.7±0.9a | 96.3% (52/54)b | 12.4±1.1a | 1.7±0.1a | 85.0% (51/60)a | 12.4±1.4a | 1.7±0.2b |
| Null | 93.1% (27/29)a | 7.5±1.2b | 1.9±0.3a | 92.9% (13/14)ab | 8.8±2.1a | 2.1±0.3a | 93.3% (14/15)a | 5.8±1.4b | 1.7±0.4b |
| DBC-exposed offspring stratified by diet, then genotype | |||||||||
| CON diet group | |||||||||
| Wild-type | 84.2% (16/19)a | 11.2±2.1a | 1.7±0.1a | 70.0% (7/10)a | 10.6±3.3a | 2.0±0.3a | 100.0% (9/9)a | 12.9±2.9a | 1.5±0.2a |
| Heterozygous | 96.0% (24/25)a | 14.4±2.0a | 1.7±0.2a | 100.0% (12/12)a | 11.9±2.0a | 1.8±0.3a | 92.3% (12/13)a | 16.8±3.4a | 1.5±0.1a |
| Null | 80.0% (4/5)a | 9.0±4.3a | 2.6±1.3a | 100.0% (2/2)a | 16.5±0.5a | 1.5±0.3a | 66.7% (2/3)a | 1.5±0.5b | 3.7±2.9b |
| I3C:M diet group | |||||||||
| Wild-type | 91.7% (22/24)a | 10.0±1.4a | 1.8±0.2a | 83.3% (10/12)a | 10.0±2.3ab | 2.2±0.4a | 100.0% (12/12)a | 8.6±1.6a | 1.5±0.2a |
| Heterozygous | 91.4% (53/58)a | 10.5±1.1a | 1.9±0.2a | 96.4% (27/28)a | 11.3±1.6a | 1.8±0.1a | 86.7% (26/30)a | 9.6±1.7a | 1.9±0.4a |
| Null | 92.9% (13/14)a | 4.6±1.2b | 2.0±0.3a | 87.5% (7/8)a | 3.6±1.1b | 2.5±0.5a | 100.0% (6/6)a | 5.8±2.3a | 1.4±0.2a |
| I3C:O diet group | |||||||||
| Wild-type | 83.3% (15/18)a | 14.8±2.5a | 1.4±0.1a | 87.5% (7/8)a | 20.6±3.1a | 1.4±0.1a | 80.0% (8/10)a | 9.8±2.9a | 1.5±0.1a |
| Heterozygous | 83.9% (26/31)a | 14.3±1.7a | 1.5±0.1a | 92.9% (13/14)a | 14.8±2.4a | 1.5±0.1a | 76.5% (13/17)a | 13.8±2.4a | 1.5±0.1a |
| Null | 100.0% (10/10)a | 9.9±2.4a | 1.4±0.2a | 100.0% (4/4)a | 14.0±4.5a | 1.7±0.3a | 100.0% (6/6)a | 7.2±2.2a | 1.3±0.1a |
Note: Tallies for lung tumor incidence include only animals that survived to 10 months of age. Tumor multiplicity was calculated as the average number of tumors per tumor-bearing animal ± SEM. Average tumor size was estimated by the diameter (mm) ± SEM. For all statistical analyses, different superscripts indicate a significant difference within each comparison group (as indicated by row headings). P values of the differences of least squares means for the indicated comparisons are available in Supplementary Table S3. Effects of diet, gender or genotype in sham-initiated animals were not considered as so few animals with lung tumors were observed in these groups. Analyses of lung tumor incidences in 10 month-old survivors initiated with DBC were performed using a quasi-likelihood logistic regression (SAS Genmod procedure) where the apparent variation between litters was used to account for over-dispersion in the grouped binomial data. Mixed models analyses (SAS Mixed procedure) of tumor multiplicities (log-transformed) and tumor sizes were performed with data for DBC-exposed offspring that survived to 10 months of age. The parameter litters was included as a random factor to account for data over-dispersion. Analyses were performed for all DBC-initiated offspring, and then separately for male and female offspring.
Significant main effects of Esr2 genotype on lung tumor multiplicity in 10-month survivors were detected in this study when considering DBC-exposed offspring irrespective of diet group (7.5±1.2 tumors/animal for Esr2 null offspring compared to 11.6±1.1 and 12.4±0.9 for wild-type and heterozygous offspring, respectively, P ≤0.01), although this effect was predominant in females (Supplementary Table S2). On the other hand, significant main effects of experimental diet on lung tumor multiplicity were also observed, irrespective of Esr2 genotype, primarily when both males and females were considered collectively (9.2±0.8 tumors/animal for offspring in the I3C:M diet group compared to 13.1±1.4 and 13.7±1.2 for offspring in the control and I3C:O diet groups, respectively (P=0.042 and 0.006, respectively).
A further examination of the interaction of diet and Esr2 genotype suggested a complex response. Esr2 null female offspring in the control diet group had significantly fewer lung tumors than their wild-type or heterozygous counterparts (P=0.0241 and 0.0206, respectively). However, one should accept this statistical observation with caution, given that few Esr2 null females are present in the control diet group. On the other hand, Esr2 null male offspring in the I3C:M diet group had (marginally) fewer lung tumors than Esr2 wild-type (trend for significance, P=0.0822) or heterozygous offspring (P=0.0257). Even though these observations point to an interaction between the I3C:M diet and Esr2 genotype in male and female offspring, a test for this interaction was, in fact, not significant (P = 0.8795) suggesting that the influence of Esr2 status on lung tumor multiplicity was not strictly dependent on the experimental diet group. Finally, although a significant effect of Esr2 genotype on lung tumor size was observed in control female offspring (Supplementary Table S3), this observation cannot be considered robust due to the small sample size for Esr2 null females.
Liver carcinogenesis in 10 month-old survivors
Incidence, multiplicity and size of liver tumors are presented in Supplementary Table S4. Because this study was not designed to examine liver cancer as a primary outcome, an insufficient number of offspring survived to 10 months of age to allow for a thorough statistical analysis of liver tumor data.
Discussion
Although dietary I3C has great potential in the field of cancer chemoprevention, the current lack of understanding of the mechanism(s) by which it exerts its chemopreventive properties has caused some concern regarding its possible utility as a preventive and/or therapeutic agent for human carcinogenesis and its widespread, uncontrolled use as a dietary supplement by the public. Previously, members of our research team demonstrated that I3C administered via the maternal diet during gestation and lactation conferred substantial protection to offspring against DBC-initiated transplacental cancer (14). A primary goal of the present study was examine the role of ERβ in mediating transplacental chemoprevention by I3C. Our experimental design allowed us to test multiple key hypotheses related to I3C prevention of DBC-initiated transplacental cancer, specifically regarding the timing of intervention with dietary I3C and the role of ERβ in mediating the anticancer effects of I3C in vivo.
We report herein for the first time that dietary I3C is effective at blocking DBC-initiated T-ALL only when administered via the maternal diet. Importantly, this transplacental chemoprevention requires expression of ERβ in female offspring. Moreover, the protection conferred to these female offspring was substantial, as nearly all mice that were exposed to I3C during pregnancy and lactation and that carried at least one copy of the wild-type Esr2 allele survived to adulthood. To our knowledge, this is the first demonstration that ERβ mediates the anticancer activity of I3C in vivo. On the other hand, protection by maternal dietary I3C was independent of Esr2 genotype in males, as survival in all male offspring was substantially greater in this group compared to control animals or those fed I3C directly. This apparent difference in the role of ERβ in mediating the chemopreventive activity of maternal dietary I3C against DBC-initiated T-ALL points to possible distinct modes of action for this bioactive food chemical, or its derivative(s), in males and females, although the physiological basis for such a difference is not yet clear.
Prior to this study, no information was available regarding the beneficial or deleterious effects of I3C consumption by offspring that have been exposed in utero to a chemical carcinogen. Our results show that dietary I3C is most effective when administered coincidently with the carcinogen and that I3C provided either via the maternal diet or directly to the offspring does not increase risk of T-ALL, lung or liver tumorigenesis in this transplacental cancer model. Others have shown that I3C fed long-term subsequent to carcinogen exposure in rat and rainbow trout promotes liver tumorigenesis (4, 5), although this phenomenon was not observed in our study, pointing to a likely species difference in the impact of I3C on liver tumor development. Similarly, Oganesian et al., showed that consumption of I3C reduced hepatocarcinogenesis in mice that were initiated with diethylnitrosamine as infants (25).
Several mechanisms exist by which I3C or its acid condensation products can modulate the influence of estrogens on tumorigenesis including alteration of estrogen metabolism, direct interaction with the ER and modulation of estrogen signaling pathways (reviewed by 6). Importantly, I3C and some of its derivatives are known ligands for the ER exhibiting both estrogenic and anti-estrogenic effects in human breast cancer cells (26–28). The anti-estrogenic properties of I3C and DIM in human breast and prostate cancer cells may be mediated by the AhR, demonstrating that cross talk exists between these two transcription factor signaling pathways (29–31). Alternatively, in the trout liver, I3C and DIM are potent xenoestrogens that show moderate affinity for the hepatic ER and induce a transcriptional profile similar to that of estradiol (32, 33). Finally, I3C and other acid condensation products modulate transcription of many genes that have the potential to counteract the proliferative effects of estrogen, including genes associated with growth arrest and apoptosis (15, 34–36).
Since the discovery in 1996 of two ER subtypes with unique expression patterns (37), the relative roles of ERα and ERβ in development of cancer in both estrogen target and non-target tissues has been a subject of great interest. ERβ is the predominant form expressed in the human fetal thymus, and a large portion of lymphocytes in lymph nodes and multiple human lymphoma cell lines preferentially express ERβ (20, 38). Estrogen is critical for thymus and lymphocyte development via ERα, whereas ERβ mediates estrogen-induced thymic atrophy during early adulthood and pregnancy (39). Evidence suggests that the negative effects of estrogen on the immune system are mediated by ERβ because lack of ERβ expression in the aged ERβ knockout mouse can lead to myeloproliferative disease, which resembles human chronic myeloid leukemia (40). The importance of estrogen in the immune system is also demonstrated by increased risk of non-Hodgkin lymphoma in individuals expressing a polymorphism that enhances CYP17A1 activity, a key enzyme in the estrogen biosynthetic pathway (41, 42). However, in the present study, ERβ status in offspring did not alter lymphoma-dependent survival in the absence of dietary intervention, suggesting that actions of endogenous estrogens via ERβ do not have a strong influence on the development of T-ALL in this mouse model of transplacental carcinogenesis by DBC.
Estrogen plays an important role in lung development, particularly in females (21), and is likely responsible for the greater susceptibility of women to chronic pulmonary disease, lung cancer and the deleterious effects of tobacco (43, 44). ERβ is the sole subtype expressed in human fetal lung (20) and the predominant form expressed in mouse lung (21). Moreover, immunohistological analysis of archived or surgically resected human lung tumors detected ERβ in more than half of the tumors evaluated, whereas ERα was not expressed (45, 46). In the present study, we report that lack of ERβ expression substantially reduced lung tumor multiplicity in surviving adult female offspring fed a control diet and in male offspring that were exposed to I3C via the maternal diet, although ERβ status did not impact lung tumor incidence. The apparent contradictory observation that ERβ facilitates chemoprevention of T-ALL by I3C, whereas the loss of ERβ confers modest suppression of lung carcinogenesis (depending on the dietary regimen), suggests that the role of this nuclear receptor in mediating the anticancer activity of I3C may differ depending on the cancer type. However, the observation that lung tumor multiplicity in ERβ null offspring was consistently lower compared to wild-type or heterozygous counterparts irrespective of dietary treatment (see DBC-exposed offspring stratified by genotype in Table 2 and Supplementary Table S3), points to a potential role of this steroid receptor in lung chemical carcinogenesis, while rendering any specific conclusions about the relative role of ERβ in mediating I3C-dependent suppression of lung carcinogenesis speculative at this time. Additionally, the loss of some offspring to lymphoma in this model of transplacental carcinogenesis can be considered a confounding factor when interpreting lung tumor outcome, especially considering the substantial difference in survival among some treatment groups. In order to decisively determine the role of ERβ in mediating lung tumorigenesis in this cancer model, a different dosing strategy (lower DBC dose or intermittent dosing) or alternative mouse strain (e.g., A/J mouse) could be employed to evaluate lung tumors without the confounding effects of T-ALL.
In summary, administration of I3C via the maternal diet provides substantial protection to offspring initiated in utero with the environmental carcinogen DBC, and ERβ likely mediates this chemoprevention by I3C in female offspring. Collectively, results of this and prior studies by our research team lend weight to the argument that dietary I3C is an effective anticancer agent in vivo. Furthermore, our observations point to the critical gestation/lactation window of exposure for transplacental chemoprevention by I3C. However, the fact that this compound (or its bioactive derivative) may act via ERβ, a critical component of the endocrine system, to modulate carcinogenesis may raise some concern about its suitability as a dietary supplement during pregnancy.
Supplementary Material
Acknowledgments
The authors would like to thank Mandy Louderback, Marilyn Henderson, Lisbeth Siddens, David Castro, Lyndsey Shorey, Deanna Larson, Amanda Hagman, Trevor Fish and Brittany Packard and the staff of the Laboratory Animal Research Center at Oregon State University for their excellent technical assistance. We also thank Dr. S. Clay Isom for reviewing a draft of this manuscript.
Grant Support
This research was supported, in part, by the Utah Agricultural Experiment Station, Utah State University, and approved as journal paper number 8440. Support for this study was also provided by the Linus Pauling Institute at Oregon State University. Funding was provided by the following PHS grants from NIH: F32 ES014777 to A.D.B., R21 CA135523 to A.D.B. and P01 CA090890 to D.E.W.
Abbreviations
- AhR
aryl hydrocarbon receptor
- DBC
dibenzo[def,p]chrysene
- ERβ
estrogen receptor beta
- I3C
indole-3-carbinol
- I3C:M
indole-3-carbinol via maternal diet
- I3C:O
indole-3-carbinol via offspring diet
- PAH
polycyclic aromatic hydrocarbon
- T-ALL
T-cell acute lymphoblastic lymphoma/leukemia
Footnotes
COI statement: The authors have no conflicts of interest to disclose.
References
- 1.Murillo G, Mehta RG. Cruciferous vegetables and cancer prevention. Nutr Cancer. 2001;41(1–2):17–28. doi: 10.1080/01635581.2001.9680607. [DOI] [PubMed] [Google Scholar]
- 2.Bradlow HL. Review. Indole-3-carbinol as a chemoprotective agent in breast and prostate cancer. In Vivo. 2008;22(4):441–5. [PubMed] [Google Scholar]
- 3.Weng JR, Tsai CH, Kulp SK, Chen CS. Indole-3-carbinol as a chemopreventive and anti-cancer agent. Cancer Lett. 2008;262(2):153–63. doi: 10.1016/j.canlet.2008.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoner G, Casto B, Ralston S, Roebuck B, Pereira C, Bailey G. Development of a multi-organ rat model for evaluating chemopreventive agents: efficacy of indole-3-carbinol. Carcinogenesis. 2002;23(2):265–72. doi: 10.1093/carcin/23.2.265. [DOI] [PubMed] [Google Scholar]
- 5.Oganesian A, Hendricks JD, Pereira CB, Orner GA, Bailey GS, Williams DE. Potency of dietary indole-3-carbinol as a promoter of aflatoxin B1-initiated hepatocarcinogenesis: results from a 9000 animal tumor study. Carcinogenesis. 1999;20(3):453–8. doi: 10.1093/carcin/20.3.453. [DOI] [PubMed] [Google Scholar]
- 6.Kim YS, Milner JA. Targets for indole-3-carbinol in cancer prevention. J Nutr Biochem. 2005;16(2):65–73. doi: 10.1016/j.jnutbio.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 7.Bostrom CE, Gerde P, Hanberg A, Jernstrom B, Johansson C, Kyrklund T, et al. Cancer risk assessment, indicators, and guidelines for polycyclic aromatic hydrocarbons in the ambient air. Environ Health Perspect. 2002;110 (Suppl 3):451–88. doi: 10.1289/ehp.110-1241197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buters JT, Mahadevan B, Quintanilla-Martinez L, Gonzalez FJ, Greim H, Baird WM, et al. Cytochrome P450 1B1 determines susceptibility to dibenzo[a,l]pyrene-induced tumor formation. Chem Res Toxicol. 2002;15(9):1127–35. doi: 10.1021/tx020017q. [DOI] [PubMed] [Google Scholar]
- 9.Higginbotham S, RamaKrishna NV, Johansson SL, Rogan EG, Cavalieri EL. Tumor-initiating activity and carcinogenicity of dibenzo[a,l]pyrene versus 7,12-dimethylbenz[a]anthracene and benzo[a]pyrene at low doses in mouse skin. Carcinogenesis. 1993;14(5):875–8. doi: 10.1093/carcin/14.5.875. [DOI] [PubMed] [Google Scholar]
- 10.Platt KL, Dienes HP, Tommasone M, Luch A. Tumor formation in the neonatal mouse bioassay indicates that the potent carcinogen dibenzo[def,p]chrysene (dibenzo[a,l]pyrene) is activated in vivo via its trans-11,12-dihydrodiol. Chem Biol Interact. 2004;148(1–2):27–36. doi: 10.1016/j.cbi.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 11.Reddy AP, Spitsbergen JM, Mathews C, Hendricks JD, Bailey GS. Experimental hepatic tumorigenicity by environmental hydrocarbon dibenzo[a,l]pyrene. J Environ Pathol Toxicol Oncol. 1999;18(4):261–9. [PubMed] [Google Scholar]
- 12.Yu Z, Loehr CV, Fischer KA, Louderback MA, Krueger SK, Dashwood RH, et al. In utero exposure of mice to dibenzo[a,l]pyrene produces lymphoma in the offspring: role of the aryl hydrocarbon receptor. Cancer Res. 2006;66(2):755–62. doi: 10.1158/0008-5472.CAN-05-3390. [DOI] [PubMed] [Google Scholar]
- 13.Lightfoot TJ, Roman E. Causes of childhood leukaemia and lymphoma. Toxicol Appl Pharmacol. 2004;199(2):104–17. doi: 10.1016/j.taap.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 14.Yu Z, Mahadevan B, Lohr CV, Fischer KA, Louderback MA, Krueger SK, et al. Indole-3-carbinol in the maternal diet provides chemoprotection for the fetus against transplacental carcinogenesis by the polycyclic aromatic hydrocarbon dibenzo[a,l]pyrene. Carcinogenesis. 2006;27(10):2116–23. doi: 10.1093/carcin/bgl072. [DOI] [PubMed] [Google Scholar]
- 15.Auborn KJ, Fan S, Rosen EM, Goodwin L, Chandraskaren A, Williams DE, et al. Indole-3-carbinol is a negative regulator of estrogen. J Nutr. 2003;133(7 Suppl):2470S–5S. doi: 10.1093/jn/133.7.2470s. [DOI] [PubMed] [Google Scholar]
- 16.Meng Q, Yuan F, Goldberg ID, Rosen EM, Auborn K, Fan S. Indole-3-carbinol is a negative regulator of estrogen receptor-alpha signaling in human tumor cells. J Nutr. 2000;130(12):2927–31. doi: 10.1093/jn/130.12.2927. [DOI] [PubMed] [Google Scholar]
- 17.Riby JE, Feng C, Chang YC, Schaldach CM, Firestone GL, Bjeldanes LF. The major cyclic trimeric product of indole-3-carbinol is a strong agonist of the estrogen receptor signaling pathway. Biochemistry (Mosc) 2000;39(5):910–8. doi: 10.1021/bi9919706. [DOI] [PubMed] [Google Scholar]
- 18.Sundar SN, Kerekatte V, Equinozio CN, Doan VB, Bjeldanes LF, Firestone GL. Indole-3-carbinol selectively uncouples expression and activity of estrogen receptor subtypes in human breast cancer cells. Mol Endocrinol. 2006;20(12):3070–82. doi: 10.1210/me.2005-0263. [DOI] [PubMed] [Google Scholar]
- 19.Wang TT, Milner MJ, Milner JA, Kim YS. Estrogen receptor alpha as a target for indole-3-carbinol. J Nutr Biochem. 2006;17(10):659–64. doi: 10.1016/j.jnutbio.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 20.Brandenberger AW, Tee MK, Lee JY, Chao V, Jaffe RB. Tissue distribution of estrogen receptors alpha (ER-alpha) and beta (ER-beta) mRNA in the midgestational human fetus. J Clin Endocrinol Metab. 1997;82(10):3509–12. doi: 10.1210/jcem.82.10.4400. [DOI] [PubMed] [Google Scholar]
- 21.Patrone C, Cassel TN, Pettersson K, Piao YS, Cheng G, Ciana P, et al. Regulation of postnatal lung development and homeostasis by estrogen receptor beta. Mol Cell Biol. 2003;23(23):8542–52. doi: 10.1128/MCB.23.23.8542-8552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stresser DM, Williams DE, Griffin DA, Bailey GS. Mechanisms of tumor modulation by indole-3-carbinol. Disposition and excretion in male Fischer 344 rats. Drug Metab Disposition. 1995;23(9):965–75. [PubMed] [Google Scholar]
- 23.Castro DJ, Yu Z, Lohr CV, Pereira CB, Giovanini JN, Fischer KA, et al. Chemoprevention of dibenzo[a,l]pyrene transplacental carcinogenesis in mice born to mothers administered green tea: primary role of caffeine. Carcinogenesis. 2008;29(8):1581–6. doi: 10.1093/carcin/bgm237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castro DJ, Baird WM, Pereira CB, Giovanini J, Lohr CV, Fischer KA, et al. Fetal mouse Cyp1b1 and transplacental carcinogenesis from maternal exposure to dibenzo(a,l)pyrene. Cancer Prev Res. 2008;1(2):128–34. doi: 10.1158/1940-6207.CAPR-07-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oganesian A, Hendricks JD, Williams DE. Long term dietary indole-3-carbinol inhibits diethylnitrosamine-initiated hepatocarcinogenesis in the infant mouse model. Cancer Lett. 1997;118(1):87–94. doi: 10.1016/s0304-3835(97)00235-8. [DOI] [PubMed] [Google Scholar]
- 26.Chang YC, Riby J, Chang GH, Peng BC, Firestone G, Bjeldanes LF. Cytostatic and antiestrogenic effects of 2-(indol-3-ylmethyl)-3,3′-diindolylmethane, a major in vivo product of dietary indole-3-carbinol. Biochem Pharmacol. 1999;58(5):825–34. doi: 10.1016/s0006-2952(99)00165-3. [DOI] [PubMed] [Google Scholar]
- 27.Liu H, Wormke M, Safe SH, Bjeldanes LF. Indolo[3,2-b]carbazole: a dietary-derived factor that exhibits both antiestrogenic and estrogenic activity. J Natl Cancer Inst. 1994;86(23):1758–65. doi: 10.1093/jnci/86.23.1758. [DOI] [PubMed] [Google Scholar]
- 28.Riby JE, Chang GH, Firestone GL, Bjeldanes LF. Ligand-independent activation of estrogen receptor function by 3,3′-diindolylmethane in human breast cancer cells. Biochem Pharmacol. 2000;60(2):167–77. doi: 10.1016/s0006-2952(00)00307-5. [DOI] [PubMed] [Google Scholar]
- 29.Chen I, McDougal A, Wang F, Safe S. Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis. 1998;19(9):1631–9. doi: 10.1093/carcin/19.9.1631. [DOI] [PubMed] [Google Scholar]
- 30.Wang TT, Schoene NW, Milner JA, Kim YS. Broccoli-derived phytochemicals indole-3-carbinol and 3,3′-diindolylmethane exerts concentration-dependent pleiotropic effects on prostate cancer cells: comparison with other cancer preventive phytochemicals. Mol Carcinog. 2012;51(3):244–56. doi: 10.1002/mc.20774. [DOI] [PubMed] [Google Scholar]
- 31.Marconett CN, Sundar SN, Poindexter KM, Stueve TR, Bjeldanes LF, Firestone GL. Indole-3-carbinol triggers aryl hydrocarbon receptor-dependent estrogen receptor (ER)alpha protein degradation in breast cancer cells disrupting an ERalpha-GATA3 transcriptional cross-regulatory loop. Mol Biol Cell. 2010;21(7):1166–77. doi: 10.1091/mbc.E09-08-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shilling AD, Carlson DB, Katchamart S, Williams DE. 3,3′-Diindolylmethane, a major condensation product of indole-3-carbinol, is a potent estrogen in the rainbow trout. Toxicol Appl Pharmacol. 2001;170(3):191–200. doi: 10.1006/taap.2000.9100. [DOI] [PubMed] [Google Scholar]
- 33.Tilton SC, Givan SA, Pereira CB, Bailey GS, Williams DE. Toxicogenomic profiling of the hepatic tumor promoters indole-3-carbinol, 17β-estradiol and β-naphthoflavone in rainbow trout. Toxicol Sci. 2006;90(1):61–72. doi: 10.1093/toxsci/kfi341. [DOI] [PubMed] [Google Scholar]
- 34.Cover CM, Hsieh SJ, Tran SH, Hallden G, Kim GS, Bjeldanes LF, et al. Indole-3-carbinol inhibits the expression of cyclin-dependent kinase-6 and induces a G1 cell cycle arrest of human breast cancer cells independent of estrogen receptor signaling. J Biol Chem. 1998;273(7):3838–47. doi: 10.1074/jbc.273.7.3838. [DOI] [PubMed] [Google Scholar]
- 35.Carter TH, Liu K, Ralph W, Jr, Chen D, Qi M, Fan S, et al. Diindolylmethane alters gene expression in human keratinocytes in vitro. J Nutr. 2002;132(11):3314–24. doi: 10.1093/jn/132.11.3314. [DOI] [PubMed] [Google Scholar]
- 36.Shorey LE, Hagman AM, Williams DE, Ho E, Dashwood RH, Benninghoff AD. 3,3′-Diindolylmethane induces G1 arrest and apoptosis in human acute T-cell lymphoblastic leukemia cells. PLoS One. 2012;7(4):e34975. doi: 10.1371/journal.pone.0034975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A. 1996;93(12):5925–30. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shim GJ, Gherman D, Kim HJ, Omoto Y, Iwase H, Bouton D, et al. Differential expression of oestrogen receptors in human secondary lymphoid tissues. J Pathol. 2006;208(3):408–14. doi: 10.1002/path.1883. [DOI] [PubMed] [Google Scholar]
- 39.Erlandsson MC, Ohlsson C, Gustafsson JA, Carlsten H. Role of oestrogen receptors alpha and beta in immune organ development and in oestrogen-mediated effects on thymus. Immunology. 2001;103(1):17–25. doi: 10.1046/j.1365-2567.2001.01212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shim GJ, Wang L, Andersson S, Nagy N, Kis LL, Zhang Q, et al. Disruption of the estrogen receptor beta gene in mice causes myeloproliferative disease resembling chronic myeloid leukemia with lymphoid blast crisis. Proc Natl Acad Sci U S A. 2003;100(11):6694–9. doi: 10.1073/pnas.0731830100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skibola CF, Lightfoot T, Agana L, Smith A, Rollinson S, Kao A, et al. Polymorphisms in cytochrome P450 17A1 and risk of non-Hodgkin lymphoma. Br J Haematol. 2005;129(5):618–21. doi: 10.1111/j.1365-2141.2005.05505.x. [DOI] [PubMed] [Google Scholar]
- 42.Skibola CF, Bracci PM, Paynter RA, Forrest MS, Agana L, Woodage T, et al. Polymorphisms and haplotypes in the cytochrome P450 17A1, prolactin, and catechol-O-methyltransferase genes and non-Hodgkin lymphoma risk. Cancer Epidemiol Biomarkers Prev. 2005;14(10):2391–401. doi: 10.1158/1055-9965.EPI-05-0343. [DOI] [PubMed] [Google Scholar]
- 43.Prescott E, Bjerg AM, Andersen PK, Lange P, Vestbo J. Gender difference in smoking effects on lung function and risk of hospitalization for COPD: results from a Danish longitudinal population study. Eur Respir J. 1997;10(4):822–7. [PubMed] [Google Scholar]
- 44.Zang EA, Wynder EL. Differences in lung cancer risk between men and women: examination of the evidence. J Natl Cancer Inst. 1996;88(3–4):183–92. doi: 10.1093/jnci/88.3-4.183. [DOI] [PubMed] [Google Scholar]
- 45.Wu CT, Chang YL, Shih JY, Lee YC. The significance of estrogen receptor beta in 301 surgically treated non-small cell lung cancers. J Thorac Cardiovasc Surg. 2005;130(4):979–86. doi: 10.1016/j.jtcvs.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz AG, Prysak GM, Murphy V, Lonardo F, Pass H, Schwartz J, et al. Nuclear estrogen receptor beta in lung cancer: expression and survival differences by sex. Clin Cancer Res. 2005;11(20):7280–7. doi: 10.1158/1078-0432.CCR-05-0498. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.