

Different types of neurons are differentially susceptible to West Nile virus (WNV) infection. Michael Diamond and colleagues now show that cerebellar granule cell neurons (GCN) have a higher basal level of expression of type I interferon–inducible genes than cortical neurons, making GCN more resistant to infection by a variety of positive-stranded RNA viruses, including WNV.
Supplementary information
The online version of this article (doi:10.1038/nm.3108) contains supplementary material, which is available to authorized users.
Subject terms: Virus-host interactions, Viral infection, Innate immunity, Neuroimmunology
Abstract
Although susceptibility of neurons in the brain to microbial infection is a major determinant of clinical outcome, little is known about the molecular factors governing this vulnerability. Here we show that two types of neurons from distinct brain regions showed differential permissivity to replication of several positive-stranded RNA viruses. Granule cell neurons of the cerebellum and cortical neurons from the cerebral cortex have unique innate immune programs that confer differential susceptibility to viral infection ex vivo and in vivo. By transducing cortical neurons with genes that were expressed more highly in granule cell neurons, we identified three interferon-stimulated genes (ISGs; Ifi27, Irg1 and Rsad2 (also known as Viperin)) that mediated the antiviral effects against different neurotropic viruses. Moreover, we found that the epigenetic state and microRNA (miRNA)-mediated regulation of ISGs correlates with enhanced antiviral response in granule cell neurons. Thus, neurons from evolutionarily distinct brain regions have unique innate immune signatures, which probably contribute to their relative permissiveness to infection.
Supplementary information
The online version of this article (doi:10.1038/nm.3108) contains supplementary material, which is available to authorized users.
Main
Virus infection of mammalian cells induces signaling pathways that promote an antiviral state. RNA intermediates of virus replication are recognized by pathogen recognition receptors (PRR) such as Toll-like receptors (TLR) and RIG-I–like receptors (RLR), which signal specific transcription factors to induce innate defense programs, including type I interferon (IFN) expression and secretion. Binding of IFN to the IFN-αβ receptor (Ifnar) on infected and uninfected cells results in a signaling cascade that restricts the infection of many viruses through expression of ISGs. Several studies have reported that type I IFN receptor knockout (Ifnar−/−) mice show enhanced mortality after viral infection as a result of increased viral burden and expanded tropism in neurons of the brain1,2.
Neurotropic viruses use many strategies to enter the central nervous system (CNS) and spread between different subtypes of neurons to establish acute or persistent infection3. Neurons are not passive targets, as they participate in the host immune response against viruses by producing and responding to type I IFN4 through signal transducer and activator of transcription 1 (Stat1)-dependent signaling pathways5,6,7. An autopsy study reported differential susceptibility of neurons to West Nile virus (WNV) in the human brain with sparing of infection in granule cell neurons of the cerebellum compared to adjacent Purkinje neurons or neurons in the cerebral cortex8. Analogous to this, herpes simplex virus localizes primarily to the temporal lobes of the cerebral cortex and causes encephalitis and cognitive impairment9.
A comprehensive understanding of innate immune programs against viral infection in specific neurons, and how this relates to regional susceptibility in the CNS, remains unexplored. Moreover, the function of specific antiviral ISGs in neuronal subtypes also has not been established. Here we defined innate immune gene programs in response to viral infection or IFN-β treatment of two developmentally disparate populations of neurons with differential susceptibility to replication of WNV and other positive-strand RNA viruses. We show that specific neurons of the cerebellum and cerebral cortex have unique host defense signatures, including the expression of newly identified antiviral genes, which confer differential permissiveness to infection by multiple neurotropic viruses.
Results
Differential antiviral effects of IFN-β in neuronal subtypes
Type I IFN restricts WNV infection in the brain10,11 in part by limiting viral replication in neurons. Granule cell neurons of the cerebellum and cortical neurons from the cerebral cortex are differentially permissive for WNV infection in humans8,12. To determine whether these neurons respond uniquely to antiviral cytokines and infectious challenge, we infected granule cell neurons and cortical neuron cultures with WNV in the presence or absence of IFN-β pretreatment (Fig. 1). IFN-β pretreatment (100 IU ml−1, 24 h) inhibited WNV replication in cortical neurons by 15-fold (P < 0.0001) and reduced infection ∼100-fold (P < 0.0001) in granule cell neurons at 24–48 h after infection (Fig. 1a). We also found a greater responsiveness to IFN-β in granule cell neurons compared to cortical neurons when cells were infected with other positive-stranded RNA viruses, including another flavivirus (Saint Louis encephalitis virus (SLEV), up to 95-fold), an alphavirus (Venezuelan equine encephalitis virus (VEEV), up to 10,000-fold, P < 0.0001) and a coronavirus (mouse hepatitis virus (MHV), up to 200-fold, P < 0.0005) (Fig. 1b–d). Granule cell neurons were also less permissive for WNV replication at baseline in the absence of IFN-β pretreatment (15- to 110-fold lower virus titers at 24 and 48 h, P < 0.0001) (Fig. 1a), and we found similar results with SLEV (up to 700-fold, P < 0.005), VEEV (up to 200-fold, P < 0.05) and MHV (up to 20-fold at 24 and 36 h, P < 0.0001) (Fig. 1b–d). The percentage of cells infected by WNV was greater in cultures of cortical neurons compared to granule cell neurons at baseline or after IFN-β pretreatment (Supplementary Fig. 1). Consistent with the observation of greater infection of cortical neurons, the concentration of type I IFN secreted into the supernatant after WNV infection was higher in cortical neurons compared to granule cell neurons cultures (Supplementary Fig. 2).
Figure 1. Granule cell neurons are less susceptible to virus infection and more sensitive to the antiviral effects of IFN-β than cortical neurons.

(a–e) Virus production of WNV-NY (a), SLEV (b), VEEV (c), MHV (d) and WNV-MAD (e) in primary cortical neurons (CN) and granule cell neurons (GCN) generated from wild-type mice pretreated with medium (Mock) or IFN-β (100 IU ml−1) and infected at a multiplicity of infection (MOI) of 0.1 (WNV-NY and WNV-MAD) or 0.01 (VEEV, SLEV and MHV). (f) Viral replication in granule cell and cortical neurons treated with 20 μg ml−1 of MAR1-5A3 (MAR) monoclonal antibody (mAb) to Ifnar or an isotype control mAb (GIR-208, GIR). Results are the average of three independent experiments performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.0001 determined by unpaired t test. Error bars, s.d. Dashed lines indicate the limit of sensitivity of the assay. FFU, focus-forming units.
Host defense pathways in granule cell and cortical neurons
We performed microarray and pathway analysis to model molecular networks and define whether distinct antiviral IFN responses occurred in the two subtypes of neurons. Global gene expression in granule cell neurons and cortical neurons in response to IFN-β pretreatment revealed overlapping yet distinct gene signatures (Fig. 2a). Computational analysis revealed that genes associated with type I IFN induction (PRR, such as TLR and RLR), IFN regulatory factors (for example, Irf1, Irf7 and Irf9) and effector function (for example, Mx1 and Oas) were induced in both granule cell neurons and cortical neurons (Supplementary Table 1). We speculated that the two neuronal subtypes might differentially express key host defense genes at the basal level, which could affect responsiveness to IFN and susceptibility to infection. Indeed, the basal expression and IFN-β–induced expression of genes involved in IFN induction, signaling and effector function were generally higher in granule cell neurons (Fig. 2b; P < 0.01) and included Irf7, Stat1, Ifit1 and Oas1 (Supplementary Table 2). This pattern, which we confirmed by quantitative RT-PCR (qRT-PCR) (Supplementary Fig. 3 and Supplementary Table 3), suggests that the greater IFN sensitivity of granule cell neurons may be caused by higher basal expression of PRR and IFN signaling genes. Indeed, some of the ISGs (for example, Stat1, Ifi27 and Rsad2) showed differential basal expression yet similarly induced levels of expression after IFN-β treatment. Other ISGs (for example, Ccl5 and Cxcl5) showed differences in expression patterns between IFN-β–treated and WNV-infected cells. This may reflect variation in IFN-dependent and -independent gene induction that occurs in the context of WNV replication and recognition by intracellular PRR.
Figure 2. Microarray analysis revealing differential regulation of antiviral genes in granule cell neurons and cortical neurons.
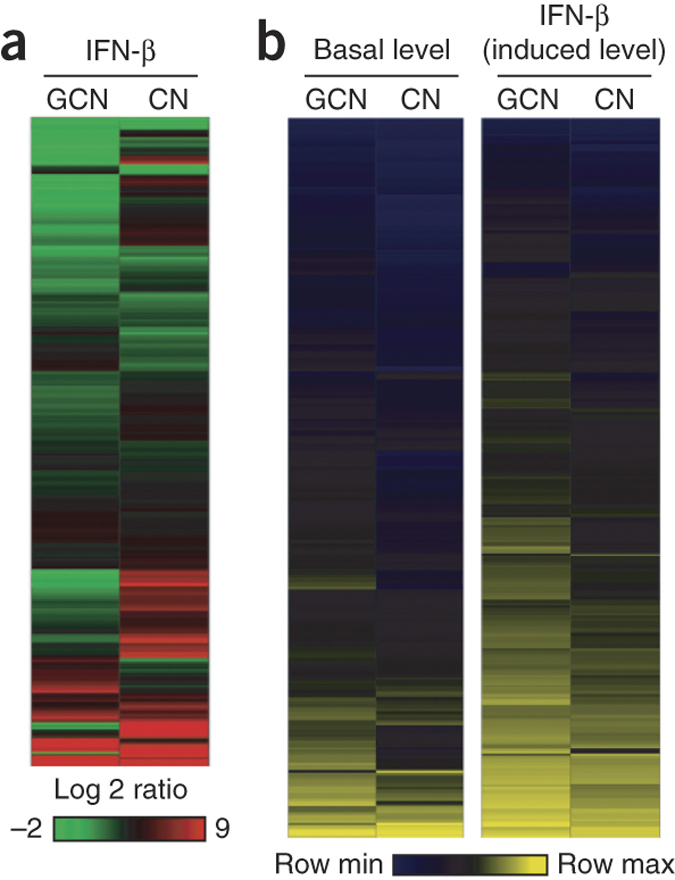
(a) Global view of gene expression 24 h after IFN-β treatment (100 IU ml−1) as determined by clustering performed using the hierarchical unweighted-pair group method using average linkages with a Euclidean distance similarity measure and average value ordering function. The data shown were compared to values from mock-treated cells (fold change). GCN, granule cell neurons; CN, cortical neurons. (b) Differential gene expression profile of IFN-β–responsive genes identified by analysis of variance (ANOVA) (within cutoff values of an at least twofold change, Benjamini-Hochberg–adjusted P < 0.01). Shown are heat maps reflecting the expression of IFN-β–responsive genes (rows) at the basal level (left columns) or after IFN-β treatment (right columns) in granule cell neurons and cortical neurons.
To identify nodes of gene regulatory network interaction that corresponded to specific antiviral or immune pathways, we performed computational analysis on genes with statistically significant expression changes. We detected an enrichment of genes with functions associated with immune responses to viral infection. Genes associated with canonical PRR signaling through TLR and RLR pathways (for example, Tlr7, Ddx58 (also called Rig-I) and Ifih1 (also called Mda5)), transcription factors (for example, NfkB, Irf7 and Irf9) and their corresponding target genes (for example, Ccl5 and Tnfa) were expressed at higher levels in granule cell neurons at the basal level (Supplementary Table 2). In particular, Stat1 and IFN signaling–dependent genes (for example, Ifit1, Ifit3, Oas and Mx1) were basally expressed in granule cell neurons at higher levels compared to their expression in cortical neurons. In resting or IFN-β–pretreated granule cell neurons, we found an enrichment of genes that have been linked to antiviral activity (for example, Ifit1, Rsad2, Oas1, Irf7, Bst2 (also called Tetherin), Unc93b1, Ifih1, Ddx58, Mx1, Ikbke (also called Ikkɛ) and Stat1), autophagy and inflammation (immune-related GTPases (IRGs), such as Irgm1, Irgm2, Igtp (also called Irgm3), Ifi47 (also called Irg47) and Tgtp1 (also called Irgb6) and leukocyte chemotaxis (Ccl5, Cxcl1, Cxcl10 and Cxcl16) (Supplementary Fig. 4 and Supplementary Table 2). This pattern of gene expression is consistent with results showing that granule cell neurons are less permissive to virus replication at baseline or in the presence of IFN-β pretreatment (Fig. 1a–d).
Some antiviral ISGs (for example, Ifit1, Rsad2 and Oas) reached peak expression levels sooner in granule cell neurons compared to cortical neurons after IFN-β pretreatment (Fig. 3). Thus, higher basal expression of host defense genes makes granule cell neurons poised for faster antiviral action in response to IFN-β than cortical neurons and ultimately affects the induced expression of ISGs. Notably, the addition of a blocking monoclonal antibody to Ifnar enhanced (∼20-fold, P < 0.0001) WNV replication in granule cell neurons yet had little effect in cortical neurons (Fig. 1f). Therefore, the lower susceptibility of granule cell neurons to viral replication compared to cortical neurons at baseline can probably not be attributed to differences in virus entry but rather to higher expression of genes with an IFN signature, which confer greater responsiveness to the antiviral effects of type I IFN.
Figure 3. Key ISGs reach peak expression sooner and at higher levels in granule cell neurons.

ΔCt values compared to the cellular levels of 18S ribosomal RNA (top) and fold changes in expression compared to untreated cells (bottom) of the indicated ISGs in granule cell neurons (GCN) and cortical neurons (CN) treated with IFN-β (100 IU ml−1), with cellular RNA harvested at the indicated time points after treatment. The results are the representative of three independent experiments performed in triplicate. Error bars, s.d.
In contrast to virulent lineage 1 WNV strains (for example, WNV-New York 1999, WNV-NY), a lineage 2 strain (WNV-Madagascar 1978, WNV-MAD) is impaired in its ability to antagonize the phosphorylation of Stat1 and the resultant IFN signaling and is thus more sensitive to antiviral actions of IFN13,14. We hypothesized that IFN-β would have a greater antiviral effect on WNV-MAD compared to WNV-NY in cortical neurons. Indeed, WNV-MAD infection was restricted to a greater degree (∼300-fold at 24 h after infection, P < 0.0001) after IFN-β pretreatment of cortical neurons (Fig. 1e). In comparison, the inhibitory effects of IFN-β against both WNV-MAD and WNV-NY were more similar (∼80-fold for WNV-MAD compared to ∼50-fold for WNV-NY at 24 h after infection, P < 0.0001) in granule cell neurons. These results support the idea that Stat1-dependent signaling proteins are expressed at greater levels basally in granule cell neurons (Supplementary Fig. 5) and contribute to the enhanced responsiveness to the antiviral activity of IFN-β in this cell type. To establish whether the differential expression of ISGs was caused by Stat1-dependent signaling, we repeated the studies in Stat1−/− and wild-type granule cell neurons. Stat1−/ − granule cell neurons showed lower basal expression of several ISGs compared to wild-type granule cell neurons (Supplementary Fig. 6).
Identification of antiviral ISGs in primary neurons
Because of the enhanced responsiveness to the antiviral actions of IFN-β in granule cell neurons, we hypothesized that at least a subset of differentially expressed ISGs would confer inhibitory effects against WNV. We attempted to create a granule cell neuron–like antiviral phenotype by ectopically expressing individual granule cell neuron–specific genes in cortical neurons. Candidate genes included those showing higher expression at the basal level (for example, Irg1, Ifi27, Ifi44, Isg15, Bst2, Rsad2, Ifitm3, Ifi204, Trim30 and Casp12) or greater induction after IFN-β pretreatment (for example, Ifit2, Cmpk2 and Lcn2) in granule cell neurons (Supplementary Table 2). We included Irf1, which was reported as a broad-acting antiviral signaling molecule15,16, as a positive control. We developed a high-efficiency (∼90%) lentivirus transduction system for ectopic expression of ISGs in primary neurons (Fig. 4a). We cloned 39 ISGs, transduced them into cortical neurons and assessed their antiviral effects in the absence or presence of exogenous IFN-β (Fig. 4 and data not shown). Three genes (Irg1, Ifi27 and Rsad2) resulted in reduced (5- to 20-fold, P < 0.05) WNV yield when expressed ectopically in cortical neurons (Fig. 4b,c); analogously, we also found reduced SLEV infection with expression of these genes (threefold to tenfold, P < 0.0001) and reduced WNV-MAD infection (5- to 20-fold, P < 0.0001) (Fig. 4f,g). Ectopic expression of Irg1 (sevenfold, P < 0.0005) and Ifi27 (threefold, P < 0.01) but not Rsad2 (P > 0.05) inhibited MHV infection (Fig. 4h). Ectopic expression of these three genes (or of any of the other ISGs tested) did not alter cell viability (Fig. 4d and data not shown) or confer an antiviral effect when tested in parallel against VEEV (Fig. 4e); thus, it is unlikely that the antiviral effect with WNV was caused by cellular stress. Moreover, transduction of shRNA against Ifi27, Irg1 or Rsad2 in granule cell neurons correspondingly resulted in enhanced infection by WNV-MAD (approximately tenfold, P < 0.0001) (Fig. 4i).
Figure 4. ISGs with antiviral activity identified in neurons using a lentivirus transduction system.

(a) Design of the bicistronic lentiviral vector. CMV, immediate early promoter from human cytomegalovirus; Pubiquitin, ubiquitin promoter; IRES, internal ribosome entry site; R/U5, HIV-1 long-terminal repeat. (b) Flow cytometry contour plots showing Irg1- and Ifi27-mediated inhibition of WNV in primary cortical neurons. The x and y axes indicate transduction efficiency (GFP intensity) and infectivity (level of WNV antigen), respectively. A packaged empty lentiviral vector was used as the negative control. (c) Results of focus-forming assays showing inhibition of WNV replication in cortical neurons transduced with Irf1, Irg1, Ifi27 and Rsad2 but not with several other candidate genes (for example, Ifi204, Pyhin1, Ifitm3, Isg15, Cmpk2, Trim30 or Lcn2). FFU, focus-forming units. (d) Cell viability after lentivirus transduction determined using a Cell-titer Glo luminescence-based assay. RLU, relative light units. (e–h) Antiviral effects of Irf1, Irg1, Ifi27 and Rsad2 in cortical neurons against VEEV (e), SLEV (f), WNV-MAD (g) and MHV (h). (i) Viral replication evaluated by focus-forming assay in granule cell neurons transduced with lentiviruses expressing shRNA targeting Irg1, Ifi27 and Rsad2. shRNA targeting GFP was used as a control. For c–i, results are the average of at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.0001 determined by unpaired t test. Error bars, s.d.
Epigenetic and miRNA-mediated regulation of ISG expression
As a recent study showed that cell type–specific differences in ISG expression can be attributed in part to epigenetic mechanisms17, and because histone acetylation of ISGs has been implicated in the regulation of an antiviral state18,19,20, we hypothesized that the innate immune phenotypes in granule cell neurons and cortical neurons might be associated with distinct patterns of histone modification. To investigate this, we pretreated cortical neurons with the histone deacetylase (HDAC) inhibitor trichostatin A (TSA), infected them with WNV and evaluated virus infection. Although treatment with the HDAC inhibitor did not alter cell viability under the conditions tested, it reduced viral yields (fivefold, P < 0.0001) (Fig. 5a) and resulted in derepression of ISGs that otherwise had low basal expression levels in cortical neurons (Fig. 5b).
Figure 5. Epigenetic and miRNA-mediated control mechanism of ISGs.

(a) Virus production evaluated by focus-forming assay 12 h after infection in cortical neurons pretreated with the HDAC inhibitor TSA (500 nM) or vehicle (0.1% (vol/vol) DMSO) for 12 h and infected with WNV (MOI of 1) (right). Results are the average of three experiments performed in triplicate. Error bars, s.d. ***P < 0.0001 determined by two-tailed Student's t test. Also shown is cell viability determined using a Cell-titer Glo luminescence-based assay 24 h after TSA treatment (right). FFU, focus-forming units; RLU, relative light units. (b) Expression levels of the indicated genes as determined by qRT-PCR in cortical neurons treated with TSA. The results are the average of three independent experiments performed in at least duplicate. The data are displayed as the ΔCt value compared to the cellular levels of 18S ribosomal RNA. Solid lines represent the median values. *P < 0.05, **P < 0.01, ***P < 0.0001 determined by two-tailed Student's t test. (c) miR-132 expression levels in total RNA isolated from granule cell neurons (GCN) and cortical neurons (CN) quantified by qRT-PCR. The result is representative of four independent experiments with technical replicates of n = 2–6 per experiment. miR-132 and miR-155 (as a control) expression levels quantified by qRT-PCR in total RNA isolated from the cerebellum and cerebral cortex from five naive C57BL/6 mice are also shown. The data are displayed as the ΔCt value compared to the cellular levels of sno202, a small RNA that is commonly used for miRNA normalization. Solid lines indicate the median values. NS, not significant. **P < 0.01, ***P < 0.0001 determined by two-tailed Student's t test. (d–f) Expression levels of miR-132 (d), ISGs (e) and non-ISGs (f) from total RNA isolated from granule cell neurons transduced with miR-132–expressing lentivirus quantified by qRT-PCR. The miR-132 level was normalized to the cellular level of sno202 RNA, and the levels of ISGs were normalized to the cellular level of 18S ribosomal RNA. Error bars, s.d. Solid lines indicate the median values. *P < 0.05, **P < 0.001, ***P < 0.0001 determined by Mann-Whitney test.
Stat1 requires its coactivator p300/CREB-binding protein (CBP), a histone acetyltransferase, to enhance ISG transcription21,22,23. Our inhibitor experiments suggested that differential histone acetylation contributes to the lower basal expression of Stat1 and key ISGs in cortical neurons. One candidate for regulating this phenotype is miRNA-132, which is required for the morphogenesis of cortical and hippocampal neurons, targets p300 and negatively regulates IFN-β and IL-1β signaling and antiviral immunity24,25,26. Expression of miRNA-132 is reportedly higher in the cerebral cortex compared to the cerebellum of rats and humans27,28. We found higher expression of miRNA-132 in cortical neurons and the cerebral cortex compared to granule cell neurons and the cerebellum (Fig. 5c). Moreover, ectopic expression of miRNA-132 in granule cell neurons (Fig. 5d) decreased the expression of some ISGs (Fig. 5e) but did not change the expression of non-ISGs (Fig. 5f). However, there was no change in viral yield when miRNA-132 was expressed ectopically in granule cell neurons (data not shown). We suggest that the distinct antiviral programs observed between granule cell neurons and cortical neurons are explained in part by differences in the epigenetic state of key genes, which may be influenced by the expression of specific miRNAs.
Differential basal expression of ISGs in brain regions
Our results with primary neurons suggested that basal expression of host antiviral genes in granule cell neurons resulted in lower permissiveness to virus replication. To establish this in vivo, we used fluorescence in situ RNA hybridization. We hybridized brain tissues from naive mice with labeled probes to visualize the relative expressions of Stat1 and key antiviral genes (for example, Irg1 and Ifi27) that showed differential basal expression ex vivo in granule cell neurons and cortical neurons (Supplementary Table 2). Although the basal expression of a housekeeping gene was equivalent, the expressions of Irg1, Ifi27 and Stat1 mRNA were in greater abundance qualitatively in neurons of the granule cell layer of the cerebellum compared to those in the cerebral cortex (Supplementary Fig. 7a–d). We validated these results quantitatively using a branched DNA amplification assay (Fig. 6a).
Figure 6. Differential WNV infection of neurons in the brains of mice and humans.
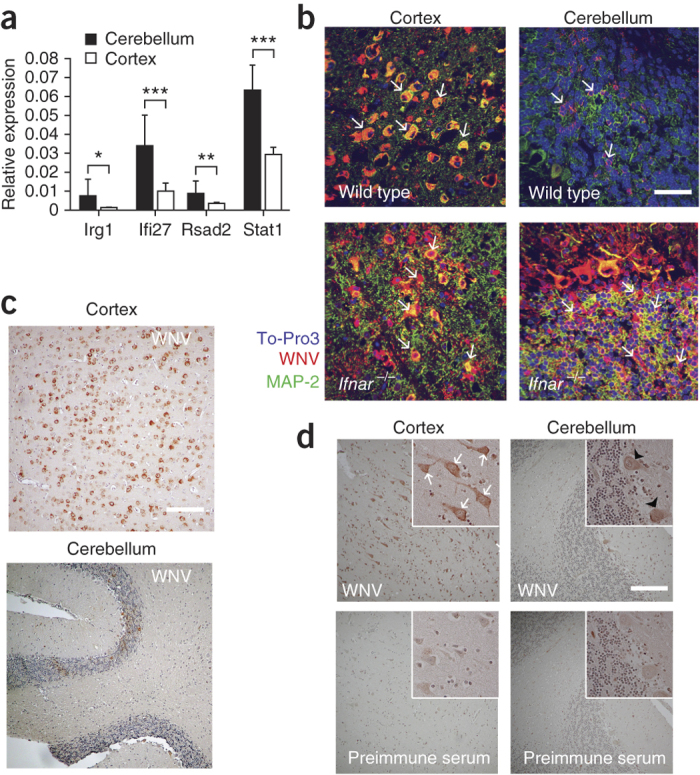
(a) Differential gene expression of Ifi27, Irg1, Rsad2 and Stat1 in the cerebellum and cortex of naive wild-type mice quantified by QuantiGene Plex branched DNA amplification assay. The data were generated from eight mice, and the analysis was performed with triplicate samples for each mouse. Error bars, s.d. *P < 0.05, **P < 0.001, ***P < 0.0001 determined by two-tailed Student's t test. (b) Representative images from the cerebellum and cerebral cortex of wild-type and Ifnar−/− (n = 5 per group) C57BL/6 mice harvested on day 6 after i.c. infection with 101 PFU of WNV and then cryoprotected, sectioned and stained with rat WNV antisera (red), an antibody to the neuronal marker MAP-2 (green) and ToPro-3 (blue) for nuclear staining. White arrows indicate infected cells. Scale bar, 20 μm. (c) Representative images from the cerebellum and cerebral cortex of wild-type C57BL/6 mice harvested at day 6 after i.c. infection with 101 PFU of WNV and then paraffin embedded sectioned, and stained with rat WNV antisera (viral antigen, brown) and haematoxylin for nuclear staining (blue). Scale bar, 80 μm. (d) Brain sections of the cerebral cortex and cerebellum from a fatal human case of WNV encephalitis after staining with rat WNV antisera (viral antigen, brown) or preimmune sera. Infected cortical neurons and Purkinje neurons are indicated by white arrows and black arrowheads, respectively. Scale bar, 80 μm.
Infection of neurons by WNV in wild-type and Ifnar−/− mice
To validate our findings in vivo, we performed immunohistochemistry for detection of WNV antigen in the brains of wild-type mice that were infected by an intracranial (i.c.) route. Granule cell neurons of the contralateral cerebellum were less vulnerable to WNV infection compared to cortical neurons of the contralateral cerebral cortex (Fig. 6b,c), with less infection in neurons of the granule cell layer even 6 d after infection. Although our primary culture data suggest this phenotype is caused by an inherent resistance of granule cell neurons because of a basal and rapidly activated antiviral program, it also is possible that productive infection of granule cell neurons occurs in vivo during the first few days but is cleared more rapidly. The low level of granule cell neurons infection, however, was not caused by inefficient viral spread to the cerebellum, as the adjacent Purkinje cell neurons were infected. In comparison, in the brains of Ifnar−/− mice, we detected comparable amounts of WNV antigen in both cortical neurons and granule cell neurons after i.c. infection (Fig. 6b). Quantification of infectious WNV from the cerebellum and the cerebral cortex of wild-type and Ifnar−/− mice supported these results (Supplementary Fig. 8).
Susceptibility of neurons to WNV in human brains
To corroborate these observations, we performed immunohistochemistry to detect viral antigen in the brains of seven human patients who succumbed to fatal WNV encephalitis. Analogous to our findings in mouse neurons, granule cell neurons were relatively spared from WNV replication compared to other neuronal populations (for example, adjacent Purkinje neurons in the cerebellum and cortical neurons in the cerebral cortex) (Fig. 6d, Supplementary Fig. 9 and data not shown).
Discussion
Comparison of neuronal cell types from functionally and evolutionarily distinct regions of the brain allowed us to identify gene signatures that correspond to unique antiviral signaling pathways. Our data reveal that genes associated with the host defense pathway are more highly expressed in granule cell neurons at both the basal level and after IFN-β treatment. This included a Stat1 and IFN-dependent signaling signature that was expressed in uninfected granule cell neurons at higher levels compared to cortical neurons, which resulted in reduced permissiveness to replication of several neurotropic viruses.
Granule cell neurons comprise the largest population of neurons in the brain and account for >90% of neurons in the cerebellum alone29. Neuronal injury associated with extensive infection of granule cell neurons might cause defects in motor movement, learning, posture and balance30. In addition to our identification of distinct host defense signatures of specific neuronal subtypes, cell-extrinsic effects of resident glial cells also could contribute to innate immune programming of neurons. A recent study demonstrated regional differences in basal chemokine and cytokine secretion by astrocytes grown from the cerebral cortex, cerebellum and spinal cord31, although no differences in type I IFN production were reported. Although it is possible that glial cells contribute to the neuron-specific infection pattern observed in vivo, our primary neuron cultures lacked astrocyte contamination, and thus cell-extrinsic regulation cannot explain the differential infectivity and innate gene expression patterns of granule cell neurons and cortical neurons observed in culture.
Characterization of the distinct antiviral programs in the two neuronal subtypes enabled us to identify antiviral genes against WNV and some (SLEV and MHV) but not other (VEEV) positive-stranded RNA viruses, including Ifi27, Irg1 and Rsad2, all of which had higher basal and IFN-β–induced expression in granule cell neurons compared to cortical neurons. Ifi27 was first suggested to have antiviral activity against Sindbis virus infection in mice32 and may also attenuate infection of hepatitis C virus33, although its mechanism of action remains unknown. Irg1 is induced in macrophages and tissues from mice infected with the intracellular parasite Toxoplasma gondii or the bacterium Listeria monocytogenes34. So far, no antimicrobial functions have been ascribed to this gene, although it associates with the mitochondria, a site of regulation for PRR adaptor molecules and the antiviral signalosome34,35. Ectopic expression of Rsad2 in cortical neurons also showed an antiviral effect against WNV infection, results that are consistent with recent studies in Rsad2−/− mice36. Rsad2 is an endoplasmic reticulum–associated protein that has reported antiviral activity against several viruses, possibly because it inhibits bulk protein secretion, lipid raft formation and virus budding, and localizes to endoplasmic reticulum–derived lipid droplets37.
Our pathway analysis identified a unique IFN-β–inducible host defense signature: IRGs. IRGs (Irgm1, Irgm2, Igtp, Ifi47 and Tgtp1) are associated with autophagy and resistance to T. gondii38,39 and were expressed preferentially at the basal level in granule cell neurons. This may be relevant, as T. gondii infection in the CNS results in lesions of the frontal and parietal cortex, with sparing of the cerebellum40,41. Although further study is warranted, the distinct basal expression of immunity-related GTPases or other host defense proteins may contribute to regional susceptibility of the brain to T. gondii42 and explain more generally neuronal subtype–specific differences to microbial pathogenesis.
Why do granule cell neurons of the cerebellum express a higher basal level of host defense genes than cortical neurons? We observed a large number of Stat1-dependent genes that were expressed at higher levels in the basal and induced states in granule cell neurons compared to cortical neurons. Unphosphorylated Stat1 can promote the expression of antiviral genes without IFN-dependent stimulation43, and higher basal levels of Stat1 can amplify cell-intrinsic immune responses by activating an amplification loop44,45. Stat1 and its downstream gene targets could be differentially regulated in granule cell neurons and cortical neurons by epigenetic control mechanisms, miRNA-dependent control mechanisms or both. The epigenetic control of ISG expression and its impact on antiviral responses has been reported17,18,19,20. Accordingly, treatment of cortical neurons with an HDAC inhibitor reduced WNV yield and resulted in derepression of the expression of ISG at the basal level. One caveat to these studies is that inconsistent ISG expression phenotypes have been observed with HDAC inhibitors46,47, although these differences may reflect variation in cell types or concentration and time of inhibitor incubation. Our experiments also revealed higher expression in cortical neurons compared to granule cell neurons of miRNA-132, which targets the p300 coactivator of Stat1; ectopic expression of miRNA-132 in granule cell neurons decreased the basal expression of ISGs. Distinct antiviral programs observed between granule cell neurons and cortical neurons may be explained by differences in the epigenetic state of key genes, which is influenced by the expression of miRNA. It is intriguing to consider that the basal expression set point of ISGs in different neuronal subtypes may vary among individuals, which could influence the level of infection and spread once encephalitic viruses reach the CNS.
In summary, we provide compelling evidence that specific neurons from disparate regions of the brain are differentially permissive to viral replication as a result of intrinsically distinct host defense programs. Our study also identifies new antiviral genes in the CNS that restrict infection by WNV and other CNS viruses. This analysis may be relevant for understanding regional infection and immune responses against other pathogens of the CNS.
Methods
Mice.
C57BL/6 wild-type and congenic Ifnar−/− mice were bred in the animal facility of Washington University School of Medicine, and experiments were performed according to the guidelines and with approval of the Washington University Animal Studies Committee. A mix of matched male and female mice were used in all experiments.
Preparation of primary neurons.
Cortical neurons and granule cell neurons were prepared from wild-type mice as described48.
Virus stocks.
The WNV-NY strain (New York 1999) was passaged once in C6/36 Aedes albopictus cells to generate an insect cell–derived stock and one additional time in Vero cells to generate a mammalian cell–derived stock. The Madagascar-AnMg798 strain of WNV (WNV-MAD) was obtained from the World Reference Center of Emerging Viruses and Arboviruses and passaged in Vero cells. The generation and propagation of MHV (strain A59) has been described elsewhere49. SLEV (strain GHA-3) was a gift of A. Barrett (Galveston, TX) and was passaged in Vero cells. VEEV (strain TC-83) was a gift of I. Frolov (Birmingham, AL) and was passaged in Vero cells. Levels of virus in the supernatant from infected cells were titrated by focus-forming or plaque assays on BHK21-15, Vero or L929 cells.
RNA preparation and microarray analysis.
Three independent cultures of cortical neurons and granule cell neurons were left untreated or were pretreated with 100 IU ml−1 of mouse IFN-β (PBL Interferon Source) using a balanced design. Twenty-four hours after treatment, total RNA was isolated according to the manufacturer's protocol with an RNeasy kit (Qiagen), and microarray analysis was performed using Agilent RM 4 × 44K microarrays using the manufacturer's one-color analysis protocol. Data were normalized with GeneData analyst 2.2.1. Differential gene expression between groups was determined by a three-way ANOVA model. The Benjamini and Hochberg algorithm was used to control the false discovery rate (FDR) of multiple testing (FDR-adjusted P < 0.01, selected for genes showing more than twofold change; Partek Genomic Suite, Partek Incorporated). Heat maps for gene expression were created using Spotfire DecisionSite 9.1.1. Functional analysis of statistically significant gene expression changes was performed using Ingenuity Pathway Analysis (IPA; Ingenuity Systems).
qRT-PCR.
Complementary DNA (cDNA) was generated from RNA using random hexamers and Multiscribe reverse transcriptase (Applied Biosystems). Transcripts were quantified on a 7500 Fast Real-time PCR system (Applied Biosystems) with Taqman primers and probe sets (Integrated DNA Technologies). Cellular levels of 18S ribosomal RNA were quantified in parallel by TaqMan analysis and used for normalization.
Lentivirus transduction and functional evaluation of ISGs.
Lentiviral constructs encoding individual ISGs were generated by PCR amplification from cDNA generated from granule cell neurons treated with IFN-β. ISGs were subcloned into the FCIV vector50 (generous gift from Y. Sasaki, St. Louis, MO). Lentivirus particles were generated, and 3 d after transduction, neurons were infected at an MOI of 1 (WNV-NY and VEEV) or 0.1 (WNV-MAD, SLEV and MHV). One day after infection, cells were stained for WNV antigen with an E protein–specific mAb (WNV E16 (ref. 51)) by flow cytometry, and viral supernatants were harvested and titered by focus-forming or plaque assay.
shRNA gene silencing.
Lentiviruses encoding shRNA targeting a gene of interest were obtained from the RNAi Core facility at Washington University School of Medicine. Granule cell neurons were transduced with lentivirus particles as described above. shRNA knockdown efficiency was confirmed by qRT-PCR (data not shown). WNV replication in shRNA-transduced cells was evaluated as described above.
miRNA experiments.
For miRNA detection, total RNA was harvested using a miRNeasy Kit (Qiagen) according to the manufacturer's protocol, reverse transcribed using miRNA-specific primers and quantified by Taqman microRNA expression assay (Applied Biosystems). sno202 levels were quantified in parallel by TaqMan analysis and used for normalization. For miRNA expression, lentiviruses expressing miR-132 or control miRNA were generated and used according to the manufacturer's protocol (Biosettia).
QuantiGene Plex 2.0 assay.
Brain tissues were harvested from naive mice, and differential RNA expression was quantified by the QuantiGene Plex 2.0 assay (Affymetrix) according to the manufacturer's protocol. The Actb and Rpl19 genes were used for normalization of data.
Immunohistochemistry.
Wild-type mice were infected with 101 PFU of WNV by an i.c. route and euthanized at day 6 after infection. Ifnar−/− mice were infected with 101 PFU of WNV by an i.c. route, treated with humanized E16 mAb to prevent spread to visceral organs and euthanized at day 4 after infection. Brains were harvested, cryoprotected or paraffin embedded, sectioned and stained with rat antisera against WNV as described elsewhere52. Human brain sections were obtained from fatal WNV encephalitis cases (generously provided by B.K. Kleinschmidt-DeMasters (Denver, CO) and C.A. Wiley (Pittsburgh, PA)) and stained with rat antisera against WNV or nonimmune sera as described above.
Statistical analyses.
Parametric and nonparametric data were analyzed by a two-tailed Student's t test and Mann-Whitney test, respectively, using Prism software (GraphPad Software). Microarray data were analyzed by ANOVA, and the Benjamini and Hochberg algorithm was used to control the FDR of multiple testing (Partek Genomic Suite, Partek Incorporated).
Additional methods.
Detailed methodology is described in the Supplementary Methods.
Accession codes.
Microarray data are deposited at NCBI under accession code GSE42727.
Supplementary information
Supplementary Figures 1–9, Supplementary Tables 1–3 and Supplementary Methods (PDF 2064 kb)
Acknowledgements
We thank O. Koues and E. Oltz for the experimental discussions and suggestions, J. Patel and R. Klein for their contribution to the initial phase of the study, B. Bradel-Tretheway for help with initial study design and microarray analysis, Y. Sasaki (Washington University) for the lentiviral plasmids, A. Barrett (University of Texas Medical Branch) for the SLEV, I. Frolov (University of Alabama, Birmingham) for the VEEV, H.W. Virgin (Washington University) for the Stat1−/− mice and B.K. Kleinschmidt-DeMasters (University of Colorado) and C.A. Wiley (University of Pittsburgh) for the human brain autopsy samples. US National Institutes of Health grants U54 AI081680 (Pacific Northwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research), U19 AI083019 (M.G. and M.S.D.) and R01 AI074973 (M.G. and M.S.D.) supported this work. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author Contributions
H.C. and K.J.S. performed the experiments. H.C. and S.C.P. analyzed the microarray data. H.C. and M.S.D. designed the experiments and wrote the initial draft of the manuscript. M.G.K. and M.G. contributed to the study design and preparation of the manuscript.
Accession codes
Primary accessions
Gene Expression Omnibus
Competing interests
The authors declare no competing financial interests.
References
- 1.Ireland DDC, Stohlman SA, Hinton DR, Atkinson R, Bergmann CC. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J. Virol. 2008;82:300–310. doi: 10.1128/JVI.01794-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schoneboom BA, Lee JS, Grieder FB. Early expression of IFN-α/β and iNOS in the brains of Venezuelan equine encephalitis virus–infected mice. J. Interferon Cytokine Res. 2000;20:205–215. doi: 10.1089/107999000312621. [DOI] [PubMed] [Google Scholar]
- 3.McGavern DB, Kang SS. Illuminating viral infections in the nervous system. Nat. Rev. Immunol. 2011;11:318–329. doi: 10.1038/nri2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delhaye S, et al. Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. USA. 2006;103:7835–7840. doi: 10.1073/pnas.0602460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J, Campbell IL. Innate STAT1-dependent genomic response of neurons to the antiviral cytokine α interferon. J. Virol. 2005;79:8295–8302. doi: 10.1128/JVI.79.13.8295-8302.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffin DE. Immune responses to RNA-virus infections of the CNS. Nat. Rev. Immunol. 2003;3:493–502. doi: 10.1038/nri1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakraborty S, Nazmi A, Dutta K, Basu A. Neurons under viral attack: victims or warriors? Neurochem. Int. 2010;56:727–735. doi: 10.1016/j.neuint.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Omalu BI, Shakir AA, Wang G, Lipkin WI, Wiley CA. Fatal fulminant pan-meningo-polioencephalitis due to West Nile virus. Brain Pathol. 2003;13:465–472. doi: 10.1111/j.1750-3639.2003.tb00477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gordon B, Selnes OA, Hart J, Jr., Hanley DF, Whitley RJ. Long-term cognitive sequelae of acyclovir-treated herpes simplex encephalitis. Arch. Neurol. 1990;47:646–647. doi: 10.1001/archneur.1990.00530060054017. [DOI] [PubMed] [Google Scholar]
- 10.Samuel MA, Diamond MS. α/β interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 2005;79:13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lazear HM, Pinto AK, Vogt MR, Gale M, Jr., Diamond MS. β interferon controls West Nile virus infection and pathogenesis in mice. J. Virol. 2011;85:7186–7194. doi: 10.1128/JVI.00396-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Armah HB, et al. Systemic distribution of West Nile virus infection: postmortem immunohistochemical study of six cases. Brain Pathol. 2007;17:354–362. doi: 10.1111/j.1750-3639.2007.00080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keller BC, et al. Resistance to α/β interferon is a determinant of West Nile virus replication fitness and virulence. J. Virol. 2006;80:9424–9434. doi: 10.1128/JVI.00768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perwitasari O, Cho H, Diamond MS, Gale M. Jr Inhibitor of κB kinase ɛ (IKKɛ), STAT1, and IFIT2 proteins define novel innate immune effector pathway against West Nile virus infection. J. Biol. Chem. 2011;286:44412–44423. doi: 10.1074/jbc.M111.285205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schoggins JW, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brien JD, et al. Interferon regulatory factor-1 (IRF-1) shapes both innate and CD8+ T cell immune responses against West Nile virus infection. PLoS Pathog. 2011;7:e1002230. doi: 10.1371/journal.ppat.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang TC, et al. Histone H3 lysine 9 di-methylation as an epigenetic signature of the interferon response. J. Exp. Med. 2012;209:661–669. doi: 10.1084/jem.20112343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agalioti T, et al. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-β promoter. Cell. 2000;103:667–678. doi: 10.1016/s0092-8674(00)00169-0. [DOI] [PubMed] [Google Scholar]
- 19.Parekh BS, Maniatis T. Virus infection leads to localized hyperacetylation of histones H3 and H4 at the IFN-β promoter. Mol. Cell. 1999;3:125–129. doi: 10.1016/s1097-2765(00)80181-1. [DOI] [PubMed] [Google Scholar]
- 20.Shestakova E, Bandu MT, Doly J, Bonnefoy E. Inhibition of histone deacetylation induces constitutive derepression of the β interferon promoter and confers antiviral activity. J. Virol. 2001;75:3444–3452. doi: 10.1128/JVI.75.7.3444-3452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horvai AE, et al. Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proc. Natl. Acad. Sci. USA. 1997;94:1074–1079. doi: 10.1073/pnas.94.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhattacharya S, et al. Cooperation of Stat2 and p300/CBP in signalling induced by interferon-α. Nature. 1996;383:344–347. doi: 10.1038/383344a0. [DOI] [PubMed] [Google Scholar]
- 23.Look DC, et al. Direct suppression of Stat1 function during adenoviral infection. Immunity. 1998;9:871–880. doi: 10.1016/s1074-7613(00)80652-4. [DOI] [PubMed] [Google Scholar]
- 24.Vo N, et al. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc. Natl. Acad. Sci. USA. 2005;102:16426–16431. doi: 10.1073/pnas.0508448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wayman GA, et al. An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc. Natl. Acad. Sci. USA. 2008;105:9093–9098. doi: 10.1073/pnas.0803072105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lagos D, et al. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat. Cell Biol. 2010;12:513–519. doi: 10.1038/ncb2054. [DOI] [PubMed] [Google Scholar]
- 27.Olsen L, Klausen M, Helboe L, Nielsen FC, Werge T. MicroRNAs show mutually exclusive expression patterns in the brain of adult male rats. PLoS ONE. 2009;4:e7225. doi: 10.1371/journal.pone.0007225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, et al. A microRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317:1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Contestabile A. Cerebellar granule cells as a model to study mechanisms of neuronal apoptosis or survival in vivo and in vitro. Cerebellum. 2002;1:41–55. doi: 10.1080/147342202753203087. [DOI] [PubMed] [Google Scholar]
- 30.Fine EJ, Ionita CC, Lohr L. The history of the development of the cerebellar examination. Semin. Neurol. 2002;22:375–384. doi: 10.1055/s-2002-36759. [DOI] [PubMed] [Google Scholar]
- 31.Fitting S, et al. Regional heterogeneity and diversity in cytokine and chemokine production by astroglia: differential responses to HIV-1 Tat, gp120, and morphine revealed by multiplex analysis. J. Proteome Res. 2010;9:1795–1804. doi: 10.1021/pr900926n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Labrada L, Liang XH, Zheng W, Johnston C, Levine B. Age-dependent resistance to lethal alphavirus encephalitis in mice: analysis of gene expression in the central nervous system and identification of a novel interferon-inducible protective gene, mouse ISG12. J. Virol. 2002;76:11688–11703. doi: 10.1128/JVI.76.22.11688-11703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itsui Y, et al. Expressional screening of interferon-stimulated genes for antiviral activity against hepatitis C virus replication. J. Viral Hepat. 2006;13:690–700. doi: 10.1111/j.1365-2893.2006.00732.x. [DOI] [PubMed] [Google Scholar]
- 34.Degrandi D, Hoffmann R, Beuter-Gunia C, Pfeffer K. The proinflammatory cytokine-induced IRG1 protein associates with mitochondria. J. Interferon Cytokine Res. 2009;29:55–67. doi: 10.1089/jir.2008.0013. [DOI] [PubMed] [Google Scholar]
- 35.Tal MC, Iwasaki A. Mitoxosome: a mitochondrial platform for cross-talk between cellular stress and antiviral signaling. Immunol. Rev. 2011;243:215–234. doi: 10.1111/j.1600-065X.2011.01038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szretter KJ, et al. The interferon-inducible gene viperin restricts West Nile virus pathogenesis. J. Virol. 2011;85:11557–11566. doi: 10.1128/JVI.05519-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fitzgerald KA. The interferon inducible gene: Viperin. J. Interferon Cytokine Res. 2011;31:131–135. doi: 10.1089/jir.2010.0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fentress SJ, et al. Phosphorylation of immunity-related GTPases by a Toxoplasma gondii–secreted kinase promotes macrophage survival and virulence. Cell Host Microbe. 2010;8:484–495. doi: 10.1016/j.chom.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steinfeldt T, et al. Phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol. 2010;8:e1000576. doi: 10.1371/journal.pbio.1000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dellacasa-Lindberg I, Hitziger N, Barragan A. Localized recrudescence of Toxoplasma infections in the central nervous system of immunocompromised mice assessed by in vivo bioluminescence imaging. Microbes Infect. 2007;9:1291–1298. doi: 10.1016/j.micinf.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 41.Strittmatter C, Lang W, Wiestler OD, Kleihues P. The changing pattern of human immunodeficiency virus–associated cerebral toxoplasmosis: a study of 46 postmortem cases. Acta Neuropathol. 1992;83:475–481. doi: 10.1007/BF00310023. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Y, et al. Virulent Toxoplasma gondii evade immunity-related GTPase-mediated parasite vacuole disruption within primed macrophages. J. Immunol. 2009;182:3775–3781. doi: 10.4049/jimmunol.0804190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheon H, Stark GR. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc. Natl. Acad. Sci. USA. 2009;106:9373–9378. doi: 10.1073/pnas.0903487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol. Rev. 2008;226:41–56. doi: 10.1111/j.1600-065X.2008.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amit I, et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–263. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nusinzon I, Horvath CM. Positive and negative regulation of the innate antiviral response and β interferon gene expression by deacetylation. Mol. Cell Biol. 2006;26:3106–3113. doi: 10.1128/MCB.26.8.3106-3113.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zupkovitz G, et al. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol. Cell Biol. 2006;26:7913–7928. doi: 10.1128/MCB.01220-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klein RS, et al. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 2005;79:11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coley SE, et al. Recombinant mouse hepatitis virus strain A59 from cloned, full-length cDNA replicates to high titers in vitro and is fully pathogenic in vivo. J. Virol. 2005;79:3097–3106. doi: 10.1128/JVI.79.5.3097-3106.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- 51.Oliphant T, et al. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat. Med. 2005;11:522–530. doi: 10.1038/nm1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shrestha B, Gottlieb D, Diamond MS. Infection and injury of neurons by West Nile encephalitis virus. J. Virol. 2003;77:13203–13213. doi: 10.1128/JVI.77.24.13203-13213.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1–9, Supplementary Tables 1–3 and Supplementary Methods (PDF 2064 kb)