

Abstract
Ewing's sarcoma (ES) is a highly malignant tumor of children and young adults. Modern therapy for Ewing's sarcoma combines high-dose chemotherapy for systemic control of disease, with advanced surgical and/or radiation therapeutic approaches for local control. Despite optimal management, the cure rate for localized disease is only approximately 70%, whereas the cure rate for metastatic disease at presentation is less than 30%. Patients who experience long-term disease-free survival are at risk for significant side-effects of therapy, including infertility, limb dysfunction and an increased risk for second malignancies. The identification of new targets for innovative therapeutic approaches is, therefore, strongly needed for its treatment. Many new pharmaceutical agents have been tested in early phases of clinical trials in ES patients who have recurrent disease. While some agents led to partial response or stable disease, the percentages of drugs eliciting responses or causing an overall effect have been minimal. Furthermore, of the new pharmaceuticals being introduced to clinical practice, the most effective agents also have dose-limiting toxicities. Novel approaches are needed to minimize non-specific toxicity, both for patients with recurrence and at diagnosis. This report presents an overview of the potential molecular targets in ES and highlights the possibility that they may serve as therapeutic targets for the disease. Although additional investigations are required before most of these approaches can be assessed in the clinic, they provide a great deal of hope for patients with Ewing's sarcoma.
Keywords: Dose-limiting toxicity, Ewing's sarcoma, molecular targets
INTRODUCTION
Ewing's sarcoma (ES) is a highly malignant bone and soft tissue tumor occurring in children and young adults. Because of its aggressive nature and early systematic spread, its treatment is highly challenging.[1–5] On introduction of VACD-IE (vincristine, actinomycin D, cyclophosphamide, doxorubicin, etoposide and ifosfamide) chemotherapy regimens, the current 5-year survival rates for patients with localized disease are about 70%.[6–8] Nevertheless, ES still has the lowest survival rates of any of the musculoskeletal tumors, with minimal improvements to patient outcomes observed over the last decade.[9] Hence, there is an urgent need to develop targeted therapeutic approaches to improve the treatment.
EWS-FLI1 AS A POTENTIAL TARGET
More than 85% of the ES patients present with a balanced t (11:22) (q24;q12) chromosomal translocation.[10,11] This reciprocal translocation generates a novel in-frame fusion gene between sequences that encode the N-terminus of the RNA binding protein EWS from chromosome 22 and the C-terminus of FLI1 transcription factor on chromosome 11.[12,13] Several evidences have shown EWS-FLI1 as a well-described oncogene and with depletion of this gene product resulting in inhibition of Ewing's sarcoma family of tumor (ESFT) growth. EWS-FLI1 fusion protein therefore is a validated tumor target functioning as an aberrant transcription factor.[14–17] EWS-FLI1 is indisputably the central player in the pathogenesis of ES.[18] EWS-FLI1 has only been identified in tumor cells thus providing a unique therapeutic target.
Strategies to target EWS-FLI1
EWS-FLI1 is not expressed in any cells except those of ESFT. Therefore, EWS-FLI1 targeting would, in theory, only affect ESFT cells. Despite this specific tumor target, there is no treatment available that directly targets EWS-FLI1 (EF).[17] However, ESFT research in recent years has been characterized by the development of some encouraging new strategies that reduce the expression or function of EF and are discussed below [Figure 1].
Figure 1.
Some of the potential molecular targets of Ewing's sarcoma described in this review include: (a) EWS-FLI1 fusion protein, (b) its target genes, (c) growth factor receptor, cell-surface receptors and (d) molecules involved in cell survival, proliferation and anti-apoptotic pathways
Using antisense/small interfering RNA (siRNA) technology
The main promise of antisense technology is its potential for highly specific and safe targeting of aberrant gene expression. It offers the possibilities to bypass resistance toward traditional chemotherapeutic drugs. Reduction of EWS-FLI1 by antisense oligodeoxynucleotides[19–21] antisense RNA expressed from a vector[21–23] and small interfering RNA (siRNA)delivered via nanoparticles[24] inhibit the proliferation of ESFT cell lines and xenografted tumors. The lack of clinical translation of these macromolecule-based strategies lies in the challenge of pharmacologic delivery.[25] The problem in delivery includes both route of administration and formulation of the drug. Successful systemic delivery needs to achieve prolonged plasma circulation, appropriate biodistribution, cellular uptake and cytoplasmic release of the therapeutic agent in an active form.
In laboratory cell line models, antisense oligonucleotides are easy to administer. In general, they need to be chemically modified to block their degradation. This is possible through encapsulation or conjugation to a suitable carrier. Nanotechnology offers some suitable encapsulation strategies like encapsulation of siRNA into lipid bilayer stable-nucleic-acid-lipid particle.[26] The cyclodextrin-based delivery system consists of cyclodextrin-based polycation that self-assemble with siRNA to form particles that are stabilized by surface coating with poly ethylene glycol (PEG).[27] To achieve targeted delivery, some of the PEGs on the surface are conjugated to a modular targeting ligand like transferrin in order to have a preferential binding to cancer cells that overexpress the transferrin receptor. Earlier in vivo studies on ES have shown marked inhibition in tumor growth and metastasis by intravenous administration of transferring-linked EWS-FLI1-targeted siRNA nanoparticles.[28] Another study has shown the same effect by using a Type 1 junction-targeted siRNA, encapsulated in polyalkylcyanoacrylate nanocapsules and injected intratumourally.[29]
In conclusion, oligonucleotides targeting EWS-FLI1 RNA have shown interesting effects in cell culture and in animal preclinical models, and have the potential to be assayed as therapeutic agents either to keep residual disease at a low level or for enhancing the activity of conventional anticancer agents. Therefore, antisense/siRNA strategies may not provide an additional benefit at this time, but suggest promise in the future [Table 1].
Table 1.
Strategies to target EWS-FLI1 gene/protein and their current status

Disrupting protein interactions
EWS-FLI1 interacts with other protein partners in various steps. These may include: (a) interaction with kinases and phosphatases during post-translational modification and (b) interaction with TATA box-binding protein, other cooperating proteins, RNA polymerase,[30] co activators like p300 protein[31] and other general transcription machinery factors. Therapeutic agents may be directed against any of these interactions.
EWS-FLI1 functions in a large multiprotein transcriptional complex and, like other transcription factors, it relies on direct protein–protein interaction as well. RNA helicase A (RHA) was identified as a protein partner of EWS-FLI1 using phage display.[32] Interaction of RHA with EWS-FLI1 suggests that RHA is necessary for EWS-FLI1 tumorigenic function. RHA was found to be present at promoters occupied by EWS-FLI1 and was required for optimal transformation of murine fibroblasts.[32] Reduced EWS-FLI1 activity and tumorigenesis was observed after using mutation, peptide and small molecule approaches to disrupt RHA from binding to EWS-FLI1.[33] One possible way to disrupt this binding is to use small molecules specifically designed to mimic certain functionally active protein domains that can keep EWS-FLI1 apart from its interacting proteins. Recently, surface plasmon resonance screening revealed that YK-4-279, a lead compound with potent anti-ES activity, blocked RHA binding to EWS-FLI1, induced apoptosis in ES cell lines and reduced growth in ES xenografts.[33] The small molecule, YK-4-279, which blocks RHA binding to EWS-FLI1 demonstrated decreased cyclin D levels in ES cells.[33] Because this small molecule is hydrophobic, it should be orally bioavailable and may be suitable for continuous dosing, an important schedule for molecularly targeted agents [Table 1].
Other strategies for targeting EWS-FLI1
It was recently reported that recombinant antibody technology was used to successfully interfere with wild type EWS protein.[34] But, the described antibody does not have any impact on the EF chimeric protein. This may be a result of the originally performed screening strategy and the difficulty in obtaining structurally unaltered and functionally active native EF. However, single-chain antibodies are a theoretically promising strategy, worthy of further investigation. In an attempt to immunologically target the breakpoint region of tumor-specific EWS-FLI1 fusion proteins, a peptide-pulsed vaccination has been investigated. However, it failed to reach clinical applicability[35] [Table 1].
TARGETING INSULIN-LIKE GROWTH FACTOR 1 RECEPTOR
Although the insulin-like growth factor (IGF) system plays a key role in processes like embryogenesis and development, consistent evidence also indicates the involvement of IGF signaling in tumorigenesis. Overexpression of insulin-like growth factor-I receptor (IGF-IR) has been implicated in many tumor models as playing a role in cell growth and tumorigenesis.[36] The interaction of IGF with IGF-IR initiates a cascade of events that starts with the activation of tyrosine kinase and results in divergent effects depending on specific cell types.[36] The IGF-IR-mediated loop is constantly present in cell lines and tissue samples of ES, which suggests a role for this autocrine circuit in the pathogenesis of ES.[37–39] Although the IGF pathway is often considered a “parallel” pathway to EWS-FLI1, recent work suggests that EWS-FLI1 may regulate IGF1 itself.[40,41] Earlier researches (both in vitro and in vivo) have shown that antibodies targeting the receptor could slow cell growth and inhibit the migratory ability of ES cells.[42,43] Both small molecule and antibody-mediated approaches to IGF pathway blockade have shown efficacy in patients in early phase clinical trials.[44–46]
Antibody-mediated approaches
Currently, more than 25 agents acting via IGF1R inhibition are in preclinical and clinical development. IGF1R-targeted monoclonal antibodies have a less-toxic safety profile and a higher patient acceptance than currently available cytotoxic chemotherapy regimens for ES.[46,47] IMC-A12 (Cixutumumab) developed by imclone, is a fully human IgG1 monoclonal antibody, specifically targeting IGF-IR with high affinity and antagonizing IGF-I and IGF-II ligand binding and signaling. It inhibits the IGF-IR pathway by effecting internalization and degradation of IGF-IR, leading to a reduction in surface receptors. In vitro and in vivo, Cixutumumab inhibits proliferation of a variety of human tumor cell lines.[48] Dalotuzumab (MK-0646), being developed by Merck and Co. Inc., is a recombinant humanized IgG1 mAb against the IGFR1 for the potential intravenous treatment of cancer. Preclinical studies have demonstrated that dalotuzumab acts by inhibiting IGF-1- and IGF-2-mediated tumor cell proliferation, IGFR1 autophosphorylation and Akt phosphorylation. In multiple cancer cell lines and in mouse xenograft models, dalotuzumab displayed significant antitumor activity. Preliminary data from Phase I clinical trials suggest that dalotuzumab is safe and well tolerated and significantly inhibits tumor proliferation. Although preliminary results appear promising, nly future clinical and translational data will clarify the best clinical setting and treatment combinations for the optimal use of dalotuzumab in clinical practice.[49] Monoclonal antibodies like BIIB022[50] and AVE-1642[51] have undergone Phase I clinical trials, showing promising results [Table 2].
Table 2.
Antibody-mediated approaches to target IGF1R and their current status

Small-molecule inhibitors
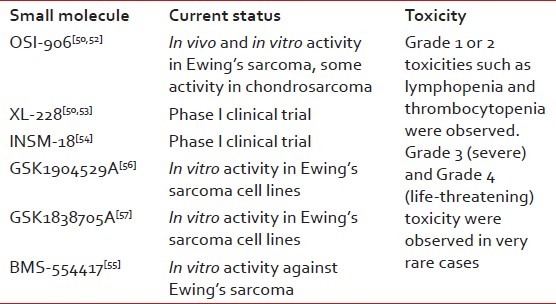
Small-molecule inhibitors of IGF1R are also in preclinical or clinical development. In addition to blocking IGF1R, some of these IGF1R inhibitors may also inhibit insulin receptor A, and several have been shown to have promising preclinical ES activity. These molecules may act more proximal with regard to IGF1R signaling and thus enable oral dosing. On the downside, such agents may have more toxicity than IGF1R monoclonal antibodies.
OSI-906 showed in vivo and in vitro activity in ES and some activity in chondrosarcoma.[50,52] XL-228 is a multitargeted protein kinase inhibitor targeting IGF1R and few other kinases.[50,53] INSM-18 is an orally bioavailable small molecule tyrosine kinase inhibitor that has demonstrated selective inhibition of IGF1R and human epidermal growth factor receptor (Her2/Neu).[54] Small-molecule inhibitors like BMS-554417, GSK1904529A and GSK1838705A have shown in vitro activity against ES.[55–57]
One potential concern is the effect of IGFR inhibition on growth in children and adolescents. IGF-I levels increase during puberty and are a critical growth mediator during this time period. Thus, careful consideration will need to be given to this approach before it is brought into clinical trials involving children. The effects of targeted therapies against IGF-IR were evaluated in combination with conventional chemotherapeutic agents to assess the best drug–drug interactions and treatment schedule. Toxic effects of these tailored therapies were also considered to offer the necessary rationale for the application of possible forthcoming clinical trials [Table 3].
Table 3.
Small-molecule inhibitors of IGF1R and their current status
TARGETING CD99
CD99 (also called MIC2) is an integral transmembrane glycoprotein that is highly expressed in ESs, and used as diagnostic marker.[58–60] ESFT express the CD99 antigen on their external cell membrane, but the function of CD99 is cryptic. Recent data suggest that CD99 is not only a marker and therapeutic target for ES but may also contribute to the disease phenotype. Knockdown of CD99 in ES cell lines resulted in decreased growth in tissue culture, diminished colony formation in soft agar assays, reduced cell motility and smaller tumors with less metastasis in xenograft models.[61] This study also suggested that CD99 inhibits full neuronal differentiation by decreasing the activity of the MAP kinase pathway. Engagement of CD99 with monoclonal antibodies can induce apoptosis through caspase-independent mechanisms in ES cell lines.[62] This finding was extended to show that CD99 engagement slowed tumor formation in athymic mice and enhanced the growth inhibitory effects of doxorubicin and vincristine in cell culture experiments.[63]
However, as the expression of CD99 has been reported in a variety of other human tissues, such as testis, prostate and gastric mucosa, we cannot completely exclude the hypothetical toxicity of anti-CD99 MAb in human ES patients. In addition, in the clinical scenario, anti-CD99 MAb will be used in combination with chemotherapeutic agents that have deleterious effects on the hematogenic progenitors. Therefore, this treatment modality needs further in vivo evaluation to assess its toxicity in an appropriate model, prior to human studies.
P53 PATHWAY AS A POTENTIAL TARGET
Earlier studies have shown that silencing of the EWS-FLI1 expression in ES cell lines increases p53 activity,[64,65] suggesting that the EWS-FLI1 fusion protein plays a role in the constitutive silencing of p53 tumor suppressor activity. It appears that EWS-FLI1 can achieve this through either an indirect mechanism, involving the Notch signaling pathway,[64] or through the formation of a protein complex involving EWS-FLI1and p53.[65] EWS-FLI1 attenuates p53 activity through physical sequestration facilitated by the EWS region of the fusion protein.[65] However, it is unclear whether interaction between p53 and EWS-FLI1 occurs directly or is mediated through other oncogenic binding partners.
ESs provide a unique tumor type in which the majority of cases retain the functionally intact p53 pathways. At present, there is no evidence of permanent suppression of the p53 pathway by specific mutation of critical components. Therefore, the most likely scenario involves abrogation of p53 function via a reversible, post-translational mechanism. This provides unique therapeutic opportunities through intervention with small molecules that directly stabilize and activate endogenous intracellular p53. Small molecules like Nutlin-3a and MI-219 antagonize the p53-MDM2 interaction by blocking the p53-binding pocket of MDM2 and, as a consequence, there is a rapid stabilization and accumulation of p53 protein levels.[66] MI-219 showed high selectivity, less toxicity and highly desirable pharmacokinetic properties, and is currently in early-phase clinical trials. Another small molecule, Tenovins, a class of p53 activators that enhance the acetylation of p53,[67] is under early-phase clinical trials. Actinomycin D, an FDA-approved drug, mimics the action of Nutlin-3a when administered at specific dosages.[68] Recent identification of actinomycin D as a p53 activator has facilitated the translation of these targeted therapies into current ESFT treatment regimens. Low-dose actinomycin D hold an exciting potential as a directed molecular-based approach to specifically activate wild-type p53 in ESFTs and the organization of clinical trials currently in progress to attest the potential of this approach. Interestingly, ES cells are highly sensitive to actinomycin D in vitro, with potent antitumor activity observed within the ranges described as “low dose,” specifically in ES cell lines that retain wild type p53.[69] Further studies are warranted to evaluate the potential of incorporation of low-dose actinomycin D with the current standard of care for the treatment of patients with wild type p53 ESFTs [Table 4].
Table 4.
Small molecules that directly stabilize and activate endogenous intracellular p53 and their current status

MAMMALIAN TARGET OF RAPAMYCIN (M-TOR)
Rapamycin, which has been used for several years as an immune suppressor in organ transplantation patients, has been shown to inhibit tumor growth and is currently in Phase I–III clinical trials. Rapamycin is an inhibitor of the mammalian target of rapamycin (mTOR). mTOR is a downstream effector of the phosphatidylinositol 3-kinase (PI3K)-AKT signaling pathway, which mediates cell survival and proliferation.[70] The PI3K/AKT pathway is thought to be overactivated in many cancers, and may account for the response of various cancers to mTOR inhibitors.[71] mTOR inhibitors block signaling to downstream pathways that control cell growth. ESFT cell lines carrying EWS-FLI1 alleles of different types expressed different levels of total and phosphorylated mTOR protein.[72] Rapamycin, an mTOR inhibitor, efficiently blocks the proliferation of cell lines by promoting cell cycle arrest at the G1 phase. This is paralleled by the downregulation of the levels of the EWS-FLI1 proteins, regardless of their fusion type, and the concomitant restoration of the expression of the transforming growth factor beta (TGF β) type 2 receptor (TGF-βRII), which is known to be repressed by several EWS-ETS fusion proteins.[72] mTOR signaling plays a central role in ES cell pathobiology and strongly suggests that the use of rapamycin as a cytostatic agent may be an efficient tool for the treatment of patients with ESFT tumors.[71,72] Concurrent administration of EWS-FLI1 antisense oligonucleotides and rapamycin efficiently induced the apoptotic death of ES cells in culture through a process involving TGF-β. Preclinical experiments conclusively showed that the combined treatment with antisense oligonucleotides and rapamycin caused a significant inhibition of tumor growth in mice.[73] These results encourage further exploration of the potential of this combined therapeutic modality as a novel strategy for the treatment of tumors of the ESFT.
TARGETING BCL2 APOPTOTIC PATHWAY
Bcl-2, an antiapoptotic family member, contributes to neoplastic progression by enhancing cell survival through repression of mitochondrial death signaling.[74–76] A spectrum of pediatric solid tumor cell lines, including neuroblastoma,[77,78] ES,[79,80] Wilm's tumor[81,82] and synovial sarcoma,[83] overexpress Bcl-2. Overexpression of Bcl-2 results in a resistance to apoptosis-inducing agents, including radiation and chemotherapy, and has been associated with poor clinical response and shorter survival.[76,84,85]
Targeted downregulation of Bcl-2 expression by antisense oligodeoxynucleotide G3139 may result in apoptosis of tumor cells, especially when coadministered with cytotoxic drugs.[86] Clinical trials using G3139 (genasense) in combination with doxorubicin and cyclophosphamide are ongoing in patients with ES. Thus, pharmacologic downregulation of Bcl-2 by G3139 may potentially enhance the chemotherapy responsiveness of tumor cells to these drugs.
CONCLUSION
ESFT is a rare cancer, afflicting mostly children. Because it is one of the rare diseases, pharmaceutical industries lack interest in investing funds in developing new drugs. The consistent characteristic chromosomal translocations of the ESFT, which result in the fusion of EWS-FLI1 critical for the maintenance of the tumor phenotype and the subsequent formation of novel chimeric proteins, offer promising molecular targets for developing new therapies. The recent availability of biologically active recombinant EWS-FLI1 makes it possible to screen potential small molecules that influence the EWS-FLI1 function. Deeper understanding of tumorigenicity pathways involved in ES will be able to enhance the progress in developing potential therapeutic agents. The challenge is how to harness these molecular targets for creating specific therapies. Knowledge of this will lead to novel ways that specifically and effectively treat patients. Progress in the near future is aimed at combining small molecules with standard chemotherapy. This is expected to synergistically increase tumor death and to decrease the development of resistance by interfering with essential pathways for tumor growth. The parallel increase in technology, such as delivery systems, will likewise benefit future patients. Patients with ESFT will benefit from ongoing investigations that will hopefully allow for more successful and less-toxic therapies.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Sailer SL, Harmon DC, Mankin HJ, Truman JT, Suit HD. Ewing's sarcoma: surgical resection as a prognostic factor. Int J Radiat Oncol Biol Phys. 1988;15:43–52. doi: 10.1016/0360-3016(88)90345-8. [DOI] [PubMed] [Google Scholar]
- 2.Arai Y, Kun LE, Brooks MT, Fairclough DL, Fontanesi J, Meyer WH, et al. Ewing's sarcoma: Local tumor control and patterns of failure following limited-volume radiation therapy. Int J Radiat Oncol Biol Phys. 1991;21:1501–8. doi: 10.1016/0360-3016(91)90325-x. [DOI] [PubMed] [Google Scholar]
- 3.Donaldson SS, Torrey M, Link MP, Glicksman A, Gilula L, Laurie F, et al. A multidisciplinary study investigating radiotherapy in Ewing's sarcoma: end results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys. 1998;42:125–35. doi: 10.1016/s0360-3016(98)00191-6. [DOI] [PubMed] [Google Scholar]
- 4.Dunst J, Jürgens H, Sauer R, Pape H, Paulussen M, Winkelmann W, et al. Radiation therapy in Ewing's sarcoma: an update of the CESS 86 trial. Int J Radiat Oncol Biol Phys. 1995;32:919–30. doi: 10.1016/0360-3016(95)00016-r. [DOI] [PubMed] [Google Scholar]
- 5.Hayes FA, Thompson EI, Meyer WH, Kun L, Parham D, Rao B, et al. Therapy for localized Ewing's sarcoma of bone. J Clin Oncol. 1989;7:208–13. doi: 10.1200/JCO.1989.7.2.208. [DOI] [PubMed] [Google Scholar]
- 6.Bacci G, Mercuri M, Longhi A, Bertoni F, Barbieri E, Donati D, et al. Neoadjuvant chemotherapy for Ewing's tumour of bone: recent experience at the Rizzoli Orthopaedic Institute. Eur J Cancer. 2002;38:2243–51. doi: 10.1016/s0959-8049(02)00148-x. [DOI] [PubMed] [Google Scholar]
- 7.Iwamoto Y. Diagnosis and Treatment of Ewing's Sarcoma. Jpn J Clin Oncol. 2007;37:79–89. doi: 10.1093/jjco/hyl142. [DOI] [PubMed] [Google Scholar]
- 8.Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348:694–701. doi: 10.1056/NEJMoa020890. [DOI] [PubMed] [Google Scholar]
- 9.Arndt CA, Crist WM. Common musculoskeletal tumors of childhood and adolescence. N Engl J Med. 1999;341:342–52. doi: 10.1056/NEJM199907293410507. [DOI] [PubMed] [Google Scholar]
- 10.Riggi N, Stamenkovic I. The Biology of Ewing sarcoma. Cancer Lett. 2007;254:1–10. doi: 10.1016/j.canlet.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 11.Arvand A, Denny CT. Biology of EWS/ETS fusions in Ewing's family tumors. Oncogene. 2001;20:5747–54. doi: 10.1038/sj.onc.1204598. [DOI] [PubMed] [Google Scholar]
- 12.Zucman J, Melot T, Desmaze C, Ghysdael J, Plougastel B, Peter M, et al. Modern diagnostic methods in the Ewing's sarcoma family: Six patients with histologic soft tissue tumors. EMBO J. 1993;12:4481–7. doi: 10.1002/j.1460-2075.1993.tb06137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumors. Nature. 1992;359:162–5. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- 14.May WA, Lessnick SL, Braun BS, Klemsz M, Lewis BC, Lunsford LB, et al. The Ewing's Sarcoma EWS/FLI-1 Fusion Gene Encodes a More Potent Transcriptional Activator and Is a More Powerful Transforming Gene than FLI-1. Mol Cell Biol. 1993;13:7393–8. doi: 10.1128/mcb.13.12.7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.May WA, Gishizky ML, Lessnick SL, Lunsford LB, Lewis BC, Delattre O, et al. Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLII for transformation. Proc Natl Acad Sci. 1993;90:5752–6. doi: 10.1073/pnas.90.12.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohno T, Rao VN, Reddy ES. EWS/Fli-1 Chimeric Protein Is a Transcriptional Activator1. Cancer Res. 1993;53:5859–63. [PubMed] [Google Scholar]
- 17.Uren A, Toretsky JA. Ewing's sarcoma oncoprotein EWS-FLI1: the perfect target without a therapeutic agent. Future Oncol. 2005;1:521–8. doi: 10.2217/14796694.1.4.521. [DOI] [PubMed] [Google Scholar]
- 18.Jedlicka P. Ewing sarcoma, an enigmatic malignancy of likely progenitor cell origin, driven by transcription factor oncogenic fusions. Int J Clin Exp Pathol. 2010;3:338–47. [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka K, Iwakuma T, Harimaya K, Sato H, Iwamoto Y. EWS-Fli1 antisense oligodeoxynucleotide inhibits proliferation of human Ewing's sarcoma and primitive neuroectodermal tumor cells. J Clin Invest. 1997;99:239–47. doi: 10.1172/JCI119152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toretsky JA, Connell Y, Neckers L, Bhat NK. Inhibition of EWS-FLI-1 fusion protein with antisense oligodeoxynucleotides. J Neurooncol. 1997;31:9–16. doi: 10.1023/a:1005716926800. [DOI] [PubMed] [Google Scholar]
- 21.Ouchida M, Ohno T, Fujimura Y, Rao VN, Reddy ES. Loss of tumorigenicity of Ewing's sarcoma cells expressing antisense RNA to EWS-fusion transcripts. Oncogene. 1995;11:1049–54. [PubMed] [Google Scholar]
- 22.Kovar H, Aryee DN, Jug G, Henöckl C, Schemper M, Delattre O, et al. EWS/FLI-1 antagonists induce growth inhibition of Ewing tumor cells in vitro. Cell Growth Differ. 1996;7:429–37. [PubMed] [Google Scholar]
- 23.Maksimenko A, Lambert G, Bertrand JR, Fattal E, Couvreur P, Malvy C. Therapeutic potentialities of EWS-Fli-1 mRNA-targeted vectorized antisense oligonucleotides. Ann N Y Acad Sci. 2003;1002:72–7. doi: 10.1196/annals.1281.017. [DOI] [PubMed] [Google Scholar]
- 24.Hu-Lieskovan S, Heidel JD, Bartlett DW, Davis ME, Triche TJ. Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing's sarcoma. Cancer Res. 2005;65:8984–92. doi: 10.1158/0008-5472.CAN-05-0565. [DOI] [PubMed] [Google Scholar]
- 25.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–41. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 26.Rossi JJ. RNAi therapeutics: SNALPing siRNAs in vivo. Gene Ther. 2006;13:583–4. doi: 10.1038/sj.gt.3302661. [DOI] [PubMed] [Google Scholar]
- 27.Howard KA. Delivery of RNA interference therapeutics using polycation-based nanoparticles. Adv Drug Deliv Rev. 2009;61:710–20. doi: 10.1016/j.addr.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Choi CH, Alabi CA, Webster P, Davis ME. Mechanism of active targeting in solid tumors with transferrin-containing gold nanoparticles. Proc Natl Acad Sci. 2010;107:1235–40. doi: 10.1073/pnas.0914140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toub N, Bertrand JR, Tamaddon A, Elhamess H, Hillaireau H, Maksimenko A, et al. Efficacy of siRNA nanocapsules targeted against the EWS-Fli1 oncogene in Ewing sarcoma. Pharm Res. 2006;23:892–900. doi: 10.1007/s11095-006-9901-9. [DOI] [PubMed] [Google Scholar]
- 30.Petermann R, Mossier BM, Aryee DN, Khazak V, Golemis EA, Kovar H. Oncogenic EWS-Fli1 interacts with hsRPB7, a subunit of human RNA polymerase II. Oncogene. 1998;17:603–10. doi: 10.1038/sj.onc.1201964. [DOI] [PubMed] [Google Scholar]
- 31.Nakatani F, Tanaka K, Sakimura R, Matsumoto Y, Matsunobu T, Li X, et al. Identification of p21WAF[1]/CIP[1] as a Direct Target of EWS-Fli1 Oncogenic Fusion Protein. J Biol Chem. 2003;278:15105–15. doi: 10.1074/jbc.M211470200. [DOI] [PubMed] [Google Scholar]
- 32.Toretsky JA, Erkizan V, Levenson A, Abaan OD, Parvin JD, Cripe TP, et al. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Res. 2006;66:5574–81. doi: 10.1158/0008-5472.CAN-05-3293. [DOI] [PubMed] [Google Scholar]
- 33.Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing's sarcoma. Nat Med. 2009;15:750–6. doi: 10.1038/nm.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aryee DN, Kreppel M, Bachmaier R, Uren A, Muehlbacher K, Wagner S, et al. Single-chain Antibodies to the EWS NH2 Terminus Structurally Discriminate between Intact and Chimeric EWS in Ewing's Sarcoma and Interfere with the Transcriptional Activity of EWS In vivo. Cancer Res. 2006;66:9862–9. doi: 10.1158/0008-5472.CAN-05-4042. [DOI] [PubMed] [Google Scholar]
- 35.Dagher R, Long LM, Read EJ, Leitman SF, Carter CS, Tsokos M, et al. Pilot trial of tumor-specific peptide vaccination and continuous infusion interleukin-2 in patients with recurrent Ewing sarcoma and alveolar rhabdomyosarcoma: An inter institute NIH study. Med Pediatr Oncol. 2002;38:158–64. doi: 10.1002/mpo.1303. [DOI] [PubMed] [Google Scholar]
- 36.LeRoith D, Baserga R, Helman L, Roberts CT., Jr Insulin-like growth factors and cancer. Ann Intern Med. 1995;122:54–9. doi: 10.7326/0003-4819-122-1-199501010-00009. [DOI] [PubMed] [Google Scholar]
- 37.Yee D, Favoni RE, Lebovic GS, Lombana F, Powell DR, Reynolds CP, et al. Insulin-like growth factor I expression by tumors of neuroectodermal origin with the t(11;22) chromosomal translocation. A potential autocrine growth factor. J Clin Invest. 1990;86:1806–14. doi: 10.1172/JCI114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Valen F, Winkelmann W, Jürgens H. Type I and type II insulin-like growth factor receptors and their function in human Ewing's sarcoma cells. J Cancer Res Clin Oncol. 1992;118:269–75. doi: 10.1007/BF01208615. [DOI] [PubMed] [Google Scholar]
- 39.Scotlandi K, Benini S, Sarti M, Serra M, Lollini PL, Maurici D, et al. Insulin-like growth factor I receptor-mediated circuit in Ewing's sarcoma/peripheral neuroectodermal tumor: a possible therapeutic target. Cancer Res. 1996;56:4570–4. [PubMed] [Google Scholar]
- 40.Cironi L, Riggi N, Provero P, Wolf N, Suvà ML, Suvà D, et al. IGF1 is a common target gene of Ewing's sarcoma fusion proteins in mesenchymal progenitor cells. PLoS One. 2008;3:e2634. doi: 10.1371/journal.pone.0002634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herrero-Martín D, Osuna D, Ordóñez JL, Sevillano V, Martins AS, Mackintosh C, et al. Stable interference of EWS-FLI1 in an Ewing sarcoma cell line impairs IGF-1/IGF-1R signalling and reveals TOPK as a new target. Br J Cancer. 2009;101:80–90. doi: 10.1038/sj.bjc.6605104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamilton G, Mallinger R, Hofbauer S, Havel M. The monoclonal HBA-71 antibody modulates proliferation of thymocytes and Ewing's sarcoma cells by interfering with the action of insulin-like growth factor I. Thymus. 1991;18:33–41. [PubMed] [Google Scholar]
- 43.Scotlandi K, Benini S, Nanni P, Lollini PL, Nicoletti G, Landuzzi L, et al. Blockage of insulin-like growth factor-I receptor inhibits the growth of Ewing's sarcoma in athymic mice. Cancer Res. 1998;58:4127–31. [PubMed] [Google Scholar]
- 44.Kolb EA, Gorlick R, Houghton PJ, Morton CL, Lock R, Carol H, et al. Initial testing (stage 1) of a monoclonal antibody (SCH 717454) against the IGF-1 receptor by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50:1190–7. doi: 10.1002/pbc.21450. [DOI] [PubMed] [Google Scholar]
- 45.Kurmasheva RT, Dudkin L, Billups C, Debelenko LV, Morton CL, Houghton PJ. The insulin-like growth factor-1 receptor targeting antibody, CP-751 871, suppresses tumor-derived VEGF and synergizes with rapamycin in models of childhood sarcoma. Cancer Res. 2009;69:7662–71. doi: 10.1158/0008-5472.CAN-09-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tolcher AW, Sarantopoulos J, Patnaik A, Papadopoulos K, Lin CC, Rodon J, et al. Phase I, Pharmacokinetic, and Pharmacodynamic Study of AMG 479, a Fully Human Monoclonal Antibody to Insulin-Like Growth Factor Receptor 1. J Clin Oncol. 2009;27:5800–7. doi: 10.1200/JCO.2009.23.6745. [DOI] [PubMed] [Google Scholar]
- 47.Olmos D, Postel-Vinay S, Molife LR, Okuno SH, Schuetze SM, Paccagnella ML, et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751 871) in patients with sarcoma and Ewing's sarcoma: A phase 1 expansion cohort study. Lancet Oncol. 2009;11:129–35. doi: 10.1016/S1470-2045(09)70354-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKian KP, Haluska P. Cixutumumab. Expert Opin Investig Drugs. 2009;18:1025–33. doi: 10.1517/13543780903055049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scartozzi M, Bianconi M, Maccaroni E, Giampieri R, Berardi R, Cascinu S. Dalotuzumab, a recombinant humanized mAb targeted against IGFR1 for the treatment of cancer. Curr Opin Mol Ther. 2010;12:361–71. [PubMed] [Google Scholar]
- 50.Subbiah V, Anderson P. Targeted Therapy of Ewing's Sarcoma. Sarcoma. 2011;2011:686985. doi: 10.1155/2011/686985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hewish M, Chau I, Cunningham D. Insulin-like growth factor 1 receptor targeted therapeutics: novel compounds and novel treatment strategies for cancer medicine. Recent Pat Anticancer Drug Discov. 2009;4:54–72. doi: 10.2174/157489209787002515. [DOI] [PubMed] [Google Scholar]
- 52.Mulvihill MJ, Cooke A, Rosenfeld-Franklin M, Buck E, Foreman K, Landfair D, et al. Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med Chem. 2009;1:1153–71. doi: 10.4155/fmc.09.89. [DOI] [PubMed] [Google Scholar]
- 53.Schenone S, Brullo C, Musumeci F, Botta M. Novel dual Src/Abl inhibitors for hematologic and solid malignancies. Expert Opin Investig Drugs. 2010;19:931–45. doi: 10.1517/13543784.2010.499898. [DOI] [PubMed] [Google Scholar]
- 54.Olmos D, Tan DS, Jones RL, Judson IR. Biological rationale and current clinical experience with anti insulin-like growth factor 1 receptor monoclonal antibodies in treating sarcoma: twenty years from the bench to the bedside. Cancer J. 2010;16:183–94. doi: 10.1097/PPO.0b013e3181dbebf9. [DOI] [PubMed] [Google Scholar]
- 55.Carboni JM, Wittman M, Yang Z, Lee F, Greer A, Hurlburt W, et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol Cancer Ther. 2009;8:3341–9. doi: 10.1158/1535-7163.MCT-09-0499. [DOI] [PubMed] [Google Scholar]
- 56.Sabbatini P, Rowand JL, Groy A, Korenchuk S, Liu Q, Atkins C, et al. Antitumor activity of GSK1904529A, a small-molecule inhibitor of the insulin like growth factor-I receptor tyrosine kinase. Clin Cancer Res. 2009;15:3058–67. doi: 10.1158/1078-0432.CCR-08-2530. [DOI] [PubMed] [Google Scholar]
- 57.Sabbatini P, Korenchuk S, Rowand JL, Groy A, Liu Q, Leperi D, et al. GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Mol Cancer Ther. 2009;8:2811–20. doi: 10.1158/1535-7163.MCT-09-0423. [DOI] [PubMed] [Google Scholar]
- 58.Kovar H, Dworzak M, Strehl S, Schnell E, Ambros IM, Ambros PF, et al. Overexpression of the pseudoautosomal gene MIC2 in Ewing's sarcoma and peripheral primitive neuroectodermal tumor. Oncogene. 1990;5:1067–70. [PubMed] [Google Scholar]
- 59.Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M. MIC2 is a specific marker for Ewing's sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewing's sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer. 1991;67:1886–93. doi: 10.1002/1097-0142(19910401)67:7<1886::aid-cncr2820670712>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 60.Fellinger EJ, Garin-Chesa P, Triche TJ, Huvos AG, Rettig WJ. Immunohistochemical analysis of Ewing's sarcoma cell surface antigen p30/32MIC2. Am J Pathol. 1991;139:317–25. [PMC free article] [PubMed] [Google Scholar]
- 61.Rocchi A, Manara MC, Sciandra M, Zambelli D, Nardi F, Nicoletti G, et al. CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. J Clin Invest. 2010;120:668–80. doi: 10.1172/JCI36667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sohn HW, Choi EY, Kim SH, Lee IS, Chung DH, Sung UA, et al. Engagement of CD99 induces apoptosis through a calcineurin-independent pathway in Ewing's sarcoma cells. Am J Pathol. 1998;153:1937–45. doi: 10.1016/S0002-9440(10)65707-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scotlandi K, Baldini N, Cerisano V, Manara MC, Benini S, Serra M, et al. CD99 engagement: an effective therapeutic strategy for Ewing tumors. Cancer Res. 2000;60:5134–42. [PubMed] [Google Scholar]
- 64.Ban J, Bennani-Baiti IM, Kauer M, Schaefer KL, Poremba C, Jug G, et al. EWS-FLI1 suppresses NOTCH-activated p53 in Ewing's sarcoma. Cancer Res. 2008;68:7100–9. doi: 10.1158/0008-5472.CAN-07-6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li Y, Tanaka K, Fan X, Nakatani F, Li X, Nakamura T, et al. Inhibition of the transcriptional function of p53 by EWS-Fli1 chimeric protein in Ewing Family Tumors. Cancer Lett. 2010;294:57–65. doi: 10.1016/j.canlet.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 66.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 67.Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13:454–63. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pishas KI, Al-Ejeh F, Zinonos I, Kumar R, Evdokiou A, Brown MP, et al. Nutlin-3a is a potential therapeutic for Ewing Sarcoma. Clin Cancer Res. 2011;17:494–504. doi: 10.1158/1078-0432.CCR-10-1587. [DOI] [PubMed] [Google Scholar]
- 69.Choong ML, Yang H, Lee MA, Lane DP. Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle. 2009;8:2810–8. doi: 10.4161/cc.8.17.9503. [DOI] [PubMed] [Google Scholar]
- 70.Vignot S, Faivre S, Aguirre D, Raymond E. Review mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol. 2005;16:525–37. doi: 10.1093/annonc/mdi113. [DOI] [PubMed] [Google Scholar]
- 71.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mT OR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mateo-Lozano S, Tirado OM, Notario V. Rapamycin induces the fusion type independent downregulation of the EWS/FLI1 proteins and inhibits Ewing's sarcoma cell proliferation. Oncogene. 2003;22:9282–7. doi: 10.1038/sj.onc.1207081. [DOI] [PubMed] [Google Scholar]
- 73.Mateo-Lozano S, Gokhale PC, Soldatenkov VA, Dritschilo A, Tirado OM, Notario V. Combined Transcriptional and Translational Targeting of EWS/FLI-1in Ewing's Sarcoma. Clin Cancer Res. 2006;12:6781–90. doi: 10.1158/1078-0432.CCR-06-0609. [DOI] [PubMed] [Google Scholar]
- 74.Reed JC, Stein C, Subasinghe C, Haldar S, Croce CM, Yum S, et al. Antisense mediated inhibition of BCL2 protooncogene expression and leukemic cell growth and survival: Comparisons of phosphodiester and phosphorothioate oligodeoxynucleotides. Cancer Res. 1990;50:6565–70. [PubMed] [Google Scholar]
- 75.Smith MR, Abubakr Y, Mohammad R, Xie T, Hamdan M, al-Katib A, et al. Antisense oligodeoxyribonucleotide down-regulation of bcl-2 gene expression inhibits growth of the lowgrade non-Hodgkin's lymphoma cell line WSU-FSCCL. Cancer Gene Ther. 1995;2:207–12. [PubMed] [Google Scholar]
- 76.Reed JC. bcl-2: prevention of apoptosis as a mechanism of drug resistance. Hematol Oncol Clin North Am. 1995;9:451–73. [PubMed] [Google Scholar]
- 77.Ramani P, Lu QL. Expression of bcl-2 gene product in neuroblastoma. J Pathol. 1994;172:273–8. doi: 10.1002/path.1711720308. [DOI] [PubMed] [Google Scholar]
- 78.Dole M, Nunez G, Merchant AK, Maybaum J, Rode CK, Bloch CA, et al. Bcl-2 inhibits chemotherapy-induced apoptosis in neuroblastoma. Cancer Res. 1994;54:3253–9. [PubMed] [Google Scholar]
- 79.Fulda S, Debatin KM. Betulinic acid induces apoptosis through a direct effect on mitochondria in neuroectodermal tumors. Med Pediatr Oncol. 2000;35:616–8. doi: 10.1002/1096-911x(20001201)35:6<616::aid-mpo27>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 80.Soldatenkov VA, Dritschilo A. Apoptosis of Ewing's sarcoma cells is accompanied by accumulation of ubiquitinated proteins. Cancer Res. 1997;57:3881–5. [PubMed] [Google Scholar]
- 81.Mayo MW, Wang CY, Drouin SS, Madrid LV, Marshall AF, Reed JC, et al. WT1 modulates apoptosis by transcriptionally upregulating the bcl-2 proto-oncogene. EMBO J. 1999;18:3990–4003. doi: 10.1093/emboj/18.14.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Re GG, Hazen-Martin DJ, El Bahtimi R, Brownlee NA, Willingham MC, Garvin AJ. Prognostic significance of bcl-2 in Wilms’ tumor and oncogenic potential of Bcl-X(L) in rare tumor cases. Int J Cancer. 1999;84:192–200. doi: 10.1002/(sici)1097-0215(19990420)84:2<192::aid-ijc17>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 83.Kawauchi S, Fukuda T, Oda Y, Saito T, Oga A, Takeshita M, et al. Prognostic significance of apoptosis in synovial sarcoma: Correlation with clinicopathologic parameters, cell proliferative activity, and expression of apoptosis related proteins. Mod Pathol. 2000;13:755–65. doi: 10.1038/modpathol.3880131. [DOI] [PubMed] [Google Scholar]
- 84.Kitada S, Takayama S, De Riel K, Tanaka S, Reed JC. Reversal of chemoresistance of lymphoma cells by antisense-mediated reduction of bcl-2 gene expression. Antisense Res Dev. 1994;4:71–9. doi: 10.1089/ard.1994.4.71. [DOI] [PubMed] [Google Scholar]
- 85.Campos L, Sabido O, Rouault JP, Guyotat D. Effects of BCL-2 antisense oligodeoxynucleotides on in vitro proliferation and survival of normal marrow progenitors and leukemic cells. Blood. 1994;84:595–600. [PubMed] [Google Scholar]
- 86.Rheingold SR, Hogarty MD, Blaney SM, Zwiebel JA, Sauk-Schubert C, Chandula R, et al. Phase I Trial of G3139, a bcl-2 Antisense Oligonucleotide, Combined With Doxorubicin and Cyclophosphamide in Children With Relapsed Solid Tumors: A Children's Oncology Group Study. J Clin Oncol. 2007;25:1512–8. doi: 10.1200/JCO.2006.09.5125. [DOI] [PubMed] [Google Scholar]