

Abstract
OBJECTIVE
In diabetes, glucose toxicity affects different organ systems, including pancreatic islets where it leads to β-cell apoptosis, but the mechanisms are not fully understood. Recently, we identified thioredoxin-interacting protein (TXNIP) as a proapoptotic β-cell factor that is induced by glucose, raising the possibility that TXNIP may play a role in β-cell glucose toxicity.
RESEARCH DESIGN AND METHODS
To assess the effects of glucose on TXNIP expression and apoptosis and define the role of TXNIP, we used INS-1 β-cells; primary mouse islets; obese, diabetic BTBR.ob mice; and a unique mouse model of TXNIP deficiency (HcB-19) that harbors a natural nonsense mutation in the TXNIP gene.
RESULTS
Incubation of INS-1 cells at 25 mmol/l glucose for 24 h led to an 18-fold increase in TXNIP protein, as assessed by immunoblotting. This was accompanied by increased apoptosis, as demonstrated by a 12-fold induction of cleaved caspase-3. Overexpression of TXNIP revealed that TXNIP induces the intrinsic mitochondrial pathway of apoptosis. Islets of diabetic BTBR.ob mice also demonstrated increased TXNIP and apoptosis as did isolated wild-type islets incubated at high glucose. In contrast, TXNIP-deficient HcB-19 islets were protected against glucose-induced apoptosis as measured by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling and caspase-3, indicating that TXNIP is a required causal link between glucose toxicity and β-cell death.
CONCLUSIONS
These findings shed new light onto the molecular mechanisms of β-cell glucose toxicity and apoptosis, demonstrate that TXNIP induction plays a critical role in this vicious cycle, and suggest that inhibition of TXNIP may represent a novel approach to reduce glucotoxic β-cell loss.
Type 2 diabetes is a growing public health issue characterized by peripheral insulin resistance and decompensation of the pancreatic β-cells that can no longer keep up with the increased insulin requirements, resulting in hyperglycemia. These elevated glucose levels have detrimental effects on various tissues including the pancreatic β-cell. β-Cell glucose toxicity leads to progressive β-cell dysfunction, impaired insulin gene transcription (1,2), and irreversible β-cell loss by apoptosis (3–14), resulting in a vicious cycle with worsening hyperglycemia. However, the exact molecular mechanisms by which glucose toxicity leads to apoptotic β-cell loss are still not fully understood.
Recently, we identified thioredoxin-interacting protein (TXNIP) as a highly glucose-regulated proapoptotic factor in β-cells (15), suggesting that it may represent a potential mediator of β-cell glucose toxicity. TXNIP binds to and inhibits thioredoxin and thereby can modulate the cellular redox state and promote oxidative stress (16–20). In addition, TXNIP has been shown to exert antiproliferative effects by inducing cell-cycle arrest at the G0/G1 phase (16,21,22), and TXNIP overexpression rendered fibroblasts and cardiomyocytes more susceptible to apoptosis (16,23). Similarly, we found that TXNIP overexpression also induces apoptosis in pancreatic β-cells (15). Moreover, we demonstrated that glucose stimulates TXNIP transcription through a carbohydrate response element in the TXNIP promoter, resulting in elevated TXNIP mRNA expression (15). Together, these findings raised the possibility that TXNIP might play a role in the glucotoxic β-cell death associated with diabetes. The present study was therefore aimed at determining whether glucose and/or diabetes upregulate TXNIP protein levels in vivo and if so how this affects β-cell apoptosis. To further investigate the causal relationship and address the question of whether TXNIP is critical for glucose toxicity–induced β-cell apoptosis, we used TXNIP-deficient primary HcB-19 islets.
RESEARCH DESIGN AND METHODS
All mouse studies were approved by the University of Wisconsin Animal Care and Use Committee, and the National Institutes of Health principles of laboratory animal care were followed. Obese, insulin-resistant, and diabetic BTBR.ob mice (The Jackson Laboratories, Bar Harbor, ME) homozygous for the leptinob mutation were used as a model of type 2 diabetes (24 –26). Diabetes was confirmed by blood glucose measurements using a OneTouch Ultra glucometer. Lean and normoglycemic BTBR.lean mice were used as controls. The C3H congenic TXNIP-deficient HcB-19 (HcB) mice harboring a naturally occurring nonsense mutation in the TXNIP gene and the control C3H/DiSnA (C3H) strain have been described previously (27–29). C57BL/6 mice were purchased from The Jackson Laboratory.
Islet isolation
Mouse pancreatic islets were isolated by a collagenase digestion procedure as described previously (25,26). In brief, immediately after the mice were killed, pancreata were inflated with 5 ml collagenase solution (0.40 mg/ml type XI collagenase; Sigma, St. Louis, MO) in Hanks’ balanced salt solution (HBSS) (Invitrogen, Carlsbad, CA) with 0.02% radioimmunoassay-grade BSA (Sigma) and placed in 25 ml of the same solution, gassed with 95%O2/5%CO2 for 5 min, and vigorously shaken at 37°C for 14 min. After a quick spin, the tissue pellet was washed twice with 10 ml cold HBSS, passed through a 925-micron Spectra mesh filter (Fisher, St. Louis, MO) to remove large debris, and resuspended in 5 ml of 25% Ficoll (type 400-DL; Sigma) prepared with HBSS in a 50-ml conical tube. A total of 2.5 ml of 23, 20.5, and 11% Ficoll were layered carefully on top of each other, and the gradient was centrifuged for 15 min at 800g. Layers above the 25% Ficoll containing the isolated islets were collected, washed with HBSS, and the islets pelleted by a 5-min centrifugation at 800g. To further exclude contamination by exocrine tissue, islets were handpicked under stereomicroscopic observation and used immediately for protein extraction or incubation at low or high glucose. Human pancreatic islets were isolated from brain-dead organ donors under a protocol approved by the University of Wisconsin Institutional Review Board and were a generous gift of Drs. Matthew Hanson and Luis A. Fernandez.
Tissue culture
INS-1 cells were grown in RPMI-1640 (Invitrogen) supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, 1 mmol/l sodium pyruvate, 2 mmol/l L-glutamine, 10 mmol/l HEPES, and 0.05 mmol/l 2-mercaptoethanol. INS-1 cells with constitutive TXNIP overexpression (INS-TXNIP) and control cells overexpressing LacZ (INS-LacZ) were generated and selected as described previously (15). INS-1 cells and isolated HcB-19, C3H, C57BL/6, or human islets were incubated in RPMI supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin and containing 5 or 25 mmol/l glucose for 24 h before protein extracts were prepared or cells were fixed for immunohistochemistry.
Western blotting
Protein extracts were prepared using a lysis buffer containing HEPES (50 mmol/l), Nonidet P-40 (10%), sodium fluoride (100 mmol/l), sodium pyrophosphate (10 mmol/l), EDTA (4 mmol/l), phenylmethanesulphonyl fluoride (1 mmol/l), leupeptin (2 μmol/l), activated sodium orthovanadate (2 mmol/l), and okadaic acid (100 nmol/l). INS-1 cells were grown in T75 flasks and scraped into 0.2 ml of lysis buffer. Isolated primary mouse islets of two animals were pooled and suspended in lysis buffer (20 μl/200 islets). Proteins were separated by 4 –20% SDS-PAGE, blotted onto polyvinylidene difluoride membranes, and detected using the following primary antibodies: TXNIP (1:400) (JY2; MBL International, Woburn, MA), monoclonal cleaved caspase-3 (1:200) (Cell Signaling, Boston, MA), and β-actin (1:200) (Abcam, Cambridge, MA) and the following secondary antibodies: anti-mouse IgG (1:5,000) (Amersham, Piscataway, NJ) and anti-rabbit IgG (Bio-Rad, Hercules, CA). Bands were visualized by Lumigen PS-3 detection reagent (Amersham) and quantified by ImageQuant version 5.1 (GE Healthcare Lifesciences).
Immunohistochemistry and terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
For immunohistochemistry and transferase-mediated dUTP nick-end labeling (TUNEL), ~100 isolated mouse islets were mixed with 15 μl of Affi-Gel Blue Gel (Bio-Rad), fixed in 4% formaldehyde, washed in PBS, and the pellets resuspended in 0.5 ml of warm 2% Difco-agar in an Eppendorf tube and centrifuged for 10 s at 10,000 rpm. After solidification, the agar containing the islet pellet was removed from the tube, trimmed, refixed, and processed in an automated Shandon Citadel 100 machine before paraffin embedding and preparation of 5-μm sections.
For TUNEL, the DeadEnd Fluorometric TUNEL System kit (Promega, Madison, WI) was used according to the manufacturer’s instructions, but a permeabilization step was included (5 min in a 1% Triton X-100 PBS solution). β-Cells were visualized by insulin staining using guinea pig anti-insulin antibody (Zymed, San Francisco, CA) and Cy3-conjugated anti-guinea pig IgG (1:500; Jackson ImmunoResearch, Westgrove, PA). The Vectashield with 4,6-diamidino-2-phenylindole mounting solution (Vector, Burlingame, CA) was used for visualization of nuclei. β-Cell proliferation was assessed by staining 10-μm pancreatic sections for Ki67 (1:200; Abcam).
Whole-pancreas insulin content
Sixteen-week-old male HcB-19 (n = 5) and C3H control mice (n = 4) were killed after a 4-h fast, and their whole-pancreas insulin content (μg/pancreas) was assessed by acid-ethanol extraction, and insulin enzyme-linked immunosorbent assay was used as a measure of β-cell mass.
Insulin secretion
Insulin secretion and glucose-induced insulin secretion of isolated C3H and HcB-19 islets were assessed after an overnight incubation at 5 mmol/l glucose as described previously (30,31). All experiments were performed in triplicates using three islets per tube.
Cytochrome C release
To obtain cytosolic and mitochondrial cell fractions, cells from two T-75 flasks per sample were trypsinized and collected by centrifugation at 1,000g for 2 min. Cells were then washed with PBS and the cell pellet resuspended in 200 μl of buffer A (20 mmol/l Hepes-KOH, pH 7.5; 10 mmol/l KCl; 1.5 mmol/l MgCl2; 1 mmol/l EGTA; 1 mmol/l dithiothreitol; 250 mmol/l sucrose; 100 mmol/l phenylmethylsulfonyl fluoride; 1 μg/ml pepstatin A; 2 μg/ml leupeptin; and 2 μg/ml aprotinin). The homogenate was then centrifuged at 1,000g for 10 min at 4°C, and the supernatant containing the cytosolic and mitochondrial fractions was centrifuged again for 15 min at 10,000g at 4°C. The resulting supernatant contained the cytosolic fractions, and the pellet contained the mitochondrial fractions. The cytosolic fractions were boiled in SDS sample buffer and frozen at −80°C until further analysis. Pellets containing mitochondria were treated with buffer B (1 × PBS; 1% Nonidet P-40; 0.5% sodium deoxycholate; 0.1% SDS; 250 mmol/l sucrose; 20 mmol/l Tris HCl, pH 7.4; 1 mmol/l dithiothreitol; and protease inhibitors) and were incubated on ice for 20 min. The lysate was centrifuged at 10,000g for 30 min at 4°C. The pellet was solubilized in buffer C (300 mmol/l sucrose; 1 mmol/l EGTA; 20 mmol/l MOPS, pH 7.4; 0.5 mg/ml BSA; and protease inhibitors), and the resulting mitochondria-enriched fractions were boiled in SDS sample buffer and stored at −80°C until further use. Cytochrome C protein levels in the mitochondrial and cytosolic fractions were determined by Western blotting using a mouse monoclonal cytochrome C antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA) and anti-mouse IgG (1:2,000; Amersham, Piscataway, NJ).
Quantitative real-time RT-PCR
RNA was extracted using Trizol, converted to cDNA with the First Strand cDNA synthesis kit (Roche), and analyzed on a Prism 7000 Sequence Detection System (Applied Biosystems). BiP was amplified using the forward primer 5′-ACGTCCAACCCGGAGAACA-3′ and the reverse primer 5′-TTCCAAGTGCGTCCGATGA-3′ and ChOP with 5′-TGGCACAGCTTGCTGAAGAG-3′ and 5′-TCAGGCGCTCGATTTCCT-3′, respectively. Total TXNIP was measured using primers recognizing both human and rat TXNIP (forward: 5′-ACAGAAAAGGATTCTGTGAAGGTGAT-3′, reverse: 5′-GCCATTGGCAAGGTAAGTGTG-3′). All samples were corrected for the 18S ribosomal subunit (Applied Biosystems) run as an internal standard.
Statistical analysis
To calculate the significance of a difference between two means, we used two-sided Student’s t tests. A P value of <0.05 was considered statistically significant.
RESULTS
TXNIP protein levels and cleaved caspase-3 expression are elevated in response to glucose in INS-1 β-cells
We previously found that glucose induces TXNIP transcription. To now determine whether the observed glucose effects are also translated into differential expression of TXNIP at the protein level, we performed Western blot experiments using INS-1 β-cells incubated at low (5 mmol/l) or high (25 mmol/l) glucose for 24 h. We observed that incubation at high versus low glucose induced TXNIP protein expression 18-fold (Fig. 1). Interestingly, we found that these elevated TXNIP protein levels were accompanied by a 12-fold increase in cleaved (activated) caspase-3 (Fig. 1C), suggesting increased apoptosis.
FIG. 1.
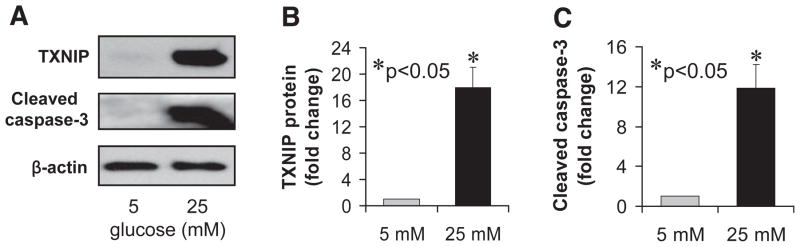
Glucose effects on TXNIP protein levels and apoptosis in INS-1 β-cells. INS-1 cells were incubated at low (5 mmol/l) or high (25 mmol/l) glucose for 24 h and analyzed by immunoblotting for the expression of TXNIP and cleaved caspase-3. Bars represent mean fold change in protein levels corrected for β-actin ± SE (n = 3 independent experiments).
These findings are consistent with our previous demonstration of glucose increasing β-cell TXNIP mRNA levels (15,32) and the observation that TXNIP overexpression promotes β-cell apoptosis (15,30) as well as with the proapoptotic properties of TXNIP described in extrapancreatic tissues (16,23). They also suggest that TXNIP-mediated β-cell apoptosis does not require constitutive overexpression of TXNIP but rather that the glucose-induced increase in endogenous TXNIP is sufficient for this effect. This raised the possibility that TXNIP might be involved in the pathophysiology of type 2 diabetes and the associated β-cell loss.
Diabetic mice demonstrate increased levels of TXNIP and cleaved caspase-3 in their pancreatic islets
To test whether diabetes alters in vivo TXNIP expression in pancreatic islets, we analyzed islets isolated from obese, insulin-resistant, and diabetic BTBR.ob mice harboring the leptinob mutation as a model of type 2 diabetes (24 –26). These analyses revealed that TXNIP protein levels were elevated eightfold in islets of obese diabetic BTBR.ob mice compared with lean normoglycemic controls (BTBR.lean), and this increase in TXNIP was again accompanied by an equal rise in cleaved caspase-3 levels (Fig. 2). These data are in agreement with the elevated TXNIP mRNA expression observed in islets of nonobese, insulin-resistant C57BL/6.azip mice as another model of diabetes (15) and further support an in vivo role of TXNIP in diabetes and β-cell death.
FIG. 2.

Expression of TXNIP and cleaved caspase-3 in islets of diabetic mice. Body weight (A) and blood glucose (B) in diabetic BTBR.ob and normoglycemic BTBR.lean mice are shown for comparison. Islets of 8-week-old BTBR.ob and control BTBR.lean mice were isolated and analyzed by immunoblotting for TXNIP protein levels (C) and cleaved caspase-3 (D). Three independent experiments were performed, and bars represent mean fold change ± SE in protein levels corrected for β-actin.
However, we also wanted to assess the causal relationship between glucose-induced TXNIP expression and β-cell apoptosis and determine whether TXNIP is critical for this effect. To this end, we studied TXNIP-deficient HcB-19 mice. HcB-19 mice are an inbred congenic C3H mouse strain (33,34) that harbors a spontaneous inactivating nonsense mutation in exon 2 at codon 97, resulting in dramatically reduced TXNIP mRNA and protein levels (29).
Glucose induces TXNIP expression and apoptosis in primary islets
To determine the role of TXNIP in glucose toxicity–mediated β-cell apoptosis, we first analyzed wild-type islets with normal TXNIP expression isolated from control C3H mice. In analogy to the INS-1 cell experiments, we incubated isolated islets at low or high glucose for 24 h and then assessed them for the expression of TXNIP, cleaved caspase-3, and by TUNEL. Again, we observed that incubation at high glucose resulted in elevated TXNIP protein levels and increased apoptosis, as measured by the sevenfold increase in cleaved caspase-3 (Fig. 3) and the ninefold higher percentage of TUNEL-positive β-cells (Fig. 4) compared with C3H islets incubated at low glucose. We also obtained very similar findings in islets isolated from C57BL/6 mice as well as in human islets (Fig. 5). In addition, incubation of human islets at 25 mmol/l glucose for 24 h also led to an ~2-fold increase in TXNIP protein levels and a >10-fold increase in cleaved caspase-3.
FIG. 3.

Effects of high glucose exposure on caspase-3 activation in TXNIP-deficient HcB-19 and C3H control islets. Isolated primary islets of control C3H and TXNIP-deficient HcB-19 mice were incubated at low (5 mmol/l) or high (25 mmol/l) glucose for 24 h and assessed for TXNIP expression and apoptosis. A: Representative immunoblot. B: Quantification of TXNIP protein levels in C3H and HcB-19 islets. (As expected, no TXNIP protein was detected in HcB-19 islets.) *P < 0.005. C: Quantification of cleaved caspase-3 in C3H and HcB-19 islets. Bars represent mean fold change ± SE in protein levels corrected for β-actin (n = 3 independent experiments). *P < 0.05 high vs. low glucose.
 , 5 mmol/l glucose; ■, 25 mmol/l glucose.
, 5 mmol/l glucose; ■, 25 mmol/l glucose.
FIG. 4.

Protection against glucose toxicity–induced β-cell apoptosis in TXNIP-deficient HcB-19 islets. Isolated primary islets of control C3H mice (A and B) and TXNIP-deficient HcB-19 mice (C and D) were again incubated at low (5 mmol/l) or high (25 mmol/l) glucose for 24 h and then analyzed by TUNEL. Representative pictures (×40) are shown, and white arrows point at bright appearing TUNEL-positive nuclei. E: For quantification >500 nuclei and at least 10 different islets were analyzed per group and condition and the percentage of TUNEL-positive β-cells per islet was calculated. Bars represent means ± SE. *P < 0.001 high vs. low glucose.
, 5 mmol/l glucose; ■, 25 mmol/l glucose.
FIG. 5.

Glucose toxicity in C57BL/6 and human islets. Islets of wild-type C57BL/6 mice (A) or isolated human islets (B) were incubated at low (5 mmol/l) or high (25 mmol/l) glucose for 24 h and then analyzed by TUNEL. At least seven different islets were analyzed per group and condition and >1,000 mouse or human β-cell nuclei were evaluated. Bars represent means ± SE.
TXNIP deficiency protects against glucose toxicity–induced β-cell apoptosis
In contrast, parallel experiments using isolated islets of TXNIP-deficient HcB-19 mice revealed no increase in TXNIP expression (as expected) but interestingly also failed to demonstrate any increase in cleaved caspase-3 (Fig. 3) or TUNEL-positive β-cells in response to glucose (Fig. 4). These findings strongly suggest that TXNIP expression is critical for the increase in β-cell apoptosis observed in response to high glucose exposure and provide direct evidence for the protective role of TXNIP deficiency against β-cell glucose toxicity.
To address the issue of whether other factors in the mutant HcB-19 islets may have contributed to the observed protection, we repeated the experiments using isolated islets of mice with β-cell–specific deletion of TXNIP created by the Cre-loxP system and of control lox/lox littermates. While incubation at high glucose led again to a significant increase in TUNEL-positive β-cells in the control islets (P = 0.001) similar to C3H, C57BL/6, and human islets, this effect was completely blunted in islets from β-cell–specific TXNIP knockout mice. These findings confirm the results obtained with the HcB-19 islets and demonstrate that it is specifically the lack of TXNIP that protects β-cells against glucose toxicity–induced apoptosis.
To further investigate the contribution of TXNIP to β-cell loss in vivo, we compared the β-cell mass in TXNIP-deficient HcB-19 and control C3H mice. Interestingly, we found that functional β-cell mass was more than twofold higher in the TXNIP-deficient mice as indicated by a whole-pancreas insulin content of 71.2 ± 1.8 μg/pancreas in HcB-19 mice compared with 35.0 ± 3.9 μg/ pancreas in control C3H mice (P = 0.001). At the same time, TXNIP deficiency did not seem to affect β-cell proliferation, as no difference in the number of Ki67-positive β-cells was observed between HcB-19 and C3H pancreata in these initial experiments (data not shown). Together with our findings of TXNIP-induced β-cell apoptosis, this suggests that the net increase in β-cell mass observed in response to TXNIP deficiency was primarily due to a decrease in β-cell apoptosis.
TXNIP deficiency does not alter islet insulin secretion
To assess whether TXNIP deficiency also affects β-cell function, we performed insulin secretion studies using isolated islets from HcB-19 and C3H mice. The results of these experiments demonstrated that basal and glucose-stimulated insulin secretion remained unchanged in TXNIP-deficient islets, suggesting that TXNIP does not interfere with normal islet function (Table 1).
TABLE 1.
Insulin secretion from C3H and HcB-19 islets
| Insulin
|
|||
|---|---|---|---|
| C3H | HcB-19 | P value | |
| Low glucose (1.7 mmol/l)* | 1.99 ± 0.18 | 2.49 ± 0.41 | 0.3 (NS) |
| High glucose (16.7 mmol/l)* | 5.89 ± 0.68 | 5.72 ± 0.75 | 0.9 (NS) |
| Glucose-stimulated insulin secretion (fold-change) | 2.96 ± 0.34 | 2.30 ± 0.30 | 0.2 (NS) |
Data are means ± SE. Numbers represent means of five independent experiments performed in triplicate.
Nanograms per milliliter per 3 islets.
TXNIP induces the intrinsic mitochondrial pathway of apoptosis
We demonstrated that TXNIP deficiency protects β-cells against glucose-induced cell death and that induction of TXNIP promotes β-cell apoptosis, but the mechanisms involved have remained unknown. Major mechanisms leading to apoptosis consist of the extrinsic pathway activated by extracellular signals and mediated by death receptors and the intrinsic pathways mediated by mitochondrial damage and/or endoplasmic reticulum (ER) stress. As the extrinsic pathway is unlikely to be involved in TXNIP-mediated apoptosis, we focused on the mitochondrial and the ER stress pathways, both of which have been implicated in pancreatic β-cell death (35).
The ER stress–mediated pathway involves depletion of ER calcium stores, accumulation of unfolded proteins, and a stress response including upregulation of ER chaperones such as BiP. Severe or persistent ER stress further activates the apoptosis cascade, which includes upregulation of transcription factors such as ChOP and activation of caspase-12.
In the case of mitochondria-mediated apoptosis, the apoptotic stimulus leads to disequilibrium of proapoptotic Bax and antiapoptotic Bcl-2 and disruption of the mitochondrial membrane potential resulting in release of cytochrome C into the cytosol. This represents a hallmark of this pathway and activates caspase-9.
Both pathways converge at the level of caspase-3 activation, which we consistently observed in our TXNIP studies. To determine which of these pathways is mediating the TXNIP effects, we now used stably transfected INS-TXNIP or INS-LacZ as a control and measured markers of mitochondrial or ER stress–mediated apoptosis.
In control cells, cytochrome C was practically exclusively localized to the mitochondria as expected, whereas INS-TXNIP cells showed a dramatic release of cytochrome C into the cytosolic fractions (Fig. 6A and B). In contrast, measurements of ER stress markers BiP and ChOP failed to demonstrate any increase in the INS-TXNIP cells (Fig. 6C and D) despite proven TXNIP overexpression in the same samples (Fig. 6E). Together, these results suggest that TXNIP activates primarily the mitochondrial pathway of apoptosis. This notion is further supported by our previous observation of increased Bax–to–Bcl-2 ratio and activated caspase-9 in response to TXNIP (15).
FIG. 6.

TXNIP effects on mitochondrial damage and ER stress. A: Immunoblot of cytochrome C in mitochondrial (mito) and cytosolic (cyto) cell fractions obtained from INS-1 β-cells overexpressing TXNIP (INS-TXNIP) and control cells (INS-LacZ). A total of 25 μg of protein were loaded per lane, and β-actin is shown as a loading control. One representative of seven independent experiments is shown. B: Quantification of cytochrome C in the different cell fractions. Bars represent mean fold change ± SE of seven independent experiments corrected for β-actin. Expression of BiP (C), ChOP (D), and TXNIP (E) as measured by quantitative real-time RT-PCR in INS-TXNIP and INS-LacZ cells. Bars represent means ± SE of three independent experiments corrected for 18S.
DISCUSSION
The results of the present study reveal for the first time that a protein (TXNIP) is essential for glucose toxicity–induced β-cell apoptosis. While the effects of glucose toxicity on β-cell dysfunction and death have been described extensively (1–14), the molecular mechanisms involved have remained largely elusive. In regard to β-cell dysfunction, small heterodimer partner has recently been identified as a factor inhibiting insulin gene transcription in response to glucose toxicity in INS-1 cells (36) but was not reported to have any effects on β-cell death.
In the present study, we have identified TXNIP as a critical factor mediating glucose toxicity–induced β-cell death and have elucidated the pathways by which TXNIP induces β-cell apoptosis. We found that TXNIP overexpression leads to activation of the intrinsic mitochondrial pathway of apoptosis, while ER stress–mediated processes of cell death remained unaffected by TXNIP (Fig. 6). These findings not only shed new light on the role of TXNIP in β-cell biology and the mechanisms involved but also identify TXNIP as a potential target to interfere with the deleterious effects of glucose toxicity. Such modification could help prevent β-cell apoptosis and the gradual β-cell loss that characterizes type 2 diabetes and ultimately makes type 2 diabetic patients require daily insulin injections. In fact, we found that TXNIP reduction mediates the antiapoptotic effects of exenatide, an antidiabetes drug recently approved for type 2 diabetes (37). The idea of TXNIP deficiency potentially enhancing β-cell mass is further supported by the phenotype of the TXNIP-deficient HcB-19 mice that, aside from elevated plasma triglycerides and ketone bodies, includes hyperinsulinemia and hypoglycemia (27,28). Recent work (38) suggests that lack of TXNIP may cause impaired hepatic glucose production, which could lead to hypoglycemia. However, since we have found that TXNIP induces β-cell apoptosis, it is also possible that lack of TXNIP may slow the natural process of β-cell death and turnover, lead to accumulation of β-cells, and result in increased basal insulin production, which would contribute to the observed hypoglycemia. Indeed, our initial results now demonstrate significantly elevated whole-pancreas insulin content in TXNIP-deficient HcB-19 mice compared with C3H controls, indicating increased pancreatic β-cell mass. These findings strongly support the importance of TXNIP not only as a factor conferring glucose toxicity–induced β-cell apoptosis but also as a novel protein controlling overall pancreatic β-cell loss in vivo.
In the context of diabetes treatment, lowering TXNIP expression may also have beneficial effects in tissues other than pancreatic β-cells, where it again may protect against glucose toxicity. TXNIP expression was found to be increased in human aortic smooth muscle cells in response to incubation at high glucose, and diabetic rats had elevated vascular TXNIP levels (39). Moreover, overexpression of TXNIP induced apoptosis in cardiomyocytes (23) and proliferation of smooth muscle cells (21), suggesting that controlling TXNIP expression might help reduce cardiovascular risk and morbidity in patients with diabetes. In addition, a recent human study (40) demonstrated that TXNIP regulates glucose uptake in adipocytes and skeletal muscle and suggested that TXNIP might play a role in the pathogenesis of type 2 diabetes by altering glucose homeostasis. Decreased TXNIP expression might therefore also enhance glucose uptake and clearance in peripheral tissues and thereby further improve glucose control.
In in vitro experiments, transfection and constitutive TXNIP overexpression has been shown to reduce thioredoxin activity (23), raising the possibility that the opposite might also be true and that the effects of TXNIP deficiency might be mediated by activation of thioredoxin. However, more recent studies using TXNIP-deficient HcB-19 mice revealed no difference in thioredoxin protein levels and no increase in thioredoxin activity compared with control mice (28). This suggests that other thioredoxin-independent pathways might be involved in the protective effects of TXNIP deficiency observed in our experiments using HcB-19 islets.
Taken together, the results of the present study have revealed TXNIP as a critical mediator of glucose toxicity–induced β-cell apoptosis. Moreover, they have demonstrated that β-cell TXNIP protein levels are dramatically induced in response to glucose and that TXNIP expression is elevated in islets of different mouse models of diabetes. This suggests that TXNIP may play an important role in diabetes progression and the associated decline in β-cell mass. Controlling TXNIP expression may therefore represent a novel approach to protect pancreatic β-cells from progressive destruction and to preserve a sufficient amount of insulin-producing β-cell mass in patients with type 2 diabetes.
Acknowledgments
This work was supported by American Diabetes Association Grants 7-03-JF-37 and 7-07-CD-22 and Juvenile Diabetes Research Foundation Grant 1-2007-790 to A.S.
We thank Anna Szabo for technical assistance and Dr. Susan Bonner-Weir for helpful suggestions with regard to the islet-embedding technique.
Glossary
- ER
endoplasmic reticulum
- INS-LacZ
INS-1 cells overexpressing LacZ
- INS-TXNIP
INS-1 cells with constitutive TXNIP overexpression
- HBSS
Hanks’ balanced salt solution
- TUNEL
transferase-mediated dUTP nick-end labeling
- TXNIP
thioredoxin-interacting protein
References
- 1.Lawrence MC, McGlynn K, Park BH, Cobb MH. ERK1/2-dependent activation of transcription factors required for acute and chronic effects of glucose on the insulin gene promoter. J Biol Chem. 2005;280:26751–26759. doi: 10.1074/jbc.M503158200. [DOI] [PubMed] [Google Scholar]
- 2.Khoo S, Gibson TB, Arnette D, Lawrence M, January B, McGlynn K, Vanderbilt CA, Griffen SC, German MS, Cobb MH. MAP Kinases and their roles in pancreatic beta-cells. Cell Biochem Biophys. 2004;40:191–200. doi: 10.1385/cbb:40:3:191. [DOI] [PubMed] [Google Scholar]
- 3.Kaiser N, Leibowitz G, Nesher R. Glucotoxicity and beta-cell failure in type 2 diabetes mellitus. J Pediatr Endocrinol Metab. 2003;16:5–22. doi: 10.1515/jpem.2003.16.1.5. [DOI] [PubMed] [Google Scholar]
- 4.Poitout V, Robertson RP. Minireview: secondary beta-cell failure in type 2 diabetes: a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143:339–342. doi: 10.1210/endo.143.2.8623. [DOI] [PubMed] [Google Scholar]
- 5.Rhodes CJ. Type 2 diabetes-a matter of beta-cell life and death? Science. 2005;307:380–384. doi: 10.1126/science.1104345. [DOI] [PubMed] [Google Scholar]
- 6.Wang X, Li H, De Leo D, Guo W, Koshkin V, Fantus IG, Giacca A, Chan CB, Der S, Wheeler MB. Gene and protein kinase expression profiling of reactive oxygen species–associated lipotoxicity in the pancreatic β-cell line MIN6. Diabetes. 2004;53:129–140. doi: 10.2337/diabetes.53.1.129. [DOI] [PubMed] [Google Scholar]
- 7.Wu L, Nicholson W, Knobel SM, Steffner RJ, May JM, Piston DW, Powers AC. Oxidative stress is a mediator of glucose toxicity in insulin-secreting pancreatic islet cell lines. J Biol Chem. 2004;279:12126–12134. doi: 10.1074/jbc.M307097200. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka Y, Tran PO, Harmon J, Robertson RP. A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci U S A. 2002;99:12363–12368. doi: 10.1073/pnas.192445199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci U S A. 1999;96:10857–10862. doi: 10.1073/pnas.96.19.10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robertson RP, Harmon J, Tran PO, Tanaka Y, Takahashi H. Glucose toxicity in β-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes. 2003;52:581–587. doi: 10.2337/diabetes.52.3.581. [DOI] [PubMed] [Google Scholar]
- 11.Kajimoto Y, Kaneto H. Role of oxidative stress in pancreatic beta-cell dysfunction. Ann N Y Acad Sci. 2004;1011:168–176. doi: 10.1007/978-3-662-41088-2_17. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi H, Tran PO, LeRoy E, Harmon JS, Tanaka Y, Robertson RP. D-glyceraldehyde causes production of intracellular peroxide in pancreatic islets, oxidative stress, and defective beta cell function via non-mitochondrial pathways. J Biol Chem. 2004;279:37316–37323. doi: 10.1074/jbc.M403070200. [DOI] [PubMed] [Google Scholar]
- 13.Kaneto H, Kajimoto Y, Miyagawa J, Matsuoka T, Fujitani Y, Umayahara Y, Hanafusa T, Matsuzawa Y, Yamasaki Y, Hori M. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic β-cells against glucose toxicity. Diabetes. 1999;48:2398–2406. doi: 10.2337/diabetes.48.12.2398. [DOI] [PubMed] [Google Scholar]
- 14.Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, Torri S, Pollera M, Boggi U, Mosca F, Del Prato S, Marchetti P. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes. 2005;54:727–735. doi: 10.2337/diabetes.54.3.727. [DOI] [PubMed] [Google Scholar]
- 15.Minn AH, Hafele C, Shalev A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology. 2005;146:2397–2405. doi: 10.1210/en.2004-1378. [DOI] [PubMed] [Google Scholar]
- 16.Junn E, Han SH, Im JY, Yang Y, Cho EW, Um HD, Kim DK, Lee KW, Han PL, Rhee SG, Choi I. Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J Immunol. 2000;164:6287–6295. doi: 10.4049/jimmunol.164.12.6287. [DOI] [PubMed] [Google Scholar]
- 17.Nishiyama A, Masutani H, Nakamura H, Nishinaka Y, Yodoi J. Redox regulation by thioredoxin and thioredoxin-binding proteins. IUBMB Life. 2001;52:29–33. doi: 10.1080/15216540252774739. [DOI] [PubMed] [Google Scholar]
- 18.Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, Takagi Y, Sono H, Gon Y, Yodoi J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem. 1999;274:21645–21650. doi: 10.1074/jbc.274.31.21645. [DOI] [PubMed] [Google Scholar]
- 19.Yamanaka H, Maehira F, Oshiro M, Asato T, Yanagawa Y, Takei H, Nakashima Y. A possible interaction of thioredoxin with VDUP1 in HeLa cells detected in a yeast two-hybrid system. Biochem Biophys Res Commun. 2000;271:796– 800. doi: 10.1006/bbrc.2000.2699. [DOI] [PubMed] [Google Scholar]
- 20.Patwari P, Higgins LJ, Chutkow WA, Yoshioka J, Lee RT. The interaction of thioredoxin with Txnip: evidence for formation of a mixed disulfide by disulfide exchange. J Biol Chem. 2006;281:21884–91. doi: 10.1074/jbc.M600427200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schulze PC, De Keulenaer GW, Yoshioka J, Kassik KA, Lee RT. Vitamin D3-upregulated protein-1 (VDUP-1) regulates redox-dependent vascular smooth muscle cell proliferation through interaction with thioredoxin. Circ Res. 2002;91:689– 695. doi: 10.1161/01.res.0000037982.55074.f6. [DOI] [PubMed] [Google Scholar]
- 22.Han SH, Jeon JH, Ju HR, Jung U, Kim KY, Yoo HS, Lee YH, Song KS, Hwang HM, Na YS, Yang Y, Lee KN, Choi I. VDUP1 upregulated by TGF-beta1 and 1,25-dihydorxyvitamin D3 inhibits tumor cell growth by blocking cell-cycle progression. Oncogene. 2003;22:4035– 4046. doi: 10.1038/sj.onc.1206610. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, De Keulenaer GW, Lee RT. Vitamin D(3)-up-regulated protein-1 is a stress-responsive gene that regulates cardiomyocyte viability through interaction with thioredoxin. J Biol Chem. 2002;277:26496–26500. doi: 10.1074/jbc.M202133200. [DOI] [PubMed] [Google Scholar]
- 24.Stoehr JP, Nadler ST, Schueler KL, Rabaglia ME, Yandell BS, Metz SA, Attie AD. Genetic obesity unmasks nonlinear interactions between murine type 2 diabetes susceptibility loci. Diabetes. 2000;49:1946–1954. doi: 10.2337/diabetes.49.11.1946. [DOI] [PubMed] [Google Scholar]
- 25.Lan H, Rabaglia ME, Stoehr JP, Nadler ST, Schueler KL, Zou F, Yandell BS, Attie AD. Gene expression profiles of nondiabetic and diabetic obese mice suggest a role of hepatic lipogenic capacity in diabetes susceptibility. Diabetes. 2003;52:688–700. doi: 10.2337/diabetes.52.3.688. [DOI] [PubMed] [Google Scholar]
- 26.Minn AH, Lan H, Rabaglia ME, Harlan DM, Peculis BA, Attie AD, Shalev A. Increased insulin translation from an insulin splice-variant overexpressed in diabetes, obesity, and insulin resistance. Mol Endocrinol. 2005;19:794– 803. doi: 10.1210/me.2004-0119. [DOI] [PubMed] [Google Scholar]
- 27.Hui TY, Sheth SS, Diffley JM, Potter DW, Lusis AJ, Attie AD, Davis RA. Mice lacking thioredoxin-interacting protein provide evidence linking cellular redox state to appropriate response to nutritional signals. J Biol Chem. 2004;279:24387–24393. doi: 10.1074/jbc.M401280200. [DOI] [PubMed] [Google Scholar]
- 28.Sheth SS, Castellani LW, Chari S, Wagg C, Thipphavong CK, Bodnar JS, Tontonoz P, Attie AD, Lopaschuk GD, Lusis AJ. Thioredoxin-interacting protein deficiency disrupts the fasting-feeding metabolic transition. J Lipid Res. 2005;46:123–134. doi: 10.1194/jlr.M400341-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Bodnar JS, Chatterjee A, Castellani LW, Ross DA, Ohmen J, Cavalcoli J, Wu C, Dains KM, Catanese J, Chu M, Sheth SS, Charugundla K, Demant P, West DB, de Jong P, Lusis AJ. Positional cloning of the combined hyperlipidemia gene Hyplip1. Nat Genet. 2002;30:110–116. doi: 10.1038/ng811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minn AH, Pise-Masison CA, Radonovich M, Brady JN, Wang P, Kendziorski C, Shalev A. Gene expression profiling in INS-1 cells overexpressing thioredoxin-interacting protein. Biochem Biophys Res Commun. 2005;336:770–778. doi: 10.1016/j.bbrc.2005.08.161. [DOI] [PubMed] [Google Scholar]
- 31.Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1– derived cell lines with robust ATP-sensitive K+ channel– dependent and –independent glucose-stimulated insulin secretion. Diabetes. 2000;49:424– 430. doi: 10.2337/diabetes.49.3.424. [DOI] [PubMed] [Google Scholar]
- 32.Shalev A, Pise-Masison CA, Radonovich M, Hoffmann SC, Hirshberg B, Brady JN, Harlan DM. Oligonucleotide microarray analysis of intact human pancreatic islets: identification of glucose-responsive genes and a highly regulated TGFbeta signaling pathway. Endocrinology. 2002;143:3695–3698. doi: 10.1210/en.2002-220564. [DOI] [PubMed] [Google Scholar]
- 33.Stassen AP, Groot PC, Eppig JT, Demant P. Genetic composition of the recombinant congenic strains. Mamm Genome. 1996;7:55–58. doi: 10.1007/s003359900013. [DOI] [PubMed] [Google Scholar]
- 34.Groot PC, Moen CJ, Dietrich W, Stoye JP, Lander ES, Demant P. The recombinant congenic strains for analysis of multigenic traits: genetic composition. Faseb J. 1992;6:2826–2835. doi: 10.1096/fasebj.6.10.1634045. [DOI] [PubMed] [Google Scholar]
- 35.Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54 (Suppl 2):S97–S107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 36.Park KG, Lee KM, Seo HY, Suh JH, Kim HS, Wang L, Won KC, Lee HW, Park JY, Lee KU, Kim JG, Kim BW, Choi HS, Lee IK. Glucotoxicity in the INS-1 rat insulinoma cell line is mediated by the orphan nuclear receptor small heterodimer partner. Diabetes. 2007;56:431– 437. doi: 10.2337/db06-0753. [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Couto FM, Minn AH, Shalev A. Exenatide inhibits beta-cell apoptosis by decreasing thioredoxin-interacting protein. Biochem Biophys Res Commun. 2006;346:1067–1074. doi: 10.1016/j.bbrc.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 38.Chutkow WA, Patwari P, Yoshioka J, Lee RT. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J Biol Chem. 2008;283:2397– 406. doi: 10.1074/jbc.M708169200. [DOI] [PubMed] [Google Scholar]
- 39.Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, Lee RT. Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J Biol Chem. 2004;279:30369–30374. doi: 10.1074/jbc.M400549200. [DOI] [PubMed] [Google Scholar]
- 40.Parikh H, Carlsson E, Chutkow WA, Johansson LE, Storgaard H, Poulsen P, Saxena R, Ladd C, Schulze PC, Mazzini MJ, Jensen CB, Krook A, Bjornholm M, Tornqvist H, Zierath JR, Ridderstrale M, Altshuler D, Lee RT, Vaag A, Groop LC, Mootha VK. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007;4:e158. doi: 10.1371/journal.pmed.0040158. [DOI] [PMC free article] [PubMed] [Google Scholar]