

Abstract
PURPOSE
To characterize the interactions of Non-small Cell Lung Cancer (NSCLC) tumors with the immune system at the level of mRNA and microRNA (miRNA) expression and to define expression signatures that characterize the presence of a malignant tumor vs. a non-malignant nodule.
EXPERIMENTAL DESIGN
We have examined the changes of both mRNA and miRNA expression levels in peripheral blood mononuclear cells (PBMC) between paired samples collected from NSCLC patients before and after tumor removal using Illumina gene expression arrays.
RESULTS
We found that malignant tumor removal significantly changes expression of more than 3,000 protein-coding genes, especially genes in pathways associated with suppression of the innate immune response, including NK cell signaling and apoptosis-associated ceramide signaling. Binding sites for the ETS-domain transcription factors ELK1, ELK4 and SPI1 were enriched in promoter regions of genes upregulated in the presence of a tumor. Additional important regulators included five miRNAs expressed at significantly higher levels before tumor removal. Repressed protein-coding targets of those miRNAs included many transcription factors, several involved in immunologically important pathways. While there was a significant overlap in the effects of malignant tumors and benign lung nodules on PBMC gene expression, we identified one gene panel which indicates a tumor or nodule presence and a second panel that can distinguish malignant from non-malignant nodules.
CONCLUSIONS
A tumor presence in the lung influences mRNA and miRNA expression in PBMC and this influence is reversed by tumor removal. These results suggest that PBMC gene expression signatures could be used for lung cancer diagnosis.
Keywords: lung cancer, pbmc, peripheral immune system
INTRODUCTION
Lung cancer is the most common cause of cancer mortality worldwide, accounting for 222,520 new cases and 157,300 cancer deaths in the United States alone in 2010 (1). The overall five-year survival for lung cancer is 16%, and prognosis is strongly correlated with the stage of disease at diagnosis (2). Only 15% of patients with lung cancer have localized disease at the time of diagnosis, leading to generally poor outcomes. Although chemotherapy and radiation therapy can be used, these modalities are primarily palliative producing only small increases in survival.
Understanding the role of the immune system in controlling tumor development, and harnessing its capabilities for target detection and cell killing in order to develop new human cancer treatments, are major challenges of substantial interest to researchers (3–5). Most proposed therapeutic approaches have been based on findings that the immune system can discriminate cancer cells from normal cells via tumor antigen recognition and secondary immune responses mediated by B- or T-cells associated with the tumors (4, 6–8). These studies focus primarily on antigen-specific T-cell responses (3–4, 6–8). Aspects of the innate immune response could potentially also be used for cancer immunotherapy but this approach has been similarly ineffective in part because tumors down regulate surface major histocompatibility complex (MHC) molecules targeted in a primary response. Further knowledge of the effect of a tumor on gene expression by the immune system is needed to effectively apply immune-therapeutic approaches.
Gene expression profiling provides a powerful approach to detecting the nature of interactions between the immune system and tumor components in patients, capturing information useful for cancer diagnosis, prognosis or new therapy development. While this approach cannot be applied to human studies of immune cell interactions that sample tumors, the peripheral immune system can provide continuous information on these interactions. For example, we recently showed that a diagnostic gene expression signature of 29 genes measured in PBMC was able to distinguish early-stage NSCLC patients from control patients that had non-malignant lung disease, with 86% accuracy. We also found that lung squamous cell carcinomas (LSCC) have a distinguishable and stronger PBMC-derived cancer signature than lung adenocarcinomas (LAC), indicating there is a specificity in the immune response to the presence of these 2 NSCLC tumor types (9). Detailed characterization of such responses might lead to new measures for diagnosis and prognosis and new therapeutic modalities.
To directly assess the effects of lung tumors on gene expression in the peripheral immune system, we initially measured the effect of surgical removal of lung tumors on the intensity of the 29-gene NSCLC signature in paired samples from 18 NSCLC patients that were taken before and then after tumor resection. We found that the tumor associated signature was significantly reduced post-surgery in a majority of the paired samples tested (14 of 18 pairs). This signature reduction was independent of patient demographics including post-surgery time of sampling. This observation supported our hypothesis that the 29 gene PBMC cancer signature was indeed a surrogate marker for the tumor presence. In the present study we have directly analyzed the nature of the global changes in PBMC gene expression that occur as a result of lung tumor removal. We have identified alterations in coding and non-coding gene expression profiles using paired pre- vs. post-tumor excision surgery PBMC samples. We identify changes associated with important immune functions that characterize the lung tumor presence and that distinguish malignant tumors from benign lung nodules. These studies show that in addition to the effects of a lung tumor on its associated immune cells, effects of the tumor in the lung can also be detected in the peripheral immune system. Our study addresses the changes in gene expression on this immune global-environment that occur with surgical removal of the tumor.
MATERIALS AND METHODS
Study populations
Study participants were recruited from the University Of Pennsylvania Medical Center (Penn) from 2003 through 2007 from the Penn Institutional Review Board’s approved lung cancer study. Patients were newly diagnosed with histopathologically confirmed NSCLC and no prior history of cancer or cancer treatment.
PBMC collection
Blood was collected from lung cancer patients pre-operatively and 1–5 months after surgery. Blood was drawn into two CPT tubes (BD), PBMC isolated within 90 min, washed in PBS, transferred into RNAlater® (Ambion) and stored at 4° C overnight before transfer to −80° C.
Microarrays for mRNA and miRNA
RNA purification was carried out at the Wistar Genomics facility as previously reported (9), controlled for quality using the Bioanalyzer (Agilent). For mRNA gene expression, 400ng of total RNA was amplified as recommended by Illumina and hybridized to the Illumina WG-6v2 human arrays. For miRNA expression, 500ng of RNA was analyzed on the Illumina MI-12v2 arrays. Expression levels were extracted with BeadStudio v.3.0 software. Arrays were quantile-normalized and filtered to remove non-informative probes as previously described (9). MRNA data is available in GEO as GSE13255 and miRNA as GSE27606.
Pairwise t-test
mRNA and miRNA expression data for 18 pre/post sample pairs (11 pairs for miRNA) was tested for differential expression using two-tail pairwise t-test, with significance set to <0.05. FDR was calculated according to the Storey procedure (10)
SVM-RFE
A list of ranked genes was obtained using linear kernel SVM-RFE (11) with 10-fold and 10x resampling cross-validation. Each cross-validation iteration started with the 1,000 genes most significant by t-test, and the number of genes was reduced by 10% at each feature elimination step. Final ranking of the genes was done using a Borda count procedure (12).
Quantitative Real-Time PCR
Validation of array results was carried out using the I TaqMan system (Applied Biosystems) as recommended, in an ABI 7900HT PCR System. Each sample was analyzed in duplicate and samples with CVs between replicates that were more than 0.5Δ Ct were repeated.
Ingenuity pathway analysis
Pathway and functional analysis was carried out with Ingenuity Pathways Analysis software (http://www.ingenuity.com/) using Ingenuity Core Analysis (IPA 6.0, Ingenuity® Systems), corrected for multiple comparisons by Benjamini-Hochberg, using P<0.05 as a significance threshold.
DAVID enrichment analysis
Enrichments of gene ontology (GO) terms, KEGG, and BIOCARTA pathways along with Swiss-Prot, INTERPRO, and SMART keywords in a gene list were done with DAVID software. Results were filtered to satisfy FDR <5% and fold enrichment >1.5 criteria.
miRNA target detection
Computationally predicted targets for a miRNA were derived from the union of results of target scanner software provided by Sanger (http://microrna.sanger.ac.uk) or Sloan-Kettering (http://www.microrna.org) databases (scans based on miRanda software), as well as Pictar (http://pictar.mdc-berlin.de) and TargetScan (http://www.targetscan.org). Overlap with gene expression data was done using Entrez Gene (http://www.ncbi.nlm.nih.gov/gene). A computationally predicted target gene was accepted only if it was significantly downregulated in pre-surgery samples as assessed by one-tailed, paired t-test with significance threshold of P<0.05.
Transcription factor motifs
The web interface of Pscan was used to find enriched TRANSFAC motifs in −450 to +50bp region from the transcriptional start site of genes from the SVM list. Bonferroni-correction was used to rank the results, using a significance threshold of P<0.05. For each motif, the top 500 scored genes were used to determine whether enrichment of up- vs. downregulated genes occurred in the list.
Additional statistical tests
The following tests were used to determine significance of results, using a threshold of P<0.05. Spearman correlation: correlation of ranks between two gene lists generated by SVM-RFE and paired t-test. Pearson correlation: correlation between expression ratios in pre-/post-surgery sample pairs vs. cancer/non-cancer samples. Fisher exact test: enrichment of an annotation term within a gene list; over-representation of up- or down-regulated genes in a list of cell type-specific genes from IRIS database. Hypergeometric test: overlap between significantly-expressed genes in pre-/post-surgery, cancer/non-cancer, and cancer/benign nodule patient sample comparisons.
RESULTS
Patient samples and demographics
PBMC from the 18 patients were collected in the University of Pennsylvania Medical Center. Clinical and demographic variables for those patients are shown in Table 1. Samples were collected with IRB approval, as previously described (9). Pre-surgery samples were collected on the day of resection just prior to surgery. Times of post-surgery sample collection ranged from 1–5 months. Most (n=10) patients were sampled 2 months after surgery and the others sampled at 1, 3, 4, or 5 months post-surgery. All samples were collected before any additional therapy was started. A more complete description of patients and staging information is shown in Supplementary Table 1.
Table 1.
Pre-post patient demographics.
| Category | N |
|---|---|
| Age (yrs) | |
| Average | 68 |
| Max | 87 |
| Min | 47 |
| Gender | |
| Male | 10 |
| Female | 8 |
| Race | |
| Caucasian | 17 |
| AA | 1 |
| Tobacco Use | |
| Former | 15 |
| Current | 1 |
| Never | 2 |
| Diagnosis | |
| LAC | 10 |
| LSCC | 6 |
| NSCLC | 2 |
| Cancer Stage | |
| Stage I/II | 11 |
| Stage III/IV | 7 |
| Sampling time from surgery | |
| 1–2 months | 12 |
| 3–5 months | 6 |
| COPD | |
| Yes | 7 |
| No | 11 |
AA=African American, LAC=adenocarcinoma, LSCC=lung squamous cell carcinoma, NSCLC=Unclassified non-small cell lung cancer, COPD=chronic obstructive pulmonary disease. All tumors, including Stage 3 and 4 were completely resected.
PBMC gene expression is altered by lung tumor removal
Expression data for the 18 pairs of pre-/post-surgery PBMC samples were generated using Illumina HGv2 bead arrays (Illumina, Inc.). The data was analyzed using the support vector machine with recursive feature elimination (SVM-RFE) and 10-fold cross-validation repeated 10 times as previously described (9). This approach generated a list of 3,271 genes ranked by ability to discriminate pre- from post-surgery samples. Sixty-seven percent of those genes were more highly expressed pre-surgery (i.e. in the presence of tumor). An expression heatmap of the 50 highest-ranked genes is shown in Figure 1. Expression values of only the four genes with the highest ranking were needed to separate pre and post surgery samples with 100% accuracy (sensitivity and specificity =100%) as determined by 10-fold cross validation. We also analyzed the data by paired t-test and identified 2,897 genes showing significant differential expression (p<0.05, false discovery rate [FDR] of 17%). When ranked by P-value, 81% of the t-test significant genes overlapped with the SVM-generated gene list, and their ranks correlated at rho=0.59 with the SVM ranking. The 3 genes highest-ranked by SVM-RFE were also those highest-ranked by paired t-test, and included CYP2R1 (a p-450 microsomal vitamin D hydroxylase), MYO5B (mitochondrial 3-oxoacyl-coenzyme A thiolase), and DGUOK (mitochondrial deoxyguanosine kinase). All three were expressed at higher levels pre- vs. post-surgery. Expression patterns of CYP2R1 and DGUOK were validated by quantitative real-time PCR on 10 of the pre-/post-surgery sample pairs (Supplementary Table 2). We used the gene list identified using SVM-RFE for all further analyses.
Figure 1. Hierarchical clustering of the 50 most-informative genes that distinguish pre- vs. post-surgery paired PBMC samples from 18 NSCLC patients.

Samples are labeled PRE- or POST-followed by the corresponding pair number (e.g. PRE-01, POST-01 indicate matched pair from patient 1). The number preceding each gene symbol is the rank assigned by SVM-RFE based on its importance in distinguishing pre- from post-surgery samples. FC = Fold-change (heatmap scale).
Because 2 of the 3 most-significant genes identified by both methods have mitochondrial functions, we determined whether mitochondria-associated genes were overrepresented in our significant gene list. We found a 1.6-fold enrichment of genes having mitochondrial annotations (P=2×10−11). Eighty percent (111/139) of the differentially expressed mitochondrial genes were upregulated pre- vs. post-surgery, suggesting the PBMC had increased metabolic activity when tumors were present. Enriched functional categories identified using DAVID (http://david.abcc.ncifcrf.gov/) software included additional metabolic processes and genes associated with protein processing (Supplementary Table 3).
Tumor removal alters expression of genes associated with immune functions
We used Ingenuity Core Analysis (Ingenuity Systems Inc.) to identify the functions and pathways significantly enriched in the SVM-generated gene list. Genes and pathways associated with specific immune functions are well-represented, including genes important for the generation of memory T-cells, T-cell accumulation, and the mobilization of NK cells. Among the 6 pathways showing significant enrichment, 5 are signaling pathways, the 3 most significant of which were NK cell, ceramide and ErbB signaling pathways (P=3×10−6, 7×10−3, and 2×10−2, respectively)(Figure 2).
Figure 2. Significantly-enriched canonical pathways identified by Ingenuity Pathway Analysis of the genes differentially regulated in pre- vs. post-surgery PBMC samples of lung cancer patients.
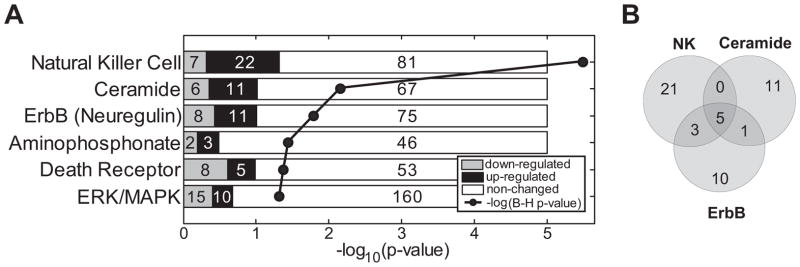
A. Significant pathways. Numbers in shaded bars indicate genes in the pathway expressed at significantly higher (black) or lower (grey) levels pre-surgery. Numbers in white bars indicate unchanged genes. All are signaling pathways, except for the aminophosphonate metabolic pathway. P-values were corrected for multiple comparisons using the Benjamini-Hochberg correction. B. Venn diagram shows the gene overlap between the 3 most-significant pathways (listed in Supplementary Table 4). Five genes (AKT3, KRAS, PIK3R1, PRKCZ, and RAF1) are shared by the three pathways.
Highly significant changes occurred in expression levels of 29 genes associated with NK cell signaling (Supplementary Table 4), including 22 that were expressed at higher levels in the pre-surgery samples. Seven of these 22 genes encode long-form members of the KIR receptor family, which are known to suppress NK signaling (13–14), suggesting that tumor presence induces an environment that suppresses NK function.
The ceramide signaling pathway was the second most-significantly enriched. Ceramide is a second messenger that affects apoptosis, metabolism, differentiation, and senescence (15–16), and is important for NK and NKT cell functions (17–19). As an inducer of apoptosis it can act through a variety of signaling molecules to induce cell death (20). Eleven of the 17 identified genes associated with ceramide signaling were upregulated in pre -surgery samples (Supplementary Table 4). These included ceramide kinase (CERK), which is specifically expressed in NK cells and T-cells (further supporting NK cell involvement), the sphingosine-1-phosphate receptor 5 (S1PR5), sphingomyelin phosphodiesterase 3 (SMPD3), and the pro-apoptotic gene BAD, also shared with the ErbB/Neuregulin pathway. Taken together, these results suggest a heightened susceptibility of one or more PBMC lineages to apoptosis in the presence of a NSCLC tumor.
The ERBb/neuregulin pathway includes 19 genes that in PBMC showed significantly altered expression (Supplementary Table 4), of which 11 were expressed at higher levels pre-surgery. Signal transduction through this pathway includes activation of the transcription factor ELK1 (21). We found binding sites for ELK1, along with those for the closely related ELK4, significantly overrepresented in the promoter regions of all genes upregulated pre-surgery in the presence of lung tumors (described below).
Enriched transcription factor binding sites in promoters regions of genes differentially expressed in PBMC after lung tumor removal
To assess whether common transcription factors might influence differential gene expression, we scanned the promoter regions of the 3,271 SVM-identified genes for enrichment in transcription factor binding motifs. We identified 13 transcription factor binding sites that were significantly overrepresented (Supplementary Table 5) within the promoters of genes that were upregulated in pre-surgery samples including sites for ETS-domain transcription factors ELK1, ELK4, and SPI1. The observed enrichment of these transcription factor binding sites in the ErbB/Neuregulin pathways was more generally found in the complete up-regulated pre-surgery gene list. The mRNAs encoding these 3 factors were not present in increased levels, suggesting an increased functional activation, likely by p38MAPK-mediated phosphorylation known to activate these factors. In this regard, we do find that the p38 mRNA expression level is significantly increased in pre-surgery samples (4, 22). ELK1 and ELK4 exhibit apparent, although not complete, redundancy in promoter binding (23) and each is implicated in important cellular growth processes (23). ELK1 has also been associated with activation of immunosuppressive TGF-β (24). The SpI1/PU.1 transcription factor plays a crucial role in myeloid cell development in vertebrates (25), and regulates basal transcription in macrophages (26). We found that 123 genes identified as having ELK1 binding sites, and which were also upregulated in pre-surgery samples, were identical to those found to bind to ELK1 and/or ELK4 in extensive ChIP/Chip and ChIP:PCR studies (23) providing further support for our identification of these transcription factors as having an important regulatory role in the phenomenon we have identified.
DAVID (27) gene enrichment analysis of the differentially expressed genes with ETS binding motifs identified 5 significant gene clusters having common functions including: 1) genes with mitochondrial functions were significantly enriched in (111/139) in our pre-surgery upregulated gene list. Recent studies by Boros et al (23) confirm the presence of ELK1 and ELK4 binding sites within a significant number of mitochondrial gene promoters ELK1 has also been identified as a potential regulator of the DAP3 gene, upregulated in our pre-surgery samples and suggested to play an important role in mitochondrial maintenance (28). Other significant clusters included: 2) genes associated with protein biosynthesis machinery, 3) RNA splicing genes, 4) intracellular organelle lumen genes and 5) genes associated with the regulation of translation. Genes associated with basal transcriptional machinery, such as RNA splicing, were also shown to be enriched for ELK1 binding sites by ChIP-seq (29) supporting the hypothesis that these 3 transcription factors are important to the pre-surgery transcription profiles we have identified.
Repression/activation of genes from specific lymphoid cell types
Since PBMC include diverse cell types, we asked whether changes in gene expression that occurred after tumor removal were primarily associated with a specific immune cell subset. For this purpose we utilized the IRIS (30) database. IRIS classifies markers as cell type-specific if they are expressed in a specific type or lineage of immune cells at levels ≥3-fold in other immune cells. We found that differentially expressed lymphoid-specific IRIS genes were mainly upregulated (58 of 71 genes, P=0.002) in pre- vs. post-surgery samples. Although the individual lymphoid categories of T-cell- and B-cell-specific genes did not show significant trends of increased or decreased expression, NK-cell-specific genes were exclusively upregulated (12 of 12 genes, P=0.006) in the pre-surgery samples, in agreement with our pathway studies. Myeloid-specific genes were significantly downregulated (39 of 65 genes, P=0.0001) in pre surgery samples. Genes specific for the individual myeloid lineages, monocytes, and dendritic cells showed no significant upward or downward expression trends. Neutrophils and other granulocytes are underrepresented in our purified PBMC samples and messages specific to those lineages are not found as significantly overrepresented (Table 2).
Table 2.
Summary of differentially expressed genes specific to different classes of immune cells as defined by the IRIS database.
| Cell type | Changed | Up | Down | Enrich-up | Enrich-down | P-value |
|---|---|---|---|---|---|---|
| T Cell | 7 | 3 | 4 | 0.67 | 1.58 | 0.3 |
| Neutrophil | 10 | 5 | 5 | 0.78 | 1.39 | 0.5 |
| NK Cell | 12 | 12 | 0 | 1.56 | 0 | 0.006 |
| Myeloid | 65 | 26 | 39 | 0.63 | 1.66 | 0.0001 |
| Multiple | 152 | 89 | 63 | 0.92 | 1.15 | 0.2 |
| Monocyte | 13 | 6 | 7 | 0.72 | 1.49 | 0.2 |
| Lymphoid | 71 | 58 | 13 | 1.28 | 0.51 | 0.002 |
| Dendritic Cell | 7 | 2 | 5 | 0.45 | 1.98 | 0.1 |
| B Cell | 11 | 6 | 5 | 0.85 | 1.26 | 0.5 |
| non-Iris genes | 2923 | 1868 | 1055 | 1 | 1 | 1 |
Up = upregulated in pre-surgery samples, Down = downregulated in pre-surgery samples, Enrich = enrichment of Up or Down genes. Bold indicates significant enrichments with p-values < 0.05 shown in italic
Changes in PBMC gene expression in pre- vs. post-surgery comparisons are similar to changes found in comparison of lung cancer patients to unrelated non-cancer control patients
To determine whether the removal of tumor(s) from lung elicit alterations in gene expression in PBMC that are similar to those we identified in comparing patients with malignant tumors vs. control patients with non-malignant lung disease (NHC), we carried out the following comparison. We first determined the overlap between the most informative SVM genes (n=3,271) found in pre- vs. post-surgery comparison and the corresponding SVM list (n=2,385) found in comparing 127 samples of cancer patients to 91 non-cancer patients in our previous study (9). We identified 751 genes changed significantly and in the same direction, vs. only 254 expected by chance (P<3×10−36). There is a highly significant correlation (r=0.76) between the expression ratios derived from the 2 comparisons (Figure 3A) and the ratios were more highly correlated for the more informative genes (Supplementary Figure 1) demonstrating that the changes seen in PBMC gene expression are similar irrespective of whether the comparison is between individuals before and after tumor removal, or between unrelated lung cancer patients and controls with non-malignant lung disease. To further test the possibility that the former may provide an effective way to identify class differences we used a pre vs. post surgery SVM-RFE classifier with the 50 most informative differentially expressed genes (Figure 1) to discriminate subclasses of patients with malignant and non-malignant pulmonary disease. This classifier was applied to PBMC expression data from three independent classes of patients: a validation set with 127 malignant tumors (group 1), 41 patients with benign lung nodules (group 2) and 50 pulmonary patients without lung nodules (group 3) from our previous study (9). The results are shown in Figure 3B. The 50 genes perfectly separate the pre vs. post surgery training set; 77% of the group 1 validation samples are classified correctly as being from patients with malignant tumors, 76% of group 3 samples from controls with no tumor are also correctly classified as tumor free. However, 73% of the samples from group 2 with benign nodules surprisingly misclassify with the pre-surgery group. This observation is at odds with the benign nodule classification in our previous study (80% classified as non-cancer controls with 24 genes) and suggests that many of the pre vs. post expression changes defined by the top 50 pre vs. post genes are attributable to the presence of a growth in the lung irrespective of whether it is benign or malignant.
Figure 3. Similarities in gene expression changes and relationship between different classes of PBMC samples.
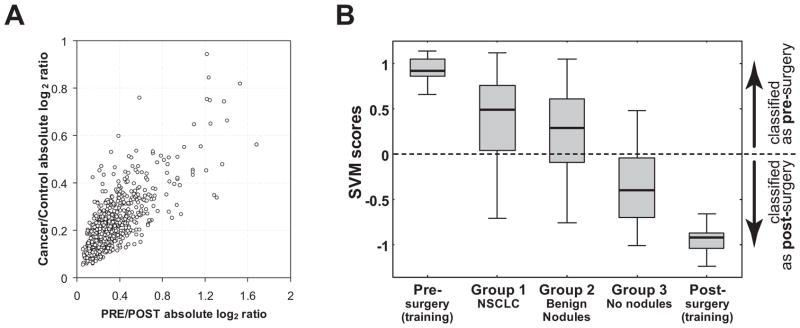
A. Correlation of expression ratios (log2 values) for 751 genes identified both in the comparison of NSCLC to non-malignant control patients (y-axis) and the pre- vs. post-surgery lung cancer patients (x-axis). This similarity in differential gene expression in both studies suggests information that distinguishes malignant and benign nodules in the previous study is also present in the pre vs. post surgery data.
B. Relationships between cancer and control sample classes. Classification (SVM) scores of various sample groups based on 50 genes (Figure 1) that were differentially expressed in pre- vs. post-surgery PBMC from NSCLC patients. Whisker ends show full range of scores for each group; grey boxes include 50% range; horizontal line within each indicates median score. Number of samples in each class (from left to right) is 18, 127, 41, 50 and 18.
To determine whether we could identify those genes specific to the presence of a benign nodule, we compared 3276 genes we previously found to be differentially expressed between patients with lung cancer and benign nodules (9) among the 3271 genes differentially expressed in this pre vs. post surgery analysis. We found a significant overlap of 789 (P=3×10−5) suggesting the pre vs. post surgery comparison should contain similar information that could distinguish malignant tumors from benign nodules. Confirmation that this is the case was obtained by using SVM-RFE 10-fold cross-validation to distinguish malignant tumors from non-malignant nodules using only the 3276 pre- vs. post-surgery genes. This identified a 135-gene signature (not shown) which separated these two groups with 78% sensitivity and 71% specificity similar to the accuracy previously reported (9).
Altered expression of 5 miRNAs in pre- vs. post-surgery samples
To assess whether miRNAs might function as diagnostic biomarkers and whether they could be responsible for some of the changes in mRNA levels detected, we compared miRNA expression levels in pre- and post-surgery PBMC samples from 11 of the pre vs. post pairs for which sufficient RNA was available. By paired t-test we determined that 46 of the 643 miRNAs detected in PBMC were significantly differentially expressed between pre- and post-surgery samples (P<0.05, FDR 42%), and that 42/46 (91%) were upregulated in the pre-surgery samples. Because our sample size was relatively small, we focused further investigation on 5 miRNAs that showed the most significant changes with a low FDR (15%). These included let-7c, miR-34a, miR-202*, miR-769-5p, and hsa-miR-642 (Supplementary Figure 2). To identify pathways and functions potentially affected by these 5 miRNAs, we identified putative target genes based on two criteria: 1) the gene was computationally predicted to be a target of a specific miRNA, and, 2) expression of the target gene changed significantly between the pre- vs. post-surgery samples, in the direction opposite to the miRNA’s expression. For let-7c, miR-34a, miR-202*, miR-769-5p, and miR-642 we found 192, 214, 49, 129, and 184 genes respectively (Supplementary Table 6), that fit both criteria, resulting in a combined list of 569 unique genes, a number significantly more than expected by chance (P=6×10−7). This supports a model that includes these 5 miRNAs as important contributors to the observed effects of tumor presence on PBMC gene expression.
We identified functional enrichments among the potential targets, using DAVID (27) and Ingenuity (IPA) software. DAVID results indicated significant numbers of the genes were involved in transcription (51 genes; FDR=0.1%) and apoptosis (21 genes; FDR=0.1%). IPA analysis revealed that many of the miRNAs target transcription factors common to immunologically important pathways. The most frequently shared target genes among the 30 significantly altered the pathways were NFkB (25 pathways), RelA (21 pathways), cFos/cJun (24 pathways), and IKK (19 pathways) (Supplementary Table 7). The most significant numbers of targets were in the gonadotropin releasing hormone pathway (GnRH), IL-10 signaling, and the immunoregulatory roles of nuclear factor of activated T-cells (NFAT) (31). Gonadotropin secreted by PBMC (32) can alter the T helper cytokine balance (33)
DISCUSSION
We show that a NSCLC elicits large-scale changes in gene expression in PBMC and that this influence on the peripheral immune cells is substantially reversible by tumor removal. The changes we find are clearly tumor-associated and not attributable to the surgery per se as the gene expression changes detected after lung tumor removal are very similar to those found when comparing lung cancer patients with tumor-naïve patients with smoking related non-malignant lung disease. Using matched post surgery samples as our control class in these comparisons eliminates much of the variability inherent in comparisons with unrelated cancer free controls. As a result, we are able to identify a set of differentially expressed genes which exhibit significant differences in expression levels, using only 18 sample pairs.
We also present evidence that non-malignant lung nodules can also influence PBMC gene expression and identify a signature from the pre vs. post differentially expressed genes that can distinguish malignant tumors from non-malignant nodules. Although validation studies are necessary, this gene signature can potentially be used to develop a non-invasive method to assess whether suspect lung nodules are indeed malignant. The effects of the tumor presence on the peripheral immune system are manifested as relatively small changes in expression levels of several thousand genes in PBMC. The majority of these PBMC genes (67%) were expressed at higher levels prior to tumor resection.
We also find that arms of the innate immune system thought to be important for tumor-directed immunity appear to be negatively influenced by the tumor presence. First, we find evidence that NK function may be suppressed, as transcript levels for repressive forms of NK cell-associated Kir receptors were consistently elevated before vs. after tumor resection in peripheral immune cells. These receptors included KLRB1 (which acts through ceramide signaling, another highly-affected pathway), KIR2DL3, KIR2DL1, KIR3DL1, KIR2DL5A, and KIR2DL2. Since inhibition is thought to be dominant over activation in the KIR system (13, 34), the combined results suggest that the functions of NK cells within the PBMC population studied are impaired in the presence of lung tumors. Second, the differentially-expressed genes of myeloid cells important for an innate response were found to be predominantly downregulated prior to tumor removal. The majority of genes associated with chemotaxis of myeloid cells and glucocorticoid receptor signaling were also expressed at significantly lower levels prior to tumor resection, providing further indication of a suppressed state of the peripheral immune system in the presence of tumor. Third, we found that components of the ceramide pathway were upregulated in patients with tumors, suggesting that the at least some PBMC cell types are primed for apoptosis which could impair an anti-tumor responses. Genes found to be upregulated in the presence of tumor included S1PR5 receptor as well as intracellular components of the ceramide pathway, reflecting multiple mechanisms driving sphingosine-dependent apoptosis (20). Fourth, members of the ErbB/neuregulin pathway genes were also significantly different in the pre v. post surgery comparison. ErbB receptors and Neuregulin-1 have also been shown to be expressed by NSCLC cell lines (35) suggesting that effects on expression of the ErbB pathway in PBMC of lung tumor patients may be caused by Neuregulin-1 synthesized by normal lung epithelium or lung tumors (36), or by the ErbB1 ligands such as amphiregulin or TGF-α, also produced by lung tumors (37).
We also find significant alterations in expression of 5 miRNAs in the presence vs. absence of NSCLC tumors. A previous study comparing miRNA in PBMC from NSCLC and healthy controls (38) identified a series of 24 miRNAs that distinguish the 2 classes, included only let-7 of the 5 miRNAs we have identified. This may be attributed to the use of a healthy and younger control class in that comparison. In our study the miRNAs most consistently differentially expressed, like the coding genes, are primarily expressed at higher levels in the presence of the cancer. We identified potential targets for the 5 most significantly changed miRNAs among the differentially expressed genes that are down regulated in pre-surgery cancer patients and show that these 5 miRNAs primarily target differentially-expressed transcription factors found in our dataset. Among the pathways showing significant tumor-associated effects that also included significant numbers of potential miRNA targets are the toll-like receptor (TLR)-associated pathways. We find a significant decrease in message levels in pre-surgery samples for TLRs 5, 6, 7, 8 and 10 as well as messages for the TLR2 and TLR4accessory molecule CD14 and the TLR signaling components MYD88 and TOLLIP generally required for all TLR signaling. Several key transcriptional regulators of TLR signaling (Figure 4) are targeted by 1 to 4 of the 5 miRNAs over-expressed pre-surgery. For example, 4 of the 5 miRNAs target IKKα and the NFkB complex, important to many immune response pathways. Targeting is via both perfect and imperfect matches suggesting both transcript degradation and translational arrest mechanisms are in play (reviewed in (39)). Although none of the miRNAs target the TLR genes directly, expression of several TLRs is also regulated by NFkB (40–41) suggesting miRNA targeting can secondarily affect TLR expression. Although the changes in mRNA expression levels for each targeted gene are generally small, the resulting effects on translation and the cumulative effects of targeting multiple points in the signal transduction pathway could be substantial.
Figure 4. Model demonstrating the integration of mRNA and miRNA expression data associated with toll-like receptor signaling pathway.

Blue symbols indicate a gene is under- and red symbols indicate a gene is over-expressed in the pre- compared to the post-surgery samples, grey symbols indicate a gene is not differentially expressed, orange circles indicate that a gene is targeted by one or more of the five miRNAs, each represented by a black symbol on the circle.
Members of the TLR family play key roles in both innate and adaptive immune responses associated with infections, autoimmune conditions, inflammatory lung diseases and in the regulation of tolerance [reviewed in (42–43)]. A decrease of TLR-mediated responses, in concert with the miRNA-mediated inhibition of key transcription factors required for TLR signal transduction pathways would impair important immune regulatory functions associated with immune defense in patients with lung tumors.
An important question raised by our findings of diagnostic lung tumor-associated biomarkers in PBMC concerns the mechanisms by which lymphocyte gene expression is influenced. There have been a large number of studies focused on the identification of LC derived proteins in serum or plasma including circulating auto-antibodies and a number of proteins with known effects on immune cells (reviewed in (44–45). A recent study by Ostroff et al (46) identified a panel of 44 plasma proteins whose levels were significantly different between mostly early stage NSCLC patients and control groups of high-risk smoker controls (46). We are likely measuring the effects in the periphery of proteins or small RNAs derived from the tumor/and or its microenvironment. Long range effects of both pre-malignant lung lesions on bone marrow macrophages that are enhanced by tumor progression have been reported previously (47–49). Previous studies have also shown that, TGF-α synthesized by lung tumors (37) can activate ErbB, which lies on a pathway to ELK1 activation. Transcription factors ELK1, ELK4, and SPI1 binding motifs were enriched, on average, by 23% within the promoters of the genes found to be upregulated in PBMC prior to tumor resection. These observations of cross-talk between tumor and the peripheral host immune system suggest mechanisms for generating the changes in biomarker expression we have detected, observations that might potentially be exploited for development of more effective immune therapy.
Many of the changes in gene expression within PBMC detected in patients bearing lung tumors were common to both benign nodules and malignant tumors. The benign nodules, histologically diagnosed as non-malignant, were typical of high-risk nodules that require biopsy to confirm their non-malignant status. Understanding the basis for the differences that distinguish PBMC from patients with malignant tumors from patients with benign nodules could provide an important additional tool for early detection and control of lung cancer. Although this study is relatively small, the increased robustness of the comparisons using the post-surgery samples as the highly matched control class and the successful application of the derived signatures to new NSCLC samples indicates the feasibility of this approach for biomarker discovery. Although the analysis of the tumors themselves remains an important pursuit, our approach provides the opportunity for continued non-invasive surveillance after tumor removal and provides important added information on the interactions of benign nodules and malignant lung tumors with the immune system.
Supplementary Material
TRANSLATIONAL RELEVANCE.
This study addresses two related clinical problems in lung cancer diagnostics — the development of an easily applied minimally invasive test for the presence of lung cancer in the large at risk population of smokers and ex-smokers, and more immediately, to distinguish malignant from non-malignant nodules that are detected by imaging approaches. We adopt a blood-based solution in both cases. In a previous study we demonstrated the PBMC contain mRNA expression patterns that can distinguish lung cancer from non-malignant lung disease in groups of current and previous smokers. In this report we show that changes in gene expression in peripheral immune cells that occur when a lung tumor is removed provide additional lung cancer signatures that can both distinguish malignant from non-malignant disease and also distinguish malignant from benign nodules.
Acknowledgments
We thank Mea Fuller for excellent editorial assistance.
GRANT SUPPORT
This project was supported by PA DOH Tobacco Settlement grant SAP 4100020718, the PA DOH Commonwealth Universal Research Enhancement Program, EDRN set-aside funds for UO1CA86137, and the Wistar Cancer Center Support Grant P30 CA010815. A.V. was supported by NCI K07 CA111952, A.V.K. is supported by NCI T32 CA 09171. The project used the Wistar Genomics and Bioinformatics facilities supported by P30 CA010815.
Non-standard abbreviations
- FDR
false discovery rate
- LAC
lung adenocarcinomas
- LC
lung cancer
- LSCC
lung squamous cell carcinomas
- NHC
controls with non-malignant lung disease
- NSCLC
non-small cell lung cancer
- SVM-RFE
support vector machine with recursive feature elimination
- TLR
toll-like receptor
Footnotes
There are no conflicts of interest
References
- 1.ACS. Cancer Facts and Figures 2010. Atlanta: American Cancer Society; 2010. [Google Scholar]
- 2.Jemal A, Ward E, Hao Y, Thun M. Trends in the Leading Causes of Death in the United States, 1970–2002. JAMA. 2005;294:1255–9. doi: 10.1001/jama.294.10.1255. [DOI] [PubMed] [Google Scholar]
- 3.Hirschowitz EA, Yannelli JR. Immunotherapy for Lung Cancer. Proc Am Thorac Soc. 2009;6:224–32. doi: 10.1513/pats.200806-048LC. [DOI] [PubMed] [Google Scholar]
- 4.Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. 2003;21:807–39. doi: 10.1146/annurev.immunol.21.120601.141135. [DOI] [PubMed] [Google Scholar]
- 5.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–74. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 6.Abu-Shakra M, Buskila D, Ehrenfeld M, Conrad K, Shoenfeld Y. Cancer and autoimmunity: autoimmune and rheumatic features in patients with malignancies. Ann Rheum Dis. 2001;60:433–41. doi: 10.1136/ard.60.5.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother. 2005;54:187–207. doi: 10.1007/s00262-004-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raez LE, Fein S, Podack ER. Lung cancer immunotherapy. Clin Med Res. 2005;3:221–8. doi: 10.3121/cmr.3.4.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Showe MK, Vachani A, Kossenkov AV, Yousef M, Nichols C, Nikonova EV, et al. Gene expression profiles in peripheral blood mononuclear cells can distinguish patients with non-small cell lung cancer from patients with nonmalignant lung disease. Cancer Res. 2009;69:9202–10. doi: 10.1158/0008-5472.CAN-09-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Storey JD, Tibshirani R. Statistical methods for identifying differentially expressed genes in DNA microarrays. Methods Mol Biol. 2003;224:149–57. doi: 10.1385/1-59259-364-X:149. [DOI] [PubMed] [Google Scholar]
- 11.Guyon I, Weston J, Barnhill S, Vapnik V. Gene Selection for Cancer Classification using Support Vector Machines. Machine Learning. 2002;46:389– 422. [Google Scholar]
- 12.Jurman G, Merler S, Barla A, Paoli S, Galea A, Furlanello C. Algebraic stability indicators for ranked lists in molecular profiling. Bioinformatics. 2008;24:258–64. doi: 10.1093/bioinformatics/btm550. [DOI] [PubMed] [Google Scholar]
- 13.Bashirova AA, Martin MP, McVicar DW, Carrington M. The killer immunoglobulin-like receptor gene cluster: tuning the genome for defense. Annu Rev Genomics Hum Genet. 2006;7:277–300. doi: 10.1146/annurev.genom.7.080505.115726. [DOI] [PubMed] [Google Scholar]
- 14.Single RM, Martin MP, Meyer D, Gao X, Carrington M. Methods for assessing gene content diversity of KIR with examples from a global set of populations. Immunogenetics. 2008;60:711–25. doi: 10.1007/s00251-008-0331-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitatani K, Idkowiak-Baldys J, Hannun YA. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cellular Signalling. 2008;20:1010–8. doi: 10.1016/j.cellsig.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venable ME, Lee JY, Smyth MJ, Bielawska A, Obeid LM. Role of Ceramide in Cellular Senescence. J Biol Chem. 1995;270:30701–8. doi: 10.1074/jbc.270.51.30701. [DOI] [PubMed] [Google Scholar]
- 17.Pozo D, Vales-Gomez M, Mavaddat N, Williamson SC, Chisholm SE, Reyburn H. CD161 (human NKR-P1A) signaling in NK cells involves the activation of acid sphingomyelinase. J Immunol. 2006;176:2397–406. doi: 10.4049/jimmunol.176.4.2397. [DOI] [PubMed] [Google Scholar]
- 18.Moreno M, Molling JW, von Mensdorff-Pouilly S, Verheijen RH, von Blomberg BM, van den Eertwegh AJ, et al. In vitro expanded human invariant natural killer T-cells promote functional activity of natural killer cells. Clin Immunol. 2008;129:145–54. doi: 10.1016/j.clim.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Schmieg J, Yang G, Franck RW, Van Rooijen N, Tsuji M. Glycolipid presentation to natural killer T cells differs in an organ-dependent fashion. Proc Natl Acad Sci U S A. 2005;102:1127–32. doi: 10.1073/pnas.0408288102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki E, Handa K, Toledo MS, Hakomori S. Sphingosine-dependent apoptosis: a unified concept based on multiple mechanisms operating in concert. Proc Natl Acad Sci U S A. 2004;101:14788–93. doi: 10.1073/pnas.0406536101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huynh H, Nguyen TT, Chan E, Tran E. Inhibition of ErbB-2 and ErbB-3 expression by quercetin prevents transforming growth factor alpha (TGF-alpha)- and epidermal growth factor (EGF)-induced human PC-3 prostate cancer cell proliferation. Int J Oncol. 2003;23:821–9. [PubMed] [Google Scholar]
- 22.Gille H, Kortenjann M, Thomae O, Moomaw C, Slaughter C, Cobb MH, et al. ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 1995;14:951–62. doi: 10.1002/j.1460-2075.1995.tb07076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boros J, Donaldson I, O’Donnell A, Odrowaz Z, Zeef L, Lupien M, et al. Elucidation of the ELK1 target gene network reveals a role in the coordinate regulation of core components of the gene regulation machinery. Genome Res. 2009;19:1963– 73. doi: 10.1101/gr.093047.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin H, Chan MW, Liyanarachchi S, Balch C, Potter D, Souriraj IJ, et al. An integrative ChIP-chip and gene expression profiling to model SMAD regulatory modules. BMC Syst Biol. 2009;3:73. doi: 10.1186/1752-0509-3-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zakrzewska A, Cui C, Stockhammer OW, Benard EL, Spaink HP, Meijer AH. Macrophage-specific gene functions in Spi1-directed innate immunity. Blood. 2010;116:e1–11. doi: 10.1182/blood-2010-01-262873. [DOI] [PubMed] [Google Scholar]
- 26.Kubosaki A, Lindgren G, Tagami M, Simon C, Tomaru Y, Miura H, et al. The combination of gene perturbation assay and ChIP-chip reveals functional direct target genes for IRF8 in THP-1 cells. Mol Immunol. 2010;47:2295–302. doi: 10.1016/j.molimm.2010.05.289. [DOI] [PubMed] [Google Scholar]
- 27.Dennis G, Sherman B, Hosack D. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 28.Jacques C, Fontaine JF, Franc B, Mirebeau-Prunier D, Triau S, Savagner F, et al. Death-associated protein 3 is overexpressed in human thyroid oncocytic tumours. Br J Cancer. 2009;101:132–8. doi: 10.1038/sj.bjc.6605111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boros J, Donaldson IJ, O’Donnell A, Odrowaz ZA, Zeef L, Lupien M, et al. Elucidation of the ELK1 target gene network reveals a role in the coordinate regulation of core components of the gene regulation machinery. Genome Res. 2009;19:1963–73. doi: 10.1101/gr.093047.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abbas AR, Baldwin D, Ma Y, Ouyang W, Gurney A, Martin F, et al. Immune response in silico (IRIS): immune-specific genes identified from a compendium of microarray expression data. Genes Immun. 2005;6:319–31. doi: 10.1038/sj.gene.6364173. [DOI] [PubMed] [Google Scholar]
- 31.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–84. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- 32.Chen H-F, Jeung E-B, Stephenson M, Leung PCK. Human Peripheral Blood Mononuclear Cells Express Gonadotropin-Releasing Hormone (GnRH), GnRH Receptor, and Interleukin-2 Receptor {gamma}-Chain Messenger Ribonucleic Acids That Are Regulated by GnRH in Vitro. J Clin Endocrinol Metab. 1999;84:743–50. doi: 10.1210/jcem.84.2.5440. [DOI] [PubMed] [Google Scholar]
- 33.Dixit VD, Yang H, Udhayakumar V, Sridaran R. Gonadotropin-Releasing Hormone Alters the T Helper Cytokine Balance in the Pregnant Rat. Biology of Reproduction. 2003;68:2215–21. doi: 10.1095/biolreprod.102.012211. [DOI] [PubMed] [Google Scholar]
- 34.Carrington M, Martin MP. The impact of variation at the KIR gene cluster on human disease. Curr Top Microbiol Immunol. 2006;298:225–57. doi: 10.1007/3-540-27743-9_12. [DOI] [PubMed] [Google Scholar]
- 35.Gollamudi M, Nethery D, Liu J, Kern JA. Autocrine activation of ErbB2/ErbB3 receptor complex by NRG-1 in non-small cell lung cancer cell lines. Lung Cancer. 2004;43:135–43. doi: 10.1016/j.lungcan.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, Kern JA. Neuregulin-1 Activates the JAK-STAT Pathway and Regulates Lung Epithelial Cell Proliferation. Am J Respir Cell Mol Biol. 2002;27:306–13. doi: 10.1165/rcmb.4850. [DOI] [PubMed] [Google Scholar]
- 37.Mukohara T, Kudoh S, Matsuura K, Yamauchi S, Kimura T, Yoshimura N, et al. Activated Akt expression has significant correlation with EGFR and TGF-alpha expressions in stage I NSCLC. Anticancer Res. 2004;24:11–7. [PubMed] [Google Scholar]
- 38.Keller A, Leidinger P, Borries A, Wendschlag A, Wucherpfennig F, Scheffler M, et al. miRNAs in lung cancer - Studying complex fingerprints in patient’s blood cells by microarray experiments. BMC Cancer. 2009;9:353. doi: 10.1186/1471-2407-9-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomas M, Lieberman J, Lal A. Desperately seeking microRNA targets. Nat Struct Mol Biol. 2010;17:1169–74. doi: 10.1038/nsmb.1921. [DOI] [PubMed] [Google Scholar]
- 40.Lee J, Hayashi M, Lo JF, Fearns C, Chu WM, Luo Y, et al. Nuclear factor kappaB (NF-kappaB) activation primes cells to a pro-inflammatory polarized response to a Toll-like receptor 7 (TLR7) agonist. Biochem J. 2009;421:301–10. doi: 10.1042/BJ20090013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang T, Lafuse WP, Zwilling BS. NFkappaB and Sp1 elements are necessary for maximal transcription of toll-like receptor 2 induced by Mycobacterium avium. J Immunol. 2001;167:6924–32. doi: 10.4049/jimmunol.167.12.6924. [DOI] [PubMed] [Google Scholar]
- 42.Basu S, Fenton MJ. Toll-like receptors: function and roles in lung disease. Am J Physiol Lung Cell Mol Physiol. 2004;286:L887–92. doi: 10.1152/ajplung.00323.2003. [DOI] [PubMed] [Google Scholar]
- 43.Ehlers M, Ravetch JV. Opposing effects of Toll-like receptor stimulation induce autoimmunity or tolerance. Trends Immunol. 2007;28:74–9. doi: 10.1016/j.it.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 44.Bharti A, Ma PC, Salgia R. Biomarker discovery in lung cancer--promises and challenges of clinical proteomics. Mass Spectrom Rev. 2007;26:451–66. doi: 10.1002/mas.20125. [DOI] [PubMed] [Google Scholar]
- 45.Meyerson M, Carbone D. Genomic and Proteomic Profiling of Lung Cancers: Lung Cancer Classification in the Age of Targeted Therapy. Journal of Clinical Oncology. 2005;23:3219–26. doi: 10.1200/JCO.2005.15.511. [DOI] [PubMed] [Google Scholar]
- 46.Ostroff RM, Bigbee WL, Franklin W, Gold L, Mehan M, Miller YE, et al. Unlocking biomarker discovery: Large scale application of aptamer proteomic technology for early detection of lung cancer. Nature Precedings. 2010 doi: 10.1371/journal.pone.0015003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Redente EF, Dwyer-Nield LD, Merrick DT, Raina K, Agarwal R, Pao W, et al. Tumor progression stage and anatomical site regulate tumor-associated macrophage and bone marrow-derived monocyte polarization. Am J Pathol. 2010;176:2972–85. doi: 10.2353/ajpath.2010.090879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Redente EF, Higgins DM, Dwyer-Nield LD, Orme IM, Gonzalez-Juarrero M, Malkinson AM. Differential polarization of alveolar macrophages and bone marrow-derived monocytes following chemically and pathogen-induced chronic lung inflammation. J Leukoc Biol. 2010;88:159–68. doi: 10.1189/jlb.0609378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Redente EF, Orlicky DJ, Bouchard RJ, Malkinson AM. Tumor signaling to the bone marrow changes the phenotype of monocytes and pulmonary macrophages during urethane-induced primary lung tumorigenesis in A/J mice. Am J Pathol. 2007;170:693–708. doi: 10.2353/ajpath.2007.060566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.