

Abstract
Progressive multisystem disease should invoke consideration of potential mitochondrial etiologies. Mitochondrial disease can affect any organ system at any time, particularly involving neurologic, cardiac, muscular, gastroenterologic, and/or ophthalmologic manifestations. We report here a 19-year-old Caucasian man who was followed since birth in multiple pediatric subspecialty clinics for myelomeningocele complications. However, he progressively developed a host of additional problems that were not readily attributable to his neural tube defect but involved developmental, ophthalmologic, cardiac, muscular, endocrine, and intermediary metabolic manifestations. Clinical diagnostic testing limited to analysis for common point mutations and deletions in his blood mitochondrial DNA (mtDNA) was not revealing. Skeletal muscle biopsy revealed abnormal mitochondrial morphology and immunostaining, mitochondrial proliferation, and mildly reduced respiratory chain complex I–III activity. Whole mitochondrial genome sequencing analysis in muscle identified an apparently homoplasmic, novel, 12264C>T transition in the tRNA serine (AGY) gene. The pathogenicity of this mutation was supported by identification of it being present at low heteroplasmy load in his blood (34 percent) as well as in blood from his maternal grandmother (1 percent). Interestingly, the proband developed severe nuclear cataracts that proved to be homoplasmic for the pathogenic mtDNA 12264C>T mutation. This case highlights the value of pursuing whole mitochondrial genome sequencing in symptomatic tissues in the diagnostic evaluation of suspected mitochondrial disease. Furthermore, it is the first report to directly implicate an mtDNA mutation in the pathogenesis of ocular cataracts and clearly illustrates the important contribution of normal metabolic activity to the function of the ocular lens.
INTRODUCTION
Mitochondria are integral to basic cellular functions ranging from energy production and free radical scavenging to calcium homeostasis and apoptosis. Mitochondria are increasingly recognized to play pathogenic roles in human disease, with either isolated or multisystem organ involvement. Indeed, mitochondrial dysfunction commonly impairs the function of those tissues and organs with the highest energy requirements such as the nervous system, musculoskeletal system, heart, gastrointestinal tract, and sensory organs (Haas et al., 2007). Mitochondrial dysfunction can result from environmental factors and/or mutations in either nuclear genes or within the mitochondrial DNA (mtDNA) genome itself (Haas et al., 2008). Whereas genetic testing options for suspected mitochondrial disease was long limited to analysis of a subset of common mtDNA point mutations, it has become evident in the last five years that hundreds of distinct mutations in the mtDNA genome cause significant mitochondrial diseases (Zaragoza et al., 2011). Here, we report a case that exemplifies the multisystemic and progressive nature of mitochondrial respiratory chain disease and emphasizes the diagnostic utility of performing whole mtDNA genome sequencing in affected tissues to identify the pathogenic cause. This case further elucidates the significant contribution made by mitochondria to normal metabolic function of the ocular lens, as a novel mtDNA point mutation in the tRNA gene encoding serine caused bilateral cataracts.
CASE REPORT
A 19-year-old Caucasian male presented in the joint Ophthalmology-Genetics Clinic at The Children’s Hospital of Philadelphia (CHOP) to evaluate for an etiology of pigmentary retinopathy and bilateral cataracts. He had multiple complications from a congenital open neural tube defect, including hydrocephalus and Chiari malformation necessitating two ventriculoperitoneal shunts, bilateral lower-extremity paralysis, bowel and bladder incontinence with an irregularly thickened bladder wall, scoliosis, restrictive lung disease, osteopenia, and proximal femur and acetabulum deformities. In addition, progressive multisystem problems developed that were not readily attributable to sequelae of a neural tube defect. His cardiac manifestations included Wolff-Parkinson-White (WPW) arrhythmia, hypertrophic cardiomyopathy, and congestive heart failure. His neurologic findings included hypotonia, bilateral severe mixed hearing loss, and sleep apnea. His endocrine symptoms included truncal obesity, non-insulin dependent diabetes mellitus, hypogonadotropic hypogonadism, and short stature, the latter of which resulted from growth hormone deficiency with a full adult height of only 5 foot 2 inches achieved. His ophthalmologic manifestations included severely reduced visual acuity (20/200 O.U. best-corrected), retinitis pigmentosa (electroretinogram was flatline for rod photoreceptor retinal function), and bilateral dense nuclear cataracts that progressively worsened over time. His development was significantly delayed, and his cognition at 19 years and 7 months of age was approximated at that of a 10 to 11-year-old. He was completing a life skills program and received occupational, physical, speech, and hearing therapies. He also had intermittent lactic acidemia (2 – 3 mM range, normal <2 mM) and hyperalaninemia (492 nmol/mL, normal range 88–440 nmol/mL). The proband’s family history was essentially non-contributory. He lived with his father, who at age 50 was in good health. His mother had been in apparent good health but died in a motor vehicle accident at age 30. His maternal grandmother was in overall good health (Figure 1).
Figure 1. Pedigree of proband with multisystem mitochondrial disease and cataracts.
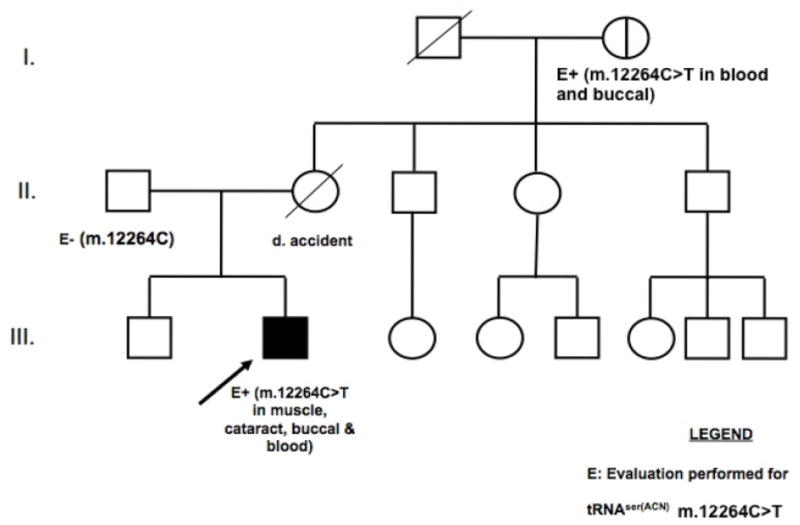
Arrow indicates proband. “E” indicates individuals in whom molecular evaluation was performed for the tRNASer(AGY) m.12264C>T mutation. No tissue was available for testing from the proband’s mother. The heteroplasmy load of various tissues of the proband, his father, and his maternal grandmother are detailed in Table 1.
Physical examination at the time of initial assessment was significant for short stature (<3rd percentile) and macrocephaly (OFC at the 95th percentile). He had bilateral lower extremity paralysis. Dysmorphic facial features included hypertelorism, a flat nasal bridge, flat midface, and a midfacial chin cleft. In addition, he had bilateral inverted nipples and bilateral fifth finger contractures at the proximal interphalangeal joints. He had truncal obesity and a buffalo hump, but no acanthosis nigricans.
Previous cardiac studies included an echocardiogram that showed significant left ventricular hypertrophy and an electrocardiogram that confirmed changes consistent with WPW. Prior radiology films showed osteopenia and a delayed bone age by approximately two years.
METHODS
Clinical diagnostic testing
Skeletal muscle and skin biopsies were performed at CHOP. Biopsy was performed in the proband’s biceps muscle, since he had previously received botox injections for spasticity in both quadriceps muscles. Muscle histologic and immunohistochemical analyses were performed in the Department of Pathology at CHOP. Muscle electron transport chain activity analysis was performed in frozen muscle sent to the Mitochondrial Diagnostic Laboratory at Baylor College of Medicine (BCM). Fibroblast cell line analysis for pyruvate dehydrogenase deficiency was performed at the Center for Inherited Disorders of Energy Metabolism (CIDEM, Cleveland, OH). Standard biochemical analyses in blood and urine were performed at the CHOP Biochemical Laboratory. Leukocyte Coenzyme Q content analysis was performed at Horizon Medical Laboratory (Atlanta, GA). Mitochondrial DNA (mtDNA) analyses were performed on a clinical diagnostic basis at BCM. These included mtDNA common point mutations and deletions in muscle, MitoMet® oligonucleotide comparative genomic hybridization array analysis for nuclear gene deletions as well as mtDNA deletions and mtDNA copy number (in muscle), and whole mitochondrial genome sequence analysis by PCR and Sanger sequencing (in muscle). Genetic testing for Bardet Biedl syndrome caused by mutations in BBS1 and BBS2 was performed at Correlagen (MA).
Heteroplasmy quantitation
mtDNA tRNA-serine gene mutational confirmation and percent heteroplasmy determination were performed in additional tissues from the proband (blood, buccal swab, muscle, cataract) and his relatives (blood, buccal swab) on a research basis by a newly developed TaqMan MGB (Minor Groove Binding) real-time quantitative PCR (qPCR) method to simultaneously measure in one reaction tube both wild-type (m.12264C) and mutant (m.12264T) levels. The MGB moiety stabilizes the hybridized probe and effectively raises the melting temperature (Tm), thereby allowing for MGB probes to be shorter than traditional dual-labeled probes and better suited for allelic discrimination applications. qPCR primers used were: mtF12232, 5′-TCATGCCCCCATGTCTAACAA-3′; and mtR12466, 5′-AGGTGGATGCGACAATGGATT-3′. MGB probes used were: 5′-6FAM-TGGCTTTCTcAACTTT-NFQ-3′ for m.12264C and 5′-TET-CATGGCTTTCTtAACTTTTA-NFQ-3′ for m.12264T. Primers were designed using ABI Primer Express Software V2.0 and synthesized by Applied Biosystems (Foster City, CA). TaqMan qPCR analysis was performed on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA). Relative expression was calculated as previously described (Ji et al., 2003; Nicklas & Buel, 2006).
Cataract analysis
Cataract extraction was performed per surgical routine (WA), with extracted material from one eye collected in 6 ml of saline and flash frozen in liquid nitrogen. DNA was extracted from the combined lens tissue by using QIAamp DNA mini Kit (Qiagen, Valencia, CA) and studied by qPCR analysis to determine heteroplasmy levels, as detailed above.
RESULTS
Metabolic analyses
Leukocyte coenzyme Q content was within normal limits. Pyruvate dehydrogenase enzyme analysis in skin fibroblasts was in the normal range. Increased citrate synthase in skeletal muscle was suggestive of mitochondrial proliferation. Enzymatic activity analyses for respiratory chain complexes I through IV in skeletal muscle were within the control range, although rotenone-sensitive I–III activity was mildly reduced to 54% of the mean after correction for citrate synthase.
Histological and histochemical examination of cryomicrotomy sections from frozen biceps muscle tissue
Gomori trichrome staining identified ragged red fibers (Figure 2A) and reduced nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR) staining confirmed the presence of basophilic rims (Figure 2B), which were indicative of mitochondrial proliferation. Combined succinic dehydrogenase and cytochrome oxidase (SDH/COX) staining showed fibers that were devoid of cytochrome oxidase (COX, complex IV) activity (Figure 2C). Electron microscopy analysis revealed irregular and branched mitochondrial morphology with focal mitochondrial collections, as well as lipid deposits (data not shown).
Figure 2. Immunohistochemical analysis of proband’s biceps muscle.
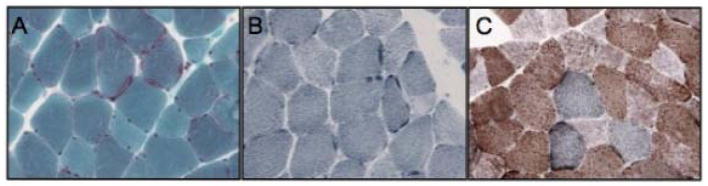
(A) Modified Gomori trichrome stain (40X). (B) Reduced nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR) stain (40X). (C) Combined succinic dehydrogenase and cytochrome oxidase (SDH/COX) stain (40X). Ragged red and blue fibers are seen in panels A and B, respectively. COX-negative fibers are evident in panel C, where fibers devoid of COX activity (C) appear blue.
Genetic diagnostic testing
BBS1 and BBS2 gene sequencing analysis did not detect any mutations. Common mtDNA point mutations and mtDNA large deletions were not detected in skeletal muscle DNA. Sequencing analysis of the whole mitochondrial genome (BCM) identified a novel, apparently homoplasmic m.12264C>T change in the tRNASer(AGY) gene in the proband’s muscle mtDNA. This mutation was subsequently quantified and found to be homoplasmic in the proband’s extracted cataract, muscle, and buccal cells, but heteroplasmic in his blood (34% mutant load) (Table 1). Familial analyses showed the proband’s father had only the wild-type m.12264C allele, whereas his maternal grandmother had low level heteroplasmy for the mutant m.12264T allele (1% in blood and 18% in buccal swab) (Table 1). m.12264T is a novel single nucleotide variant that is complementary to a previously reported m.12207G>A mutation in tRNASer(AGY) (Wong et al., 2006). Both tRNASer(AGY) mutations (m.12207G>A and m.12264C>T) disrupt G–C pairing at the last base of the amino acid acceptor stem that is presumed to be structurally important for precursor RNA processing and amino acylation (Figure 3).
Table 1.
Average m.12264C>T mutation load in various tissues isolated from the proband, his father, and his maternal grandmother.
| INDIVIDUAL | Sample | Average Wildtype load percentage m.12264C | Average Mutant load percentage m.12264T |
|---|---|---|---|
| Proband | Blood | 66±2% | 34±1% |
| Proband | Muscle | 0 | 100 |
| Proband | Cataract | 0 | 100 |
| Proband | Buccal Swab | 0 | 100 |
| Father | Blood | 100 | 0 |
| Maternal Grandmother | Blood | 99±7% | 1±0.1% |
| Maternal Grandmother | Buccal Swab | 82±0.5% | 18±0.1% |
Note: The increased heteroplasmy load for the m.12264C>T in the proband’s clinically symptomatic tissues (muscle and cataract) relative to his blood, and in the proband with severe multisystem disease as compared to his healthy maternal grandmother, supports the pathogenesis of the m.12264C>T mtDNA mutation in tRNA Ser(AGY).
Figure 3. Structure of the tRNASer(AGY) residue.
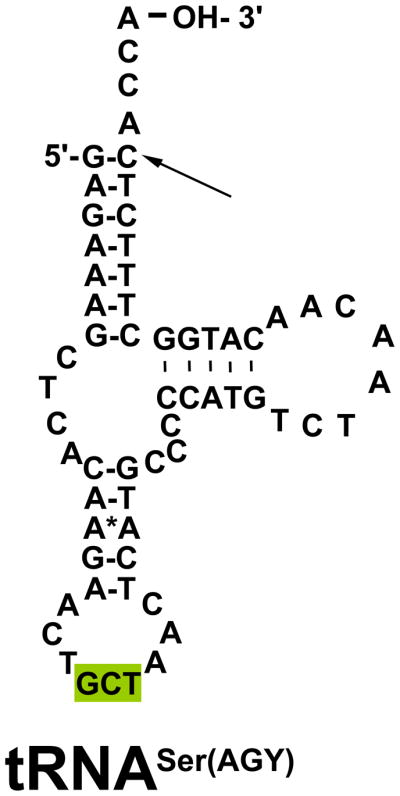
tRNASer(AGY) has a unique structure, as it is the only tRNA with two clover-leaf structures, rather than three. While there are two tRNA serine-encoding mtDNA genes, pathogenic mutations in tRNASer(AGY) are not common. The highlighted area is the anticodon region of the tRNA. The arrow indicates the location of the tRNASer(AGY) m.12264C>T point mutation detected in the proband. This mutation is the last base before the addition of the 3′ CCA tail at the amino acid acceptor stem.
DISCUSSION
Cataracts in mitochondrial disease
The eye is a highly energetic organ system, with mitochondrial dysfunction known to potentially result in a myriad of varied pathologies in any and all ocular components. Ocular pathology may occur in isolation or in conjunction with other systemic disease manifestations. Indeed, pigmentary retinopathy, optic atrophy, ophthalmoplegia, and/or strabismus are common manifestations of primary (genetic-based) mitochondrial diseases (Haas et al., 2007). In addition, “classical” eye diseases such as glaucoma, diabetic retinopathy, age-related macular degeneration, congenital cataracts, and cataracts of senility have been increasingly linked to various aspects of mitochondrial dysfunction (Schrier & Falk, 2011). Extraocular muscle tissue commonly discarded during strabismus surgery has recently been shown to offer a reliable tissue in which to perform diagnostic investigations for mitochondrial disease (Almousa et al, 2009). Similarly, histologic studies of the ocular lens and extracted cataract tissue have previously been reported to reveal evidence of mitochondrial disease (Ciulla et al., 1995; Babizhayev & Yegorov, 2010; Vinson, 2006). However, it is not common clinical practice to routinely investigate either the general histopathology or genetic composition of extracted cataracts. The presence of cataracts in our proband with a pathogenic tRNASer(AGY) mutation illustrates the importance of considering mtDNA as a potential cause of diverse ophthalmologic pathology. Here, we have reported the first case, to our knowledge, in which a pathogenic mtDNA mutation was verified within an extracted cataract. Remarkably, the novel mtDNA mutation was homoplasmic in the cataract tissue despite its only being present at a low heteroplasmy level in blood. Such low heteroplasmy levels might potentially be missed by some blood-based mtDNA genome sequencing methodologies, with this report supporting the utility of pursuing mtDNA genome sequencing in clinically affected tissues.
Cataracts have only been previously described in a few systemic mitochondrial disease syndromes. Sengers disease (OMIM 212350), characterized by congenital cataracts, hypertrophic cardiomyopathy, mitochondrial myopathy, and lactic acidosis (Sengers et al., 1975), has been postulated to result in at least some cases from post-translational changes to adenosine nucleotide translocator 1 (ANT1) (Jordens et al., 2002). 3-methylglutaconic aciduria (3-MGA) is a biomarker of several mitochondrial disease subtypes, including Barth syndrome (OMIM 302060) that manifests hypertrophic cardiomyopathy, bone marrow failure, growth retardation, and myopathy, as well as Costeff syndrome (OMIM 258501) that manifests optic atrophy, cognitive deficits, and central nervous system abnormalities. A variant subtype of 3-MGA has been proposed that appears to overlap with Sengers syndrome and involves hypertrophic cardiomyopathy, neurologic symptoms, lactic acidosis, as well as congenital cataracts (Di Rosa et al., 2006). Cataracts have also been associated with Pearson syndrome (OMIM 557000), a mitochondrial disease caused by large deletions in the mtDNA genome that presents in the newborn period with pancytopenia, pancreatic dysfunction, growth retardation, kidney tubulopathy, and skin findings. Specifically, a 6-year-old boy with Pearson syndrome was previously reported to also have bilateral zonular cataracts (Cursiefen et al., 1998) and corneal opacities were described in one other affected individual (Ribes et al., 1993). Overall, it is clear that mutations in either the mtDNA genome or in nuclear genes that affect mitochondrial function can result in the development of cataracts.
The pathologic role of mitochondria in cataract formation has been extensively studied as it relates to senescence and free radical damage. Damage to mitochondria from either external factors or genetic mutations can disrupt the balance of reactive oxygen species production and scavenging within mitochondria, with deleterious clinical sequelae. External factors that subject the mitochondria of ocular tissues to damage include UV light, environmental toxins, drugs, inflammation, and ischemia. Gene mutations in either the nuclear or mtDNA genomes can disrupt mitochondrial energy production and free radical balance (Brennan & Kantorow, 2009; Babizhayev, 2011; Fletcher, 2010). Multiple antioxidant systems exist to temper the overproduction of free radicals, including reducing agents such as glutathione, manganese superoxide dismutase (MnSOD), and amino acid moieties including methionine and cysteine (Brennan & Kantorow, 2009). The lens appears particularly susceptible to disruption of these antioxidant defenses, as free radical overproduction impairs the transparency of the lens (Lou, 2003).
Other genetic and metabolic causes of cataracts
Known causes of cataract formation are vast, encompassing both genetic and non-genetic etiologies. Over 17 genes and 30 loci have been implicated in either autosomal dominant or recessive forms of congenital cataracts, including those related to the γ-, α-, and β-crystallins, as well as other membranous and filament-related proteins (Schmidt et al., 2008; Graw et al., 2009). Schmidt et al. (2008) postulated that autosomal recessive triangular cataracts result from mutations in GJA8, which encodes a protein involved in the gap-junctions that are crucial to maintaining membrane homeostasis (Reddy et al., 2004). However, the GJA8 mutation in question was subsequently shown to be a rare polymorphism rather than a disease-causing mutation (Graw et al., 2009). Non-genetic causes of cataracts include infection, malnutrition, drug exposure, trauma, aging, and radiation (Churchill & Graw, 2011).
Cataract formation can also result from disruption of normal intermediary metabolic flux within the ocular lens. The metabolic differential diagnosis of cataracts is commonly broken down by age at presentation. Metabolic causes are common in cataracts presenting in the newborn and childhood periods, including galactosemia, peroxisomal disease, disorders of carbohydrate metabolism, amino acid disorders, lipid disorders, as well as mitochondrial disease (Fernandes et al, 2006). The wide range of disorders that can cause cataract formation demonstrates the sensitivity of the ocular lens to genetic changes that impair its metabolic activity.
The lens itself is indeed a highly metabolically active tissue. It is comprised of three separate layers named the epithelium, the cortex, and the lens nucleus (Vinson, 2006), each of which is susceptible to cataract formation. The cells of the lens age and become less metabolically active as they progress centrally toward the nucleus. The outermost epithelial layer has the most frequent and rapid cell turnover and is subject to subcapsular cataract formation. Cortical cataracts can form in the inner and outer cortical layers. Nuclear cataracts can form in the innermost and metabolically quiet nuclear core of the lens. Nonetheless, some metabolic activity must still be integral to normal nuclear lens function as evidenced by our proband with a pathogenic mtDNA mutation in the tRNASer(AGY) gene developing progressively dense nuclear cataracts in his late teenage years.
Mitochondrial tRNASer mutations
Nuclear and mtDNA genes both contribute to normal mitochondrial function. The mtDNA genome is comprised of 37 genes, which encode 13 structural protein components of the five complex energy-generating mitochondrial respiratory chain, as well as 2 ribosomal RNA (rRNA) genes and 22 mitochondrial transfer RNA (tRNA) genes. One tRNA gene exists in the mtDNA genome for each amino acid except for leucine and serine, each of which has two distinct tRNAs. The two tRNASer genes differ only by the nucleotide of the third position, designated as either serine-UCN (tRNASer(UCN)) or serine-AGY (tRNASer(AGY)). The tRNASer(AGY) m.12264C>T point mutation that we identified in our patient is a novel mtDNA mutation that disrupts G–C pairing at the last base of the amino acid acceptor stem, which is presumably important in the maintenance of the conserved tRNA structure for the recognition of precursor RNA processing, the addition of the 3′ CCA tail, and amino acylation (Figure 3). Point mutations in tRNAs can affect the molecule in many ways. Possible mechanisms include disruption in the aminoacylation process (Scaglia and Wong, 2008), instability of the tRNA due to abnormal secondary structure, post-transcriptional modifications (Yarham et al., 2010), and/or abnormal transcription and translation (Shutt and Shadel, 2010). In addition, our proband’s skeletal muscle biopsy findings of mildly reduce complex I–III activity is consistent with the known percentage of Ser(AGY) being highest in complex I and then complex III (Wong et al., 2006).
Mutations in both tRNASer genes have previously been implicated in the pathogenesis of multisystemic mitochondrial diseases with ophthalmologic manifestations. Distinct point mutations in four tRNA genes, including tRNASer(UCN), were first identified in subjects with either chronic progressive external ophthalmoplegia (CPEO) or chronic intestinal pseudoobstruction with myopathy and ophthalmoplegia (CIPO) (Lauber et al., 1991). Three different tRNASer(UCN) mutations were subsequently reported following whole mtDNA genome sequencing in a large cohort of French individuals with non-syndromic hearing loss (Leveque et al., 2007). tRNASer(UCN) mutations were similarly identified in a Han Chinese family affected with maternally inherited sensorineural hearing loss (Tang et al., 2010).
Only a handful of mutations have been reported in the tRNASer(AGY) gene that was implicated in our proband (Scaglia & Wong, 2008). A large Irish kindred with features of sensorineural deafness and retinitis pigmentosa reminiscent of Usher syndrome type III (OMIM 276902) was found to have an m.12258C>A mtDNA mutation in tRNASer(AGY) (Mansergh et al., 1999). The same m.12258C>A mutation was identified in a second family affected with cataracts, cerebellar ataxia, diabetes, and deafness (Lynn et al., 1998), where their muscle and blood were found to be heteroplasmic for the mutation but cataract tissue was not examined. In addition, an m.12207G>A mutation in tRNASer(AGY) that affected the same G–C pairing as does the m.12264C>T mutation was identified in an individual with combined clinical features of myoclonic epilepsy with ragged red fibers (MERRF) and mitochondrial encephalopathy, lactic acidosis, and stroke (MELAS), diseases which are typically associated with mtDNA mutations in the tRNA-lysine and tRNA-leucine genes, respectively (Wong et al., 2006). Similar to our proband, the subject with the m.12207G>A tRNASer(AGY) mutation had high heteroplasmy load, mitochondrial proliferation, relative complex I–III deficiency, lactic acidemia, and hearing loss. However, our proband with the m.12264C>T tRNASer(AGY) mutation also had significant cardiac, endocrine, and ophthalmologic involvement but lacked any hepatic features. Finally, an individual who presented with a progressive myopathy, deafness, and sporadic seizures was recently found by whole mitochondrial genome sequencing to harbor a novel m.12262C>A tRNASer(AGY) mutation (Cardaioli et al., 2011). Their findings reinforce the multisystem effects of tRNASer mtDNA mutations, although no ophthalmologic features were reported in that case.
CONCLUSION
Mitochondrial diseases, while individually uncommon, are an increasingly recognized and diagnosable cause of a variety of multi-systemic problems. When faced with a patient with progressive, multisystem organ involvement, mitochondrial disease should be raised in the differential diagnosis. Rather than obtaining blood-based testing solely for common point mutations in mtDNA, this report supports the utility of pursuing whole mtDNA genome sequencing in clinically affected tissues as part of the diagnostic evaluation of suspected mitochondrial disease. It further illustrates the impact of primary mitochondrial dysfunction on ophthalmologic pathology, which in our proband involved severely reduced visual acuity due to retinitis pigmentosa and progressive nuclear cataracts. Most interestingly, the novel m.12264C>T tRNASer(AGY) mutation identified at low heteroplasmy load in our proband’s blood was confirmed to be homoplasmic within his muscle, buccal cells, and extracted cataract tissue. This is the first report to our knowledge to verify a pathogenic mtDNA mutation as the cause of cataracts, and clearly illustrates the important contribution of normal metabolic activity to the function of the ocular lens.
Acknowledgments
This work was funded in part by a grant from the National Institutes of Health (R03-DK082446) to M.J.F.
Footnotes
AUTHOR CONTRIBUTIONS
MJF, EP, EAP, and WA clinically evaluated the proband. WA performed cataract extraction surgery. JG and MS performed histopathologic studies. LJW and JQJ performed molecular analyses. SAS and MJF wrote the manuscript.
DISCLOSURE
All authors have reported no conflicts of interest.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Almousa R, Charlton A, Rajesh ST, Sundar G, Amrith S. Optimizing muscle biopsy for the diagnosis of mitochondrial myopathy. Ophthal Plast Reconstr Surg. 2009;25:366–370. doi: 10.1097/IOP.0b013e3181b2fd06. [DOI] [PubMed] [Google Scholar]
- Babizhayev MA, Yegorov YE. Reactive oxygen species and the aging eye: specific role of metabolically active mitochondria in maintaining lens function and in the initiation of the oxidation-induced maturity onset cataract-a novel platform of mitochondria-targeted antioxidants with broad therapeutic potential for redox regulation and detoxification of oxidants in eye diseases. Am J Ther. doi: 10.1097/MJT.0b013e3181ea31ff. epub ahead of print, Oct. 22, 2010. [DOI] [PubMed] [Google Scholar]
- Babizhayev MA. Mitochondria induce oxidative stress, generation of reactive oxygen species and redox state unbalance of the eye lens leading to human cataract formation: disruption of redox lens organization by phospholipid hydroperoxides as a common basis for cataract disease. Cell Biochem Funct. 2011;29(3):183–206. doi: 10.1002/cbf.1737. [DOI] [PubMed] [Google Scholar]
- Brennan LA, Kantorow M. Mitochondrial function and redox control in the aging eye: role of MsrA and other repair systems in cataract and macular degenerations. Exp Eye Res. 2009;88(2):195–203. doi: 10.1016/j.exer.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardaioli E, Malfatti E, Da Pozzo P, Gallus GN, Carluccio MA, Rufa A, Volpi N, Dotti MT, Federico A. Progressive mitochondrial myopathy, deafness, and sporadic seizures associated with a novel mutation in the mitochondrial tRNASer(AGY) gene. J Neurol Sci. 2011;303(1–2):142–145. doi: 10.1016/j.jns.2010.12.020. [DOI] [PubMed] [Google Scholar]
- Churchill A, Graw J. Clinical and experimental advances in congenital and paediatric cataracts. Philos Trans R Soc Lond B Biol Sci. 2011;366(1568):1234–1249. doi: 10.1098/rstb.2010.0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciulla TA, North K, Mccabe O, Anthony DC, Korson MS, Petersen RA. Bilateral infantile cataractogenesis in a patient with deficiency of complex I, a mitochondrial electron transport chain enzyme. J Pediatr Ophthalmol Strabismus. 1995;32(6):378–382. doi: 10.3928/0191-3913-19951101-11. [DOI] [PubMed] [Google Scholar]
- Cursiefen C, Kuchle M, Scheurlen W, Naumann GO. Bilateral zonular cataract associated with the mitochondrial cytopathy of Pearson syndrome. Am J Ophthalmol. 1998;125(2):260–261. doi: 10.1016/s0002-9394(99)80105-6. [DOI] [PubMed] [Google Scholar]
- Di Rosa G, Deodato F, Loupatty FJ, Rizzo C, Carrozzo R, Santorelli FM, Boenzi S, D’Amico A, Tozzi G, Bertini E, Maiorana A, Wanders RJ, Dionisi-Vici C. Hypertrophic cardiomyopathy, cataract, developmental delay, lactic acidosis: a novel subtype of 3-methylglutaconic aciduria. J Inherit Metab Dis. 2006;29(4):546–550. doi: 10.1007/s10545-006-0279-y. [DOI] [PubMed] [Google Scholar]
- Fernandes J, Saudubray J, van den Berghe G, Walter J. Inborn Metabolic Diseases: Diagnosis and Treatment. 4. Springer-Verlag; Berlin, Germany: 2006. [Google Scholar]
- Fletcher AE. Free radicals, antioxidants and eye diseases: evidence from epidemiological studies on cataract and age-related macular degeneration. Ophthalmic Res. 2010;44(3):191–198. doi: 10.1159/000316476. [DOI] [PubMed] [Google Scholar]
- Graw J, Schmidt W, Minogue PJ, Rodriguez J, Tong JJ, Klopp N, Illig T, Ebihara L, Berthoud VM, Beyer EC. The GJA8 allele encoding CX50I247M is a rare polymorphism, not a cataract-causing mutation. Mol Vis. 2009;15:1881–1885. [PMC free article] [PubMed] [Google Scholar]
- Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, Cohen BH. Mitochondrial disease: a practical approach for primary care physicians. Pediatrics. 2007;120(6):1326–1333. doi: 10.1542/peds.2007-0391. [DOI] [PubMed] [Google Scholar]
- Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, Wong LJ, Cohen BH, Naviaux RK. The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab. 2008;94(1):16–37. doi: 10.1016/j.ymgme.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Q, Chang L, Vandenberg D, Stanczyk FZ, Stolz A. Selective reduction of AKR1C2 in prostate cancer and its role in DHT metabolism. Prostate. 2003;54(4):275–289. doi: 10.1002/pros.10192. [DOI] [PubMed] [Google Scholar]
- Jordens EZ, Palmieri L, Huizing M, Van Den Heuvel LP, Sengers RC, Dorner A, Ruitenbeek W, Trijbels FJ, Valsson J, Sigfusson G, Palmieri F, Smeitink JA. Adenine nucleotide translocator 1 deficiency associated with Sengers syndrome. Ann Neurol. 2002;52(1):95–99. doi: 10.1002/ana.10214. [DOI] [PubMed] [Google Scholar]
- Lauber J, Marsac C, Kadenbach B, Seibel P. Mutations in mitochondrial tRNA genes: a frequent cause of neuromuscular diseases. Nucleic Acids Res. 1991;19(7):1393–1397. doi: 10.1093/nar/19.7.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveque M, Marlin S, Jonard L, Procaccio V, Reynier P, Amati-Bonneau P, Baulande S, Pierron D, Lacombe D, Duriez F, Francannet C, Mom T, Journel H, Catros H, Drouin-Garraud V, Obstoy MF, Dollfus H, Eliot MM, Faivre L, Duvillard C, et al. Whole mitochondrial genome screening in maternally inherited non-syndromic hearing impairment using a microarray resequencing mitochondrial DNA chip. Eur J Hum Genet. 2007;15(11):1145–1155. doi: 10.1038/sj.ejhg.5201891. [DOI] [PubMed] [Google Scholar]
- Lou MF. Redox regulation in the lens. Prog Retin Eye Res. 2003;22(5):657–682. doi: 10.1016/s1350-9462(03)00050-8. [DOI] [PubMed] [Google Scholar]
- Lynn S, Wardell T, Johnson MA, Chinnery PF, Daly ME, Walker M, Turnbull DM. Mitochondrial diabetes: investigation and identification of a novel mutation. Diabetes. 1998;47(11):1800–1802. doi: 10.2337/diabetes.47.11.1800. [DOI] [PubMed] [Google Scholar]
- Mansergh FC, Millington-Ward S, Kennan A, Kiang AS, Humphries M, Farrar GJ, Humphries P, Kenna PF. Retinitis pigmentosa and progressive sensorineural hearing loss caused by a C12258A mutation in the mitochondrial MTTS2 gene. Am J Hum Genet. 1999;64(4):971–985. doi: 10.1086/302344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklas JA, Buel E. Simultaneous determination of total human and male DNA using a duplex real-time PCR assay. J Forensic Sci. 2006;51(5):1005–1015. doi: 10.1111/j.1556-4029.2006.00211.x. [DOI] [PubMed] [Google Scholar]
- Reddy MA, Bateman OA, Chakarova C, Ferris J, Berry V, Lomas E, Sarra R, Smith MA, Moore AT, Bhattacharya SS, Slingsby C. Characterization of the G91del CRYBA1/3-crystallin protein: a cause of human inherited cataract. Hum Mol Genet. 2004;13(9):945–953. doi: 10.1093/hmg/ddh110. [DOI] [PubMed] [Google Scholar]
- Ribes A, Riudor E, Valcarel R, Salva A, Castello F, Murillo S, Dominguez C, Rotig A, Jakobs C. Pearson syndrome: altered tricarboxylic acid and urea-cycle metabolites, adrenal insufficiency and corneal opacities. J Inherit Metab Dis. 1993;16(3):537–540. doi: 10.1007/BF00711675. [DOI] [PubMed] [Google Scholar]
- Scaglia F, Wong LJ. Human mitochondrial transfer RNAs: role of pathogenic mutation in disease. Muscle Nerve. 2008;37(2):150–171. doi: 10.1002/mus.20917. [DOI] [PubMed] [Google Scholar]
- Schmidt W, Klopp N, Illig T, Graw J. A novel GJA8 mutation causing a recessive triangular cataract. Mol Vis. 2008;14:851–856. [PMC free article] [PubMed] [Google Scholar]
- Schrier SA, Falk MJ. Mitochondrial disorders and the eye. Curr Opin Ophthalmol. 2011;22(5):325–331. doi: 10.1097/ICU.0b013e328349419d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengers RC, Trijbels JM, Willems JL, Daniels O, Stadhouders AM. Congenital cataract and mitochondrial myopathy of skeletal and heart muscle associated with lactic acidosis after exercise. J Pediatr. 1975;86(6):873–880. doi: 10.1016/s0022-3476(75)80217-4. [DOI] [PubMed] [Google Scholar]
- Shutt T, Shadel G. A compendium of human mitochondrial gene expression machinery with links to disease. Environ Mol Mutagen. 2010;51(5):360–79. doi: 10.1002/em.20571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Li R, Zheng J, Cai Q, Zhang T, Gong S, Zheng W, He X, Zhu Y, Xue L, Yang A, Yang L, Lu J, Guan MX. Maternally inherited hearing loss is associated with the novel mitochondrial tRNA Ser(UCN) 7505T>C mutation in a Han Chinese family. Mol Genet Metab. 2010;100(1):57–64. doi: 10.1016/j.ymgme.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Vinson JA. Oxidative stress in cataracts. Pathophysiology. 2006;13(3):151–162. doi: 10.1016/j.pathophys.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Wong LJ, Yim D, Bai RK, Kwon H, Vacek MM, Zane J, Hoppel CL, Kerr DS. A novel mutation in the mitochondrial tRNA(Ser(AGY)) gene associated with mitochondrial myopathy, encephalopathy, and complex I deficiency. J Med Genet. 2006;43(9):e46. doi: 10.1136/jmg.2005.040626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarham K, Elson J, Blakely E, McFarland R, Taylor R. Mitochondrial tRNA mutations and disease. Wiley Interdiscip Rev. 2010;1(2):304–24. doi: 10.1002/wrna.27. [DOI] [PubMed] [Google Scholar]
- Zaragoza MV, Brandon MC, Diegoli M, Arbustini E, Wallace DC. Mitochondrial cardiomyopathies: how to identify candidate pathogenic mutations by mitochondrial DNA sequencing, MITOMASTER and phylogeny. Eur J Hum Genet. 2011;19(2):200–207. doi: 10.1038/ejhg.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]