

Summary
Focal adhesion kinase (FAK) is a critical mediator of matrix- and growth factor-induced signaling during development. Myocyte-restricted FAK deletion in mid-gestation mice results in impaired ventricular septation and cardiac compaction. However, whether FAK regulates early cardiogenic steps remains unknown. To explore a role for FAK in multi-chambered heart formation, we utilized anti-sense morpholinos to deplete FAK in Xenopus laevis. Xenopus FAK morphants exhibited impaired cardiogenesis, pronounced pericardial edema, and lethality by tadpole stages. Spatial-temporal assessment of cardiac marker gene expression revealed that FAK was not necessary for midline migration, differentiation, fusion of cardiac precursors, or linear heart tube formation. However, myocyte proliferation was significantly reduced in FAK morphant heart tubes and these tubes failed to undergo proper looping morphogenesis. Collectively our data imply that FAK plays an essential role in chamber outgrowth and looping morphogenesis likely stimulated by fibroblast growth factors (and possibly other) cardiotrophic factors.
Keywords: cardiogenesis, integrins, development, mitogen, Xenopus
INTRODUCTION
Heart formation involves an intricate and complex series of events that must occur in a coordinated spatial and temporal manner. The heart develops from bilaterally symmetric cardiogenic primordia that migrate and fuse at the embryonic midline, proliferate, and form a primitive heart tube (Dehaan, 1963; Goetz and Conlon, 2007; Kolker et al., 2000; Mohun et al., 2000, 2003; Trinh and Stainier, 2004a). In species with multichambered hearts the primitive heart tube rapidly undergoes looping morphogenesis and maturation to form a fully functioning heart with outflow tracts aligned with the vasculature.
Appropriate cardiac morphogenesis requires regional coordinated recruitment, differentiation, and proliferation of cardiomyocytes. A large body of evidence indicates that signals secreted from the endoderm including soluble factors (e.g., fibroblast growth factors (FGFs), bone morphogenic proteins (BMPs) and extracellular matrix (ECM) proteins (e.g., fibronectin) are prominent inducers of the cardiogenic field (Ahuja et al., 2007; Chen et al., 2004; Choi et al., 2007; Lavine and Ornitz, 2008; Lavine et al., 2005). Via incompletely understood mechanisms, these signals induce the expression of critical myocardial transcription factors in discrete (often chamber-specific) locales. Notably, genetic studies in chick, zebrafish, frogs, and mice have revealed that spatiotemporal induction of myocyte enhancement factor 2 (MEF2) and GATA factors are necessary for regulated myocyte differentiation, while Nkx2.5, Tbx2, and Tbx3 are required for coordinating myocyte proliferation (Moorman and Christoffels, 2003; Srivastava and Olson, 2000; Zaffran and Frasch, 2002).
The non-receptor tyrosine kinase, focal adhesion kinase (FAK) is strongly activated by fibronectin-binding integrins (α5β1) and growth factors (Parsons, 2003) and is a likely candidate to integrate downstream signals from these diverse pathways during myocardial development. Indeed, studies by our group and others have indicated that myocyte-specific depletion of FAK (or inactivation of FAK) leads to early embryonic lethality associated with left ventricular noncompaction (DiMichele et al., 2009; Peng et al., 2008). Although these studies highlight an essential role for FAK during mid-gestational cardiac growth, a limitation of these studies is that FAK signaling is not significantly depleted in these hearts until embryonic day 13.5 (E13.5) or later, precluding determination of whether FAK activity is necessary for earlier stages of cardiac morphogenesis. Importantly, FAK is expressed in the mouse mesoderm prior to cardiogenesis (E7.5) and germ-line deletion of FAK in the mouse induces a variety of mesodermal defects and early embryonic death within a time frame either prior to or during looping morphogenesis (between E8.5-10.5 in mice) (Furuta et al., 1995). In stark contrast, depletion of the FAK ortholog FAK56 in Drosophila does not affect viability of this organism and no defects in heart formation were reported (Grabbe et al., 2004). Since the Drosophila heart consists of a linear tube that does not undergo the marked morphological changes that occur during multichambered heart formation, we speculated that looping morphogenesis may be a key developmental process regulated by FAK in higher organisms.
To study the possible requirement for FAK in this intricate morphogenetic event, we depleted FAK in Xenopus laevis by microinjection of inhibitory antisense morpholino oligonucleotides. This model system is particularly suited to studying cardiac development because of the ease of temporal analysis of morphogenetic events and because Xenopus do not require heart function to survive (at least until the late tadpole stage of development) since nutrient exchange readily occurs by diffusion. Herein, we show that FAK-depleted tadpoles exhibit abnormal myocardial morphogenesis accompanied by pericardial edema and early embryonic lethality. Our mechanistic studies reveal that FAK activation, likely by FGFs, facilitates myocyte proliferation in the prelooped heart tube, thus contributing to the complex process of looping morphogenesis.
RESULTS
Inhibition of FAK by Morpholino-Injection
FAK plays a critical function in murine development and is necessary for myocardial compaction. However, no studies to date have addressed a specific role for FAK in early cardiac development, specifically in regulating cardiac morphogenesis (DiMichele et al., 2009; Furuta et al., 1995; Peng et al., 2008). To study the time-dependent requirements for FAK during this intricate process, we depleted FAK protein in Xenopus, which develop a fully functioning three-chambered heart by 72 h post-fertilization. To this end, we designed two FAK-specific antisense morpholinos to target sequences either upstream of, or flanking, the start codon of Xenopus fak (denoted FAK Mo and xFAKst, respectively). Both morpholinos significantly reduced flag-tagged FAK protein production in an in vitro transcription/translation assay, with FAK Mo being slightly more potent (data not shown).
To establish that FAK Mo effectively blocks FAK translation in vivo, we injected standard quantities (20 and 40 ng) into single-cell fertilized Xenopus embryos. Western blot analysis of stage 22, 26, and 39 embryos confirmed that FAK protein levels were reduced in a dose-dependent manner (Fig. 1A). We next performed further temporal analysis of FAK levels during development in embryos injected with 40 ng of either control morpholino (Con Mo) or FAK Mo. As previously reported, we found that a low level of maternal FAK was apparent in fertilized eggs which persisted throughout the onset of gastrulation (stage 10.5) at which time embryonic FAK protein was markedly induced (Hens and DeSimone, 1995). As expected, maternal FAK was not depleted by FAK Mo, which was designed to block translation of nascent transcripts. However, injection of FAK Mo at the one-cell stage did reduce embryonic FAK levels by Stage 10.5 and FAK protein was nearly undetectable by Western analysis in the morphants during cardiogenesis (Stage 28–39; Fig. 1A, B). Densitometric analysis of Western blot band intensities demonstrated that FAK protein expression in FAK morphant embryos was reduced by greater than 80% as compared with controls by Stage 28 (Fig. 1C). FAK activity was also ablated in FAK-depleted embryos at these stages as assessed by Western blotting for phospho-FAK with Y-397 antibody (data not shown). Importantly, no changes in FAK expression were evident after injection of a control five-base mismatch morpholino (data not shown). Moreover, Western blot analysis for the protein tyrosine kinase, PYK2, demonstrated that FAK Mo did not disrupt the translation of this closely related FAK family member (Fig. 1D).
FIG. 1.

Depletion of FAK in Xenopus laevis leads to pericardial edema. A: FAK or Control morpholinos (20 and 40 ng) were injected into fertilized oocytes and embryonic FAK protein levels were assessed at the indicated stages by Western blotting. Levels of ERK are shown as a control for loading. B: Western blot analysis for FAK in Con Mo- and FAK Mo-injected embryos (40 ng/embryo) at the indicated stages of development. Levels of ERK are shown as a control for loading. C: Densitometric analysis of Western blots comparing FAK band intensity relative to ERK. Data are presented as FAK levels in FAK Mo-embryos relative to Con Mo-embryos (set to 1) at each developmental stage analyzed. D: Western blot analysis for PYK2 (and ERK) in stage 30 Con Mo- and FAK Mo-injected embryos. E: Gross morphology of Control and FAK morphant tadpoles at Stage 37. FAK morphants exhibit a slightly shortened anteroposterior axis and pericardial edema (arrow).
FAK-Depletion Results in Abnormal Cardiac Morphogenesis
FAK morphant embryos exhibited a slight developmental delay beginning around Stage 10 but underwent normal gastrulation and neurulation as assessed by gross morphology (data not shown). Furthermore, in situ hybridization analysis demonstrated that the mesodermal markers, chordin, brachyury, and Myo-D were expressed in the appropriate spatiotemporal pattern in both Con Mo- and FAK Mo-injected embryos at stages 10, 16, and 22 (Supporting Information Fig. 1) suggesting that early mesodermal development was also unperturbed in the FAK morphants. The lack of effect on these early developmental processes is not surprising given that heterozygous FAK null mice are viable (Ilic et al., 1995) and that FAK protein levels in our model system were not reduced by greater than 50% until after commencement of these critical developmental stages (Fig. 1C).
Interestingly, by Stage 34, a large percentage of FAK-depleted embryos (79.6%) showed pronounced pericardial edema (Fig. 1E and Table 1), indicating a morphogenetic cardiac abnormality. Indeed, whole mount immunohistochemical staining of stage 39 hearts with cardiac myosin heavy chain (MHC) or tropomyosin antibodies revealed marked dys-morphogenesis in the majority of FAK morphant embryos. Although all hearts in Con Mo-injected embryos were fully looped with three distinct chambers, FAK Mo-injected embryonic hearts exhibited bent or twisted heart tubes that failed to fully undergo looping (see Fig. 2). Other less penetrant phenotypes included anterior defects (such as decreased head size), a shortened antero-posterior axis, and developmental arrest prior to Stage 30 (Supporting Information Table 1). Embryos that exhibited signs of these less frequent abnormalities were excluded from subsequent analysis of cardiac morphology. Notably, all FAK-depleted tadpoles died by Stage 42 indicating an essential role for FAK in Xenopus development.
Table 1.
FAK Depletion Leads to Marked Pericardial Edema
| Con Mo | FAK Mo | |
|---|---|---|
| Trial 1 | 0%(n = 51) | 85.2% (n = 27) |
| Trial 2 | 2.5% (n = 79) | 92.3% (n = 78) |
| Trial 3 | 4%(n = 26) | 61.4% (n = 44) |
Embryos were scored for pericardial edema between Stages 34 and 39 in three separate trials.
FIG. 2.

FAK morphant embryos exhibit marked cardiac dysmorphogenesis. A: Lateral view of whole-mount immunohistochemistry for tropomyosin reveals a fully looped three-chambered heart in Con Mo-injected embryos while those injected with FAK Mo appear distended and partially looped (middle panel) or unlooped (right panel). Anterior is to the left and dorsal toward the top in all panels. Regions of interest are labeled as follows: i, inflow tract; v, ventricle; o, outflow tract. B: Western blot analysis for FAK in uninjected, Con Mo-injected, FAK Mo-injected, and rescue embryos at Stage 37. Levels of ERK are shown as a control for loading. C: Lateral view of whole-mount immunohisto-chemistry for MHC reveals rescue of the FAK morphant phenotype is achieved by coexpression of 2 ng chicken FAK. D: Heart morphology analysis of Con Mo-injected, FAK Mo-injected, rescue (FAK Mo and 2 ng chicken FAK co-injection), and 2 ng chicken FAK alone demonstrates that while FAK morphant embryos exhibit full looping in only 30% of embryos examined, rescue embryos exhibit full looping morphology in 67%. Total number of embryos analyzed were n = 27 (Con Mo), n = 34 (FAK Mo), n = 42 (Rescue), n = 20 (2 ng c-FAK), collected from two separate experiments.
To further confirm that looping morphogenesis was dependent on FAK, we utilized a phenotypic rescue approach. After aligning the sequences of Xenopus and chicken fak, we reasoned that FAK Mo would not interfere with chicken FAK translation, and subsequent in vitro transcription/translation assays confirmed this assertion (data not shown). Thus, we injected embryos at the one cell stage with either FAK Mo or a combination of FAK Mo and 2 ng chicken FAK capped-RNA (cFAK; denoted “rescue”) and assessed cardiac morphology at Stages 37–39. Importantly, Western blot analysis confirmed that rescued embryos expressed FAK protein at near endogenous levels (Fig. 2B). We next assessed cardiac morphology using whole-mount MHC antibody staining and quantified properly looped hearts. As shown in Figure 2C, D, expression of 2 ng cFAK induced a demonstrable rescue of the morphant phenotype, as 67% of dually injected embryos exhibited normal three-chambered hearts, compared to only 30% observed following injection of FAK Mo alone. Collectively, these results demonstrate that FAK is required for appropriate morphogenesis of the multichambered heart.
FAK Morphants Exhibit Appropriate Specification, Midline Migration, and Differentiation of Cardiac Precursors
To define the FAK-dependent mechanisms involved in chamber morphogenesis, we analyzed control and FAK-depleted hearts at various stages of development. As noted above, the initial step of heart formation involves the bilateral movement of cardiac progenitors (cardioblasts) to the ventral midline of the embryo where they fuse prior to formation of the linear heart tube, a process that occurs between stages 28 and 32 in Xenopus embryos. We performed in situ hybridization to examine the spatiotemporal expression pattern of several cardioblast marker genes to determine whether the cardiac precursors were specified and properly redistributed in the FAK morphants. The nkx2.5, tbx5, and tbx20 expression domains of Con Mo- and FAK Mo-injected embryos were comparable between Stages 26–32 and semiquantitative PCR analysis confirmed similar transcript levels of these cardioblast markers in Con Mo- and FAK Mo-injected embryos at Stages 30 and 32 (Fig. 3A, B and data not shown). Moreover, the myocyte differentiation markers tropomyosin and troponin I were expressed at similar levels in FAK Mo- and Con Mo-injected embryos as assessed by semiquantitative RT-PCR and immunohistochemistry (Fig. 3C and data not shown). Indeed, 3D rendering of MHC-stained Stage 30 FAK morphant embryos revealed a continuous sheet of myocytes, confirming appropriate differentiation and fusion at the ventral midline (Fig. 3D). Moreover, despite subsequent aberrant morphogenesis, FAK-depleted cardiac tissue exhibited normal rhythmic contractions (data not shown). Collectively, these results indicate that specified cardiac precursors migrated to the ventral mid-line and expressed markers of terminal differentiation in a FAK-independent manner.
FIG. 3.

FAK depletion does not impact cardiac specification or differentiation. A: In situ hybridization of Stage 30 embryos for TBX5 and NKX2.5. Ventral views (left panels) are oriented with anterior toward the top, lateral views (right panels) are oriented with anterior to the left and dorsal to the top. B, C: RT-PCR analysis at Stages 30 (left panels) and 32 (right panels) for TBX5, TBX20, NKX2.5 (B), and tropomyosin (tm), and Troponin T (TnT) (C). Histone H4 (H4) serves as a control. Data represent results from 10 embryos per condition and experiments were repeated at least twice. D: Ventral and front views of whole-mount immunohistochemistry for MHC reveals a continuous and fused sheet of differentiated myocytes at the ventral midline (indicated by white arrow) in FAK Mo-injected embryos. Images represent 3D reconstructions of confocal z-stack sections.
FAK Morphants Form Linear Heart Tubes but Fail to Undergo Appropriate Looping Morphogenesis
Considering that the initial stages of cardiogenesis were not affected by FAK depletion, we hypothesized that FAK may play an integral role in looping morphogenesis. To test this possibility, we performed additional whole-mount tropomyosin antibody staining and analyzed cardiac morphology using laser scanning confocal microscopy and 3D isosurfacing. By Stage 32, when the heart tube is undergoing closure at the dorsal surface, Con Mo- and FAK Mo- injected embryos appeared similar (Fig. 4A); however, analysis of the dorsal aspect of the heart revealed that complete closure of the heart tube had not occurred in FAK morphant embryos at this time (Supporting Information Fig. 2). Despite this delay, all FAK morphant embryos analyzed at later stages exhibited fully closed heart tubes (Supporting Information Fig. 2). However, analysis of FAK morphants at Stage 34 onward demonstrated a marked deficiency in heart tube looping. Indeed, the majority of Stage 34 FAK morphant hearts exhibited a straight or only slightly bent appearance (Fig. 4B and Supporting Information Fig. 2). Notably, injection with either Con Mo or five-base mismatch morpholinos did not affect cardiac morphogenesis when compared with uninjected embryos, indicating that the phenotypes observed were due to FAK depletion.
FIG. 4.

FAK depletion impairs looping morphogenesis. Whole mount immunohisto-chemical staining for tropomyosin was performed on Stage 32 (A), and 34 (B) embryos that were injected with either Con Mo (left) or FAK Mo (right) at the one-cell stage. Images represent 3D reconstructions of confocal z-stack sections (top) and Imaris isosurfacing (bottom). Stage 32 view is from the posterior looking through the heart tube toward the anterior end; dorsal is to the top. Stage 34 view is lateral with anterior to the left, dorsal to the top. Note that looping is perturbed in Stage 34 FAK morphants. Arrows point to region of interest where the heart takes on a spiral shape indicative of looping morphogenesis.
Myocyte Proliferation Is Reduced in FAK Morphant Heart Tubes
Since differential rates and/or locales of cell proliferation within the heart tube have been correlated with appropriate looping morphogenesis (Ribeiro et al., 2007), we determined whether cardiomyocyte proliferation was impaired in FAK-depleted embryos. To test this possibility, we immunostained Con Mo- and FAK Mo-injected embryonic heart tubes for tropomyosin and phospho-specific histone H3 (pH3) and imaged the hearts using laser scanning confocal microscopy. As shown in Figure 5, we observed a significant decrease in the total number of pH3-positive cardiomyocytes in pre-looped (Stage 32) FAK Mo hearts relative to controls. Notably, pH3 staining in the surrounding noncardiac tissue was comparable between Con Mo- and FAK Mo-injected embryos, indicating that FAK depletion did not induce a global reduction in cellular proliferation (Fig. 5B). Double-immunolabeling with tropomyosin and cleaved caspase 3 antibodies revealed no significant difference in cardiomyocyte apoptosis between Con Mo-and FAK Mo-injected embryos (Supporting Information Fig. 3), consistent with our previous findings that FAK depletion does not induce apoptosis in developing mouse hearts (Hakim et al., 2007). Collectively, these data indicate that the looping defect found in FAK morphant embryos may be due, at least in part, to impaired cardiomyocyte proliferation.
FIG. 5.

Myocyte mitosis is attenuated in FAK morphant heart tubes. A: Whole mount immunohistochemical staining for tropomyosin (green) and phospho-Histone H3 (red) was performed on prelooped (Stage 32) embryos that were injected with either Con Mo (left) or FAK Mo (right) at the one-cell stage. Images represent 3D reconstructions of confocal z-stack sections (top panels) or a single optical section (bottom panels). B: Total number of pH3 positive myocytes and noncardiac cells were counted in each optical section of Con Mo- and FAK Mo-injected embryos (19 embryos were analyzed per condition, collected from at least three separate experiments).
FAK Regulates FGF-Dependent Myocyte Proliferation
Proper development of the myocardium is dependent on the close interaction with (and signaling from) the endo- and epicardium. FGFs are potent regulators of embryonic myocyte proliferation (Lavine and Ornitz, 2008; Lavine et al., 2008), and given evidence from our lab that FAK activity is required for FGF-induced Map kinase signaling (DiMichele et al., 2009) and that inactivation of FAK phenocopies the noncompaction defects observed in FGF Receptor 1 (FGFR1) knock-out mice, we reasoned that FAK may play a major role in regulating cardiac morphogenesis mediated by epicardial-derived FGFs.
To test whether FGF signaling was required for Xenopus heart looping, we treated embryos with a selective FGFR1 tyrosine kinase inhibitor, SU5402 (Langdon et al., 2007; Mohammadi et al., 1997). As shown in figure 6, continuous exposure of embryos to SU5402 (50 μM) during cardiogenesis (Stages 25–37) resulted in small and dysmorphic hearts, although the embryos appeared relatively normal in size and shape (Fig. 6A, B). Importantly, the hearts of the SU5402-treated embryos resemble the FAK morphant hearts in that they fail to undergo looping (Fig. 6B and Table 2). This phenotype was highly penetrant (observed in 94% of treated embryos) while no looping defects were observed in embryos treated with DMSO (vehicle). These data indicate that both FGF- and FAK-dependent signals play an important role in coordinating chamber growth and morphogenesis.
FIG. 6.
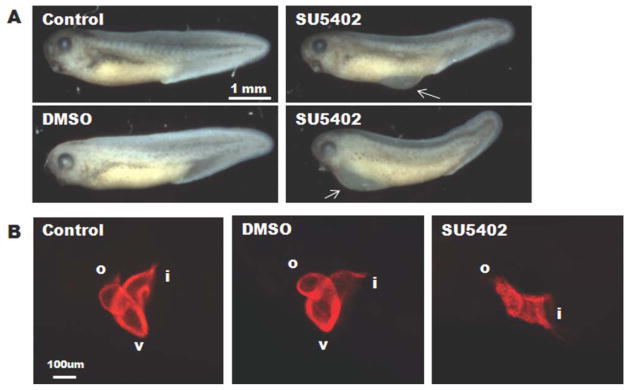
Treatment with the FGFR1-inhibitor, SU5402, impairs cardiac looping. Widefield microscopic analysis (A) of gross morphology of untreated control, DMSO-treated (2.5 μM), or SU5402-treated (50 μM) embryos at Stage 37 reveals that SU5402 treatment induces edema in the middle-ventral region of the embryo (top right panel) or pericardial edema (bottom right panel) in some embryos. Arrows point to regions of edema. B: Lateral view of whole-mount immunohistochemistry for MHC reveals a fully looped three-chambered heart in untreated and DMSO-treated embryos while those treated with SU5402 appear unlooped. Anterior is to the left and dorsal is to the top in all panels. Total number of embryos analyzed for heart morphology were: n, 22 (untreated), n = 34 (DMSO), and n = 35 (SU5402) and were collected from three separate experiments.
Table 2.
Treatment With SU5402 Causes Defects in Cardiac Looping Morphology
| Phenotypic analysis after treatment with SU5402 | |||
|---|---|---|---|
| Treatment (No. of Embryos) | Untreated (n = 22) | DMSO (n = 34) | SU5402 (n = 35) |
| Anterior defect | 0 (0%) | 1 (3%) | 1 (3%) |
| Middle–ventral region edema | 0 (0%) | 0 (0%) | 4 (11%) |
| Pericardial edema | 0 (0%) | 0 (0%) | 12 (34%) |
| Unlooped heart | 0 (0%) | 0 (0%) | 33 (94%) |
Embryos were scored for gross morphology in three separate experiments. As shown, SU5402 induced edema and cardiac looping defects. Several embryos exhibited both edema and an unlooped heart, therefore the totals do not add up to 100%.
To determine whether FAK might coordinate mitogenic cues initiated by FGF, we first determined whether FGF stimulates FAK activity in rat primary cardiomyocytes in culture. Importantly, Western blot analysis showed that basic fibroblast growth factor (bFGF) induced a marked increase in FAK activity in cardiomyocytes and that SU5402, dramatically reduced this response (Fig. 7A). It is well established that the endogenous FAK inhibitor, FAK-related non-kinase (FRNK), can serve as a dominant interfering mutant that downregulates FAK-mediated signaling pathways (Richardson and Parsons, 1996). We previously generated FRNK adenoviruses that are well-suited for studying FAK-dependent processes in cultured cardiomyocytes. Therefore, to determine whether FAK was essential for FGF-stimulated myocyte proliferation, we infected embryonic rat cardiomyocytes with 10 multiplicity-of-infection (m.o.i) green fluorescent protein (GFP) or GFP-FRNK adenovirus (Fig. 7B) and examined the rate of proliferation of these cells in serum-free (SF) medium in the absence or presence of bFGF. GFP and GFP-FRNK were efficiently expressed in 100% of the cardiomyocyte population as determined by immunofluorescence (data not shown). Under these conditions, bFGF induced the rate of 5-bromo-2-deoxyuridine (BrdU) incorporation in GFP-infected cells by ~20%, whereas only 4% of FRNK-infected cells were BrdU-positive (Fig. 7C). Collectively, these data indicate that FAK activity is necessary for mediating FGFR-dependent cardiomyocyte proliferation, an event that may control both looping morphogenesis and growth of the embryonic heart.
FIG. 7.

FAK is activated by FGF and is necessary for FGF-dependent myocyte proliferation. A: Western blot analysis of cell lysates isolated from primary embryonic rat cardiomyocytes. Cells were maintained in serum free (SF) media and treated with bFGF (100 ng/mL) or vehicle (veh.) for 30 min with or without 10 min pretreatment with the specific FGFR-inhibitor, SU5402 (10 μM). Lysates were immunoblotted with antibodies directed towards phospho-specific Y397-FAK or total FAK. B: BrdU incorporation in isolated embryonic cardiomyocytes infected with GFP- or GFP-FRNK adenovirus (10 m.o.i). Cells were maintained in serum-free medium and treated with vehicle (not shown) or bFGF (100 ng/mL) for 24 h. Costaining with anticardiac troponin T (cTnT) was performed to identify cardiomyocytes. C: Quantification of BrdU- positive cardiomyocytes (means ± SEM; N = 3; minimum of 300 cells/condition). Scale bar is 20 μm.
DISCUSSION
Previous studies have demonstrated an essential role for FAK during embryonic development in mice but not flies (Grabbe et al., 2004; Ilic et al., 1995). FAK null mice exhibit general cardiovascular defects that were not reported in FAK-depleted Drosophila. A major notable difference between the cardiovascular systems of these two species is that Drosophila contains a simple linear heart tube that does not undergo looping morphogenesis. Although recent studies in myocyte-restricted FAK-depleted mouse hearts revealed that FAK is necessary for growth of the four chambered heart (DiMichele et al., 2009; Peng et al., 2008), no studies to date have addressed a role for FAK in regulating cardiac looping. Our current findings show that FAK-depleted Xenopus embryos exhibit pronounced pericardial edema, cardiac dysmorphogenesis, and embryonic death. Temporal assessment of cardiac morphogenesis revealed that FAK depletion did not affect the midline migration or differentiation of cardiac precursors. However, depletion of FAK markedly reduced proliferation in prelooped hearts in comparison to stage-matched controls and induced a profound defect in looping morphogenesis.
We found that FAK-depleted Xenopus embryos died by Stage 42, which is relatively late in development in comparison to FAK-depleted mice (E8-10.5). Prolonged survival in FAK-depleted Xenopus may be due, in part, to the enhanced nutrient exchange that is known to permit uncoupling of heart and embryonic development in this species until late tadpole stages. However, it is also possible that the presence of maternal FAK (which is refractory to the FAK morpholino) may have been sufficient to drive early FAK-dependent morphogenetic processes in the studies presented herein. Indeed, we found a low level of maternal FAK protein in both Con Mo- and FAK Mo-injected embryos until at least stage 10.5 (the onset of gastrulation), consistent with previous studies of FAK expression during Xenopus development (Hens and DeSimone, 1995). Therefore, while FAK-depleted Xenopus embryos gastrulate and neurulate normally, the role of FAK in gastrulation has not been addressed in this study. Nonetheless, the translation of embryonically expressed FAK protein was dramatically reduced in the FAK morphants from Stage 10.5 onwards, and FAK protein was nearly undetectable in these embryos during cardiogenesis. Thus, our findings that the migration of specified cardioblasts from the bilateral heart fields to the ventral midline and their differentiation into mature cardiomyocytes was not altered in the FAK morphants, indicates that these processes are both FAK-independent.
The finding that cardioblast migration was FAK-independent was somewhat surprising given that several published studies from our laboratory and others have indicated that FAK is essential for cell motility (Parsons, 2003). Notably, we previously showed that directional motility of cardiomyocytes requires FAK (Hakim et al., 2007). Moreover, previous studies in zebrafish have revealed that the coordinated motility of cardioblasts to the midline is regulated by fibronectin (Trinh and Stainier, 2004b) and studies in Drosophila revealed a strong genetic interaction between the Drosophila β1 integrin and the guidance cue, Slit, in regulating cardioblast movement (Engel et al., 2005; MacMullin and Jacobs, 2006). However, consistent with our finding that midline fusion of cardioblasts occurs normally in FAK-depleted Xenopus embryos, MacMullin et al. found no phenotypic interaction between FAK and Slit (MacMullin and Jacobs, 2006), leading us to reason that distinct integrin-induced signals regulate cardioblast and cardiomyocyte motility. Indeed, cardioblast motility involves a sheet-like movement that is known to be dependent on cell-cell interactions, whereas we have shown that myocytes exhibit directional motility that requires FAK-dependent lamellipodial protrusions (Hakim et al., 2007). While previous studies have indicated a role for FAK in scratch wound closure of a confluent monolayer of fibroblasts (Hsia et al., 2003; Sieg et al., 1999) we have found that FAK is not necessary for wound closure in smooth muscle cells, indicating that the mechanisms that regulate sheet-like movements may be cell type specific (unpublished observations, JMT).
Our mechanistic studies herein indicate that FAK is essential for regulating myocyte proliferation within the developing heart tube. We found that FAK morphant hearts exhibited a decrease in mitotically active myocytes relative to controls prior to looping. This finding is significant because recent studies in zebrafish have demonstrated that looping morphogenesis is associated with a shift from homogeneous proliferation to increased proliferation within the presumptive atrial and ventricular chambers (Ribeiro et al., 2007).
Several studies suggest that members of the FGF family (Itoh and Ornitz, 2004), including FGF-1 (Engelmann et al., 1991, 1993), bFGF (David et al., 2003; Pasumarthi et al., 1996), and FGF9 (Lavine et al., 2005) are critical endoderm-derived mediators of myoycte proliferation during myocardial development. Interestingly, our previous studies indicated that myocyte-restricted inactivation of FAK in mice phenocopies the noncompaction defects observed in mice with myocyte-restricted deletion of FGFR-1 and FGFR-2 (DiMichele et al., 2009). We now show that inhibition of either FAK or FGFR signaling in Xenopus leads to a failure of heart looping, that FGF stimulates FAK activity, and that FAK activation is necessary for FGFR-dependent myocyte proliferation in vitro. Since SU-5402 is an effective inhibitor of FGFR1, we hypothesize that FGFR1-dependent FAK activation is necessary for heart looping. However, it is formally possible that this compound may also inhibit other FGF receptor subtypes including FGFR4 and FGFR2 which have been reported to be expressed at higher levels than FGFR1 in the developing Xenopus heart (Lea et al., 2009). In terms of signaling, FGF receptors are linked to the MAPK signaling pathways which terminate in activation of ERK, c-Jun N-terminal kinase, and p38. Interestingly, several recent studies have indicated that p38 may function at the G2/M checkpoint to block cardiomyocyte cell cycle progression (Engel et al., 2005, 2006) and we recently showed that expression of FRNK (or inactivation of FAK) promotes p38 activity and regulates expression of the p38-dependent cell cycle modifier, p27kip (DiMichele et al., 2009). Thus, we hypothesize that FGFR-stimulated FAK activation leads to p38 repression and induction of the proliferative signals that may be necessary to drive looping morphogenesis.
Although FGFs provide mitogenic signals to embryonic cardiomyocytes, recent evidence in zebrafish indicates that FGFs are also necessary for recruitment and differentiation of the secondary heart field (SHF) (de Pater et al., 2009). This population of cells contributes to right ventricle and outflow tract formation in zebra-fish, chick, and mouse hearts, and was also recently identified in Xenopus (Brade et al., 2007; de Pater et al., 2009; Martinsen et al., 2004; Sadaghiani and Thiébaud, 1987). Although the precise timing and extent of SHF contribution during frog heart development remains unclear, studies performed in chick and mouse indicate that SHF is not typically recruited until after looping morphogenesis (de Pater et al., 2009; Martinsen et al., 2004; Sadaghiani and Thiébaud, 1987). Thus, while it is formally possible that the abnormal morphogenesis observed in FAK morphant hearts may, in part, arise from a defect in this FGF-dependent process, this is unlikely the primary cause of dysmorphogenesis, since FAK morphants exhibit defects in prelooped heart tubes.
In summary, our studies are the first to indicate that FAK is necessary for viability and cardiogenesis in Xenopus. FAK-depleted cardiac precursor cells migrated to the ventral midline, fused, and formed a linear heart tube. However, FAK morphant heart tubes exhibited a marked reduction in proliferating myocytes and a concomitant failure to undergo looping. Our studies in cultured cardiomyocytes confirmed that FAK regulates myocyte proliferation in a cell autonomous fashion and that FAK acts downstream of FGFRs to regulate this critical function.
MATERIALS AND METHODS
Embryo Culture and Microinjection
Preparation and injection of X. laevis embryos was carried out as previously described (Wilson and Hemmati-Brivanlou, 1995). Staging was performed according to Nieuwkoop and Faber (Nieuwkoop and Faber, 1994). Antisense morpholino oligonucleotides were designed against either the start site (xFAKst Mo) or the 5′-untranslated region (xFAKup Mo) of Fak. Sequences used were: xFAKup, 5′CTG ATG CTA GGT GTC TGT CAT ATT C 3′ and xFAKst, 5′TCC AGG TAA GCC GCA GCC ATA GCC T 3′. We utilized two control morpholinos, a five-base mismatched morpholino, in which five nucleotides of the xFAKup Mo sequence were changed such that the morpholino no longer reacted with Xenopus fak, and a standard control morpholino (Con Mo) (Gene Tools, Philomath, OR). Morpholinos were injected at a concentration of 40 ng/embryo at the one-cell stage, except where indicated. All in vivo data shown herein represent experiments performed with xFAKup (hereafter referred to as FAK Mo). Similar results were obtained with xFAKst Mo (data not shown).
Capped chicken FAK RNA used for rescue experiments was first generated using the mMessage/mMa-chine capped RNA kit (Ambion), according to the manufacturer’s instructions. RNA was quantified by spectrophotometry and diluted in RNase-free water. For overexpression, RNA was microinjected at a concentration of 2 ng/embryo in a 10 nL injection volume. For rescue experiments, FAK capped-RNA was mixed with FAK Mo in order to achieve 40 ng FAK Mo and 2 ng FAK capped-RNA per embryo (10 nL injection volume).
Inhibitor Treatments
Embryos for inhibitor experiments were cultured in normal culture media until stage 25, at which time the culture media was supplemented with 2.5 mM dimethyl sulfoxide (DMSO) or 50 μm SU5402 (Pfizer) (Langdon et al., 2007). Embryos were cultured under these conditions until Stage 37 and processed for immunohisto-chemical analysis to analyze heart morphology.
In Vitro Transcription/Translation Assays
In vitro transcription/translation assays were performed using the TnT Quick-Coupled Transcription/Translation System according to the manufacturer’s instructions (Promega, Madison, WI).
Whole Mount-Immunohistochemistry and In Situ Hybridization
Embryos were prepared for whole-mount immunohistochemistry by fixation in Dent’s fixative (80% methanol/20% dimethyl sulfoxide) or 4% paraformaldehyde and were then processed as previously described (Kolker et al., 2000), except that the rehydration steps were omitted for embryos fixed in paraformaldehyde. Fixed embryos were incubated overnight at 4°C with a primary antibody against α-myosin heavy chain (α-MHC) (Abcam, Cambridge, MA) (1:500), tropomyosin (DSHB, Iowa City, Iowa) (1:200), phospho-Histone H3 (pH3) (1:250) (Millipore, Billerica, MA), or cleaved caspase 3 (1:250) (Cell Signaling, Danvers, MA). Embryos were then washed and incubated overnight at 4°C with the appropriate Cy-3 or Alexa-488 conjugated secondary antibodies (1:200) and Topro-3 (1:1,000) (Molecular Probes, Carlsbad, CA) to stain the nuclei.
Whole-mount in situ hybridization was performed as previously described (Harland, 1991). Plasmids for MyoD, chordin, xBra, NKX2.5, TBX5, and Tbx20 were linearized and used to generate digoxigenin-UTP-labeled (Roche, Mannheim, Germany) antisense RNA probes using the appropriate restriction endonuclease and polymerase. Color detection was determined by BM Purple substrate (Roche) after incubation with alkaline-phos-phatase conjugated antidigoxigenin antibody.
Western Blot Analysis
Embryos (n = 5–10) were snap-frozen in liquid nitrogen and protein lysates were generated as previously described (Kragtorp and Miller, 2006). Briefly, 5–10 embryos were lysed by brief (1–2 s) sonication in a modified RIPA buffer (10 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 20 mM Na4P2O7, 1% Triton X-100). Cardiomyocytes were lysed in modified radioimmune precipitation assay buffer (50 mM Hepes, 0.15 M NaCl, 2 mM EDTA, 0.1% Nonidet P-40, 0.05% sodium de-oxycholate, pH 7.2). Each lysis buffer contained a cocktail of protease and phosphatase inhibitors including 1 mM Na3VO4, 40 mM NaF, 10 mM Na2 pyrophosphate, 100 μM leupeptin, 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride, 0.02 mg/mL soybean trypsin inhibitor, and 0.05 trypsin inhibitory units/mL aprotinin. Samples were clarified by centrifugation twice at 14,000g at 4°C and the supernatant was retained. Fifty micrograms of total protein was boiled in sample buffer and loaded onto a 10% SDS-acrylamide gel. Separated proteins were transferred onto nitrocellulose, blocked in 5% dry milk in Tris-Buffered Saline (TBS) + 0.1%Tween (TBST), and incubated overnight with primary antibody diluted in blocking solution. The following antibodies were utilized at a dilution of 1:1,000: FAK Clone 4.47 (Millipore), phospho-FAK Y397 (Invitrogen, Carlsbad, CA), PYK2 (Cell Signaling), and ERK-CT (Millipore). Blots were incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (1:2000 dilution) (GE Healthcare, Piscataway, NJ) and proteins were visualized by chemiluminescence (Thermo Scientific, Rockford, IL). Densitometry of Western blot band intensities was performed using ImageJ 1.37v software (NIH).
RT-PCR Analysis
RNA was isolated from 10 embryos following lysis in Trizol according to the manufacturer’s specifications (Invitrogen). Reverse transcription reactions were performed using the iScript cDNA kit (Bio-Rad, Hercules, CA) and PCR reactions were performed using ExTaq polymerase (Takara Bio, Japan) following previously published primer sets and cycling parameters (Meadows et al., 2008; Small et al., 2005). Histone H4 primers were as follows: Forward 5′GGG ATA ACA TTC AGG GTA TC 3′ and Reverse 5′CAT GGC GGT AAC TGT CTT C 3′.
Myocyte Cell Isolation, Culture, Infection, and Treatment
Cardiomyocytes were isolated from embryonic Day 13.5 (E13.5) rats with trypsin and collagenase digestion and were purified as described previously (Taylor et al., 2000). The cells were re-suspended in medium containing a 4:1 mixture of Dulbecco’s Modified Eagle Medium (DMEM):Media 199 containing 10% fetal calf serum and 1% penicillin-streptomycin and plated on tissue culture plastic for two consecutive 1-h periods to remove non-cardiomyocyte cells, resulting in cultures with >95% myocytes. The cardiomyocytes were then plated on tissue culture dishes precoated with fibronectin (10 μg/mL). For adenoviral infection, cells were infected with replication-defective Ad5-GFP or GFP-FRNK at a concentration of 10 multiplicity of infection (m.o.i) for the indicated times in serum-containing media. Cells were then serum starved overnight and treated for the indicated times with FGF-2 (100 ng/mL) or vehicle. 5-bromo-2-deoxyuridine (BrdU) labeling was performed as previously described (DiMichele et al., 2009). The FGF-receptor 1 (FGFR1) inhibitor, SU-5402, was a generous gift from Pfizer (Kalamazoo, MI).
Widefield and Laser Scanning Confocal Microscopy, Image Deconvolution, and 3D Rendering
Embryos were cleared for microscopic analysis in 2:1 benzyl benzoate:benzyl alcohol and placed on a glass coverslip. Embryos were analyzed by widefield microscopy using a Leica MZFLIII fluorescence dissecting scope or Olympus IX81 microscope or by confocal microscopy using an Olympus FV500 laser scanning confocal microscope (Olympus, USA) and Fluoview v5.1 software. Confocal z-stacks were obtained using a 1.24 μm step-size. Z-series stacks were deconvolved using Auto-deblur Gold v. X.1.4.1 software (Autoquant, Media Cybernetics, Bethesda, MD). Deconvolved images were then imported to Imaris x64 6.1.5 software (Bitplane AG, St. Paul, MN) for 3D rendering and isosurfacing.
To assess proliferation and apoptosis in Xenopus hearts, we immunostained embryos for tropomyosin (to label cardiomyocytes) and either pH3, to label proliferating cells, or cleaved caspase-3, to label cells undergoing apoptosis. Z-stack images of each heart were obtained by laser scanning confocal microscopy and each optical slice was examined individually throughout the thickness of the heart. Each cell that was immunoreactive for both tropomyosin and pH3 or cleaved caspase 3 was scored as a positive cardiomyocyte. In order to ensure that cells were not counted twice, we confirmed our results by counting all double-immunoreactive cells from optical sections that were 12 steps (14.88 μm) apart (the approximate size of the embryonic myocyte). The surface area of the heart (μm2) was calculated by isosurfacing the tropomyosin positive area using Imaris software. This calculated value was then multiplied by 28 μm (the average thickness of the heart wall at this stage of development), to determine total heart volume (μm3). Total noncardiomyocytes were scored and compared to the volume of the tissue section analyzed. Statistical analyses were performed using a two-tailed t-test (two-sample, equal variance-homoscedastic).
Supplementary Material
Acknowledgments
Contract grant sponsor: National Heart, Lung, and Blood Institute, Contract grant numbers: HL-081844, HL-071054, HL070953, HL 089641; Contract grant sponsor: American Heart Association, Contract grant numbers: 0355776U, 0555476U; Contract grant sponsor: National Institute of Dental and Craniofacial Research, Contract grant number: DE-018825; Contract grant sponsor: National Institutes of Health, Contract grant number: T32HL69768
Antibodies were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242. The authors thank the UNC Microscopy Services Laboratory for excellent technical assistance with confocal microscopy and utilization of the Imaris imaging software.
Footnotes
Additional Supporting Information may be found in the online version of this article.
LITERATURE CITED
- Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87:521–544. doi: 10.1152/physrev.00032.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brade T, Gessert S, Kuhl M, Pandur P. The amphibian second heart field: Xenopus islet-1 is required for cardiovascular development. Dev Biol. 2007;311:297–310. doi: 10.1016/j.ydbio.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Chen H, Shi S, Acosta L, Li W, Lu J, Bao S, Chen Z, Yang Z, Schneider MD, Chien KR, Conway SJ, Yoder MC, Haneline LS, Franco D, Shou W. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. 2004;131:2219–2231. doi: 10.1242/dev.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Stottmann RW, Yang YP, Meyers EN, Klingensmith J. The bone morphogenetic protein antagonist noggin regulates mammalian cardiac morphogenesis. Circ Res. 2007;100:220–228. doi: 10.1161/01.RES.0000257780.60484.6a. [DOI] [PubMed] [Google Scholar]
- David JP, Victoria LTB, Takashi M. Epicardium is required for the full rate of myocyte proliferation and levels of expression of myocyte mitogenic factors FGF2 and its receptor. FGFR-1, but not for transmural myocardial patterning in the embryonic chick heart. Dev Dynamics. 2003;228:161–172. doi: 10.1002/dvdy.10360. [DOI] [PubMed] [Google Scholar]
- de Pater E, Clijsters L, Marques SR, Lin Y-F, Garavito-Aguilar ZV, Yelon D, Bakkers J. Distinct phases of cardiomyocyte differentiation regulate growth of the zebrafish heart. Development. 2009;136:1633–1641. doi: 10.1242/dev.030924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehaan RL. Migration patterns of the precardiac mesoderm in the early chick embrvo. Exp Cell Res. 1963;29:544–560. doi: 10.1016/s0014-4827(63)80016-6. [DOI] [PubMed] [Google Scholar]
- DiMichele LA, Hakim ZS, Sayers RL, Rojas M, Schwartz RJ, Mack CP, Taylor JM. Transient expression of FRNK reveals stage-specific requirement for focal adhesion kinase activity in cardiac growth. Circ Res. 2009;104:1201–1208. doi: 10.1161/CIRCRESAHA.109.195941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel FB, Hsieh PC, Lee RT, Keating MT. FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proc Natl Acad Sci USA. 2006;103:15546–15551. doi: 10.1073/pnas.0607382103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–1187. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelmann GL, Dionne CA, Jaye MC. Acidic fibroblast growth factor, heart development, and capillary angiogenesis. Ann NY Acad Sci. 1991;638:463–466. doi: 10.1111/j.1749-6632.1991.tb49070.x. [DOI] [PubMed] [Google Scholar]
- Engelmann GL, Dionne CA, Jaye MC. Acidic fibroblast growth factor and heart development. Role in myocyte proliferation and capillary angiogenesis. Circ Res. 1993;72:7–19. doi: 10.1161/01.res.72.1.7. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Ilic D, Kanazawa S, Takeda N, Yamamoto T, Aizawa S. Mesodermal defect in late phase of gastrulation by a targeted mutation of focal adhesion kinase. FAK. Oncogene. 1995;11:1989–1995. [PubMed] [Google Scholar]
- Goetz SC, Conlon FL. Cardiac progenitors and the embryonic cell cycle. Cell Cycle. 2007;6:1974–1981. doi: 10.4161/cc.6.16.4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabbe C, Zervas CG, Hunter T, Brown NH, Palmer RH. Focal adhesion kinase is not required for integrin function or viability in Drosophila. Development. 2004;131:5795–5805. doi: 10.1242/dev.01462. [DOI] [PubMed] [Google Scholar]
- Hakim ZS, DiMichele LA, Doherty JT, Homeister JW, Beggs HE, Reichardt LF, Schwartz RJ, Brackhan J, Smithies O, Mack CP, Taylor JM. Conditional deletion of focal adhesion kinase leads to defects in ventricular septation and outflow tract alignment. Mol Cell Biol. 2007;27:5352–5364. doi: 10.1128/MCB.00068-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harland RM. In situ hybridization: an improved whole-mount method for Xenopus embryos. Methods Cell Biol. 1991;36:685–695. doi: 10.1016/s0091-679x(08)60307-6. [DOI] [PubMed] [Google Scholar]
- Hens MD, DeSimone DW. Molecular analysis and developmental expression of the focal adhesion kinase pp125FAK in Xenopus laevis. Dev Biol. 1995;170:274–288. doi: 10.1006/dbio.1995.1214. [DOI] [PubMed] [Google Scholar]
- Hsia DA, Mitra SK, Hauck CR, Streblow DN, Nelson JA, Ilic D, Huang S, Li E, Nemerow GR, Leng J, Spencer KS, Cheresh DA, Schlaepfer DD. Differential regulation of cell motility and invasion by FAK. J Cell Biol. 2003;160:753–767. doi: 10.1083/jcb.200212114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Kolker SJ, Tajchman U, Weeks DL. Confocal imaging of early heart development in Xenopus laevis. Dev Biol. 2000;218:64–73. doi: 10.1006/dbio.1999.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kragtorp KA, Miller JR. Regulation of somitogenesis by Ena/VASP proteins and FAK during Xenopus development. Development. 2006;133:685–695. doi: 10.1242/dev.02230. [DOI] [PubMed] [Google Scholar]
- Langdon YG, Goetz SC, Berg AE, Swanik JT, Conlon FL. SHP-2 is required for the maintenance of cardiac progenitors. Development. 2007;134:4119–4130. doi: 10.1242/dev.009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine KJ, Ornitz DM. Fibroblast growth factors and Hedgehogs: At the heart of the epicardial signaling center. Trends Genet. 2008;24:33–40. doi: 10.1016/j.tig.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Lavine KJ, Schmid GJ, Smith CS, Ornitz DM. Novel tool to suppress cell proliferation in vivo demonstrates that myocardial and coronary vascular growth represent distinct developmental programs. Dev Dyn. 2008;237:713–724. doi: 10.1002/dvdy.21468. [DOI] [PubMed] [Google Scholar]
- Lavine KJ, Yu K, White AC, Zhang X, Smith C, Partanen J, Ornitz DM. Endocardial and epicardial derived fgf signals regulate myocardial proliferation and differentiation in vivo. Dev Cell. 2005;8:85–95. doi: 10.1016/j.devcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Lea R, Papalopulu N, Amaya E, Dorey K. Temporal and spatial expression of FGF ligands and receptors during Xenopus development. Dev Dyn. 2009;238:1467–1479. doi: 10.1002/dvdy.21913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMullin A, Jacobs JR. Slit coordinates cardiac morphogenesis in Drosophila. Dev Biol. 2006;293:154–164. doi: 10.1016/j.ydbio.2006.01.027. [DOI] [PubMed] [Google Scholar]
- Martinsen BJ, Frasier AJ, Baker CVH, Lohr JL. Cardiac neural crest ablation alters Id2 gene expression in the developing heart. Dev Biol. 2004;272:176–190. doi: 10.1016/j.ydbio.2004.04.030. [DOI] [PubMed] [Google Scholar]
- Meadows SM, Warkman AS, Salanga MC, Small EM, Krieg PA. The myocardin-related transcription factor. MASTR, cooperates with MyoD to activate skeletal muscle gene expression. Proc Natl Acad Sci USA. 2008;105:1545–1550. doi: 10.1073/pnas.0703918105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M, McMahon G, Sun L, Tang C, Hirth P, Yeh BK, Hubbard SR, Schlessinger J. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science. 1997;276:955–960. doi: 10.1126/science.276.5314.955. [DOI] [PubMed] [Google Scholar]
- Mohun T, Orford R, Shang C. The origins of cardiac tissue in the amphibian Xenopus laevis. Trends Cardiovasc Med. 2003;13:244–248. doi: 10.1016/s1050-1738(03)00102-6. [DOI] [PubMed] [Google Scholar]
- Mohun TJ, Leong LM, Weninger WJ, Sparrow DB. The morphology of heart development in Xenopus laevis. Dev Biol. 2000;218:74–88. doi: 10.1006/dbio.1999.9559. [DOI] [PubMed] [Google Scholar]
- Moorman AF, Christoffels VM. Cardiac chamber formation: development, genes, and evolution. Physiol Rev. 2003;83:1223–1267. doi: 10.1152/physrev.00006.2003. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal table of Xenopus laevis (Daudin): A systematical and chronological survey of the development from the fertilized egg till the end of metamorphosis. New York: Garland Pub; 1994. p. 252. [Google Scholar]
- Parsons JT. Focal adhesion kinase: The first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Pasumarthi KBS, Kardami E, Cattini PA. High and low molecular weight fibroblast growth factor-2 increase proliferation of neonatal rat cardiac myocytes but have differential effects on binucleation and nuclear morphology: Evidence for both paracrine and intracrine actions of fibroblast growthfactor-2. Circ Res. 1996;78:126–136. doi: 10.1161/01.res.78.1.126. [DOI] [PubMed] [Google Scholar]
- Peng X, Wu X, Druso JE, Wei H, Park AY, Kraus MS, Alcaraz A, Chen J, Chien S, Cerione RA, Guan JL. Cardiac developmental defects and eccentric right ventricular hypertrophy in cardiomyocyte focal adhesion kinase (FAK) conditional knockout mice. Proc Natl Acad Sci USA. 2008;105:6638–6643. doi: 10.1073/pnas.0802319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro I, Kawakami Y, Buscher D, Raya A, Rodriguez-Leon J, Morita M, Rodriguez Esteban C, Izpisua Belmonte JC. Tbx2 and Tbx3 regulate the dynamics of cell proliferation during heart remodeling. PLoS ONE. 2007;2:e398. doi: 10.1371/journal.pone.0000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A, Parsons T. A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK. Nature. 1996;380:538–540. doi: 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- Sadaghiani B, Thiébaud CH. Neural crest development in the Xenopus laevis embryo, studied by interspecific transplantation and scanning electron microscopy. Dev Biol. 1987;124:91–110. doi: 10.1016/0012-1606(87)90463-5. [DOI] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Schlaepfer DD. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J Cell Sci. 1999;112(Part 16):2677–2691. doi: 10.1242/jcs.112.16.2677. [DOI] [PubMed] [Google Scholar]
- Small EM, Warkman AS, Wang D-Z, Sutherland LB, Olson EN, Krieg PA. Myocardin is sufficient and necessary for cardiac gene expression in Xenopus. Development. 2005;132:987–997. doi: 10.1242/dev.01684. [DOI] [PubMed] [Google Scholar]
- Srivastava D, Olson EN. A genetic blueprint for cardiac development. Nature. 2000;407:221–226. doi: 10.1038/35025190. [DOI] [PubMed] [Google Scholar]
- Taylor JM, Rovin JD, Parsons JT. A role for focal adhesion kinase in phenylephrine-induced hypertrophy of rat ventricular cardiomyocytes. J Biol Chem. 2000;275:19250–19257. doi: 10.1074/jbc.M909099199. [DOI] [PubMed] [Google Scholar]
- Trinh LA, Stainier DY. Cardiac development. Methods Cell Biol. 2004a;76:455–473. doi: 10.1016/s0091-679x(04)76020-3. [DOI] [PubMed] [Google Scholar]
- Trinh LA, Stainier DY. Fibronectin regulates epithelial organization during myocardial migration in zebrafish. Dev Cell. 2004b;6:371–382. doi: 10.1016/s1534-5807(04)00063-2. [DOI] [PubMed] [Google Scholar]
- Wilson PA, Hemmati-Brivanlou A. Induction of epidermis and inhibition of neural fate by Bmp-4. Nature. 1995;376:331–333. doi: 10.1038/376331a0. [DOI] [PubMed] [Google Scholar]
- Zaffran S, Frasch M. Early signals in cardiac development. Circ Res. 2002;91:457–469. doi: 10.1161/01.res.0000034152.74523.a8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.