

Abstract
Toxoplasma gondii is a protozoan parasite capable of infecting humans and animals. Surface antigen glycoproteins, SAG2C, -2D, -2X, and -2Y, are expressed on the surface of bradyzoites. These antigens have been shown to protect bradyzoites against immune responses during chronic infections. We studied structures of SAG2C, -2D, -2X, and -2Y proteins using bioinformatics methods. The protein sequence alignment was performed by T-Coffee method. Secondary structural and functional domains were predicted using software PSIPRED v3.0 and SMART software, and 3D models of proteins were constructed and compared using the I-TASSER server, VMD, and SWISS-spdbv. Our results showed that SAG2C, -2D, -2X, and -2Y are highly homologous proteins. They share the same conserved peptides and HLA-I restricted epitopes. The similarity in structure and domains indicated putative common functions that might stimulate similar immune response in hosts. The conserved peptides and HLA-restricted epitopes could provide important insights on vaccine study and the diagnosis of this disease.
1. Introduction
Toxoplasma gondii (T. gondii) is a species of parasitic protozoa in the genus Toxoplasma that can be carried by many warm-blooded animals including humans [1]. There are three infectious stages in a complex life cycle of T. gondii: the tachyzoites, the bradyzoites, and the sporozoites [2]. A bradyzoite is a slowly replicating version of the parasite, which is responsible for chronic infection of T. gondii [3]. In chronic toxoplasmosis, the parasitophorous vacuoles containing the reproductive bradyzoites form cysts in the tissues of the muscles and brain [4].
The surface antigen of T. gondii that plays roles in the processes of host cell attachment and host immune evasion is dominated by a SRS (SAG1-related sequence) family of proteins which includes the SAG1-like sequence branch and the SAG2-like sequence branch [5]. SRS proteins are expressed in a stage-specific manner. SAG1, SAG2A, SAG2B, SAG3, SRS1, SRS2, and SRS3 are mainly expressed on the tachyzoite surface [6]. Studies have indicated that SAG2 members participate in the process of parasite's invasion to the host, and their antibodies could block the further attachment of T. gondii on host cells [7, 8]. Previous studies have demonstrated that T. gondii parasites with a deletion of SAG2C, -2D, -2X, and -2Y gene cluster are less capable of maintaining a chronic infection in the brain [9]. It revealed that SAG2CDXY are important for persistence of cysts in the brain and these antigens might protect bradyzoites against an immune response. Contrary to SAG2A and SAG2B, which are expressed in tachyzoites, SAG2C, -2D, -2X, and -2Y appeared to be expressed exclusively on the surface of bradyzoites [9, 10]. However, among 160 members of the SRS family, only three proteins' structures were reported. They are (i) the tachyzoite-expressed SAG1 [11], (ii) the bradyzoite-expressed BSR4 [12], and (iii) the sporoSAG [13]. The structure and function domains of SAG2C, -2D, -2X, and -2Y are still not very clear.
In this study, we sought to predict the structure and function domains of SAG2C, -2D, -2X, and -2Y by bioinformatics methods. The protein sequence alignments were performed by the T-Coffee method. Secondary structural and functional domains were predicted using the software PSIPRED v3.0 and SMART software. The 3D structure model of each protein was mapped using the I-TASSER server. The structural similarities of these proteins were summarized and possible functions of some key amino acids were predicted using the space confrontation by VMD and SWISS-spdbv. Furthermore, HLA-restricted epitopes of SAG2C, -2D, -2X, and -2Y proteins were predicted via algorithms.
2. Methods
2.1. Data Resources
The protein sequences were derived from ToxoDB 5.1 (http://toxodb.org/toxo/). Toxoplasma gondii has three common types: type I, T. gondii GT1 (TGGT1_chrX 7,429,598); type II, T. gondii ME49 (TGME49_chrX 7,419,075); type III, T. gondii VEG (TGVEG_chrX 7,553,721). The original resources are listed in Table 1.
Table 1.
The original resources for SAG2C, D, X, and Y.
| Protein | Sizea | Coding gene location in types I, II, and III parasitesb | Derivationc |
|---|---|---|---|
| SAG2C | 365 aa | TGGT1_chrX: 7,358,316–7,360,225(−); | |
| TGME49_chrX: 7353954–7,365860 (−); | SRS49D | ||
| TGVEG_chrX: 7,440,957–7,442,866(−) | |||
| SAG2D | 196 aa | TGGT1_chrX: 7,352,615–7,353,874(−); | |
| TGME49_chrX: 7,353,328–7,354,587(−); | SRS49C | ||
| TGVEG_chrX: 7,435,249–7,436,508 (−) | |||
| SAG2X | 367 aa | TGGT1_chrX: 7,352,615–7,353,874(−); | |
| TGME49_chrX: 7,353,328–7,354,587(−); | SRS49B | ||
| TGVEG_chrX: 7,435,249–7,436,508 (−) | |||
| SAG2Y | 316 aa | TGGT1_chrX: 7,358,316–7,360,225(−); | |
| TGME49_chrX: 7,355,817–7,356,572 (−); | SRS49A | ||
| TGVEG_chrX: 7,440,957–7,442,866(−) |
aSize is the amino acid number that the protein has.
bCoding gene is the location of the gene that coded the protein.
cSRS domain-containing protein number.
2.2. Modular Architecture Identification
Multiple sequence alignment tool, T-Coffee (http://www.tcoffee.org/) [14, 15], was used to obtain the alignment analysis among SAG2C, SAG2D, SAG2X, and SAG2Y. The secondary structures were constructed using the software PSIPREDv3.0 (http://bioinf.cs.ucl.ac.uk/psipred/) [16, 17]. Simple modular architecture research identification and annotation of signaling domain sequences were analyzed via a web-based tool, SMART (http://smart.embl-heidelberg.de/) [18].
The 3D models of proteins were constructed by I-TASSER, a protein structure server on the website http://zhanglab.ccmb.med.umich.edu/I-TASSER/, which is considered to predict protein 3D structures that have more than 100 amino acids [19–21]. VMD is a molecular visualization software for displaying, animating, and analyzing large biomolecular systems using 3D graphics and built-in scripts (http://www.ks.uiuc.edu/Research/vmd/). VMD was used to read standard Protein Data Bank (PDB) files and display the contained structure [22–25]. Swiss-Pdb Viewer (http://www.expasy.org/spdbv/) is an application that provides a user friendly interface allowing analyses of several proteins at the same time. The proteins can be superimposed in order to obtain structural alignments and compare their active domains. We deduced amino acid mutations, H bonds, angles, and distances between atoms from the intuitive graphic and menu interface. 3D protein molecular fitness analysis was performed for SAG2C, -2D and SAG2X, -2Y [22, 23].
2.3. Conserved HLA-Restricted Epitopes Prediction
Consensus methods including ANN, SMM, and CombLib-Sidney in immune epitope database IEDB (http://www.immuneepitope.org/) were used to predict HLA-restricted epitopes [26–28]. We used this tool to determine each peptide sequence's ability to bind to the specific HLA class I molecule.
3. Results and Discussion
3.1. Amino Acid Sequence Alignment Analysis
SAG2C, SAG2D, SAG2X, and SAG2Y are positioned next to each other on chromosome X. The molecular masses of SAG2C, -2D, -2X, and -2Y are 32–38 kDa, 18–20 kDa 31–34 kDa, and 28–30 kDa, respectively [9]. Multiple sequence alignment for SAG2C, -2D, -2X, and -2Y shows that the four proteins sequences have 97% similarity (Figure 1). In Particular, SAG2C (184 to 364) has a 98% sequence identity to SAG2D (14 to 196) and SAG2X (184 to 367) has a 99% sequence identity to SAG2Y (128 to 300). The protein sequence alignment analysis indicated that SAG2C, -2D, -2X, and -2Y have high homologous sequences. However, when including SAG2A and SAG2B in the alignment analysis, the consensus dropped to 73%, even though the consensus between SAG2A and SAG2B has very good score 84%. It indicates that a great difference exists among SAG2A, -2B and SAG2C, -2D, -2X, -2Y.
Figure 1.
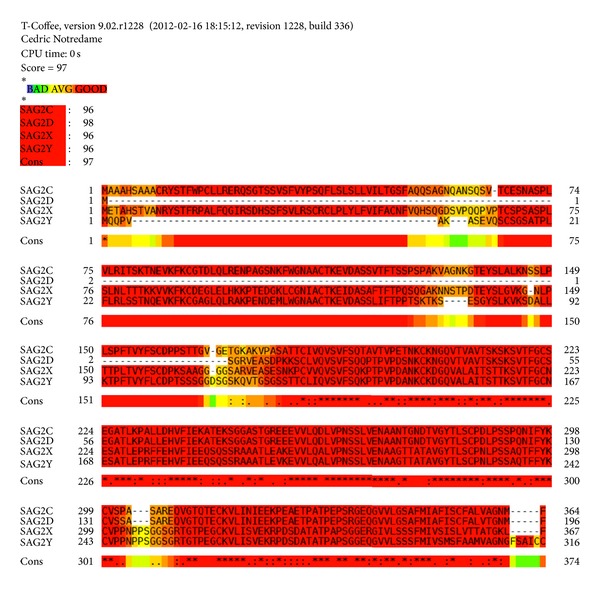
Alignment analysis for SAG2C, -2D, -2X, -2Y proteins. T-Coffee: multiple sequence alignment tools were used to obtain the alignment analysis result for SAG2C, -2D, -2X, and -2Y. Color bar indicated the identity, from bad identity to good identity.
3.2. 2-D Structure Alignment for SAG2C, -2D, -2X, and -2Y Proteins
PSIPRED v. 3.0 was used to predict the secondary structures of SAG2C, -2D, -2X, and -2Y proteins. Figure 2 showed that SAG2C protein has two α-helixes, 19 β-strands, and 20 coils; SAG2D protein has one α-helix, 9 β-strands, and 10 coils; SAG2X protein has 3 α-helixes, 14 β-strands, and 18 coils; SAG2Y protein has two α-helixes, 15 β-strands, and 18 coils. Obviously, there was a long α-helix on the C-terminal of all the proteins. SAG2D protein has similar secondary structure elements as SAG2C protein resides from 169 to 364. SAG2X and SAG2Y also have quite similar secondary structures except for a little discrepancy: SAG2X have one more helix than SAG2Y and one strand less than SAG2Y.
Figure 2.
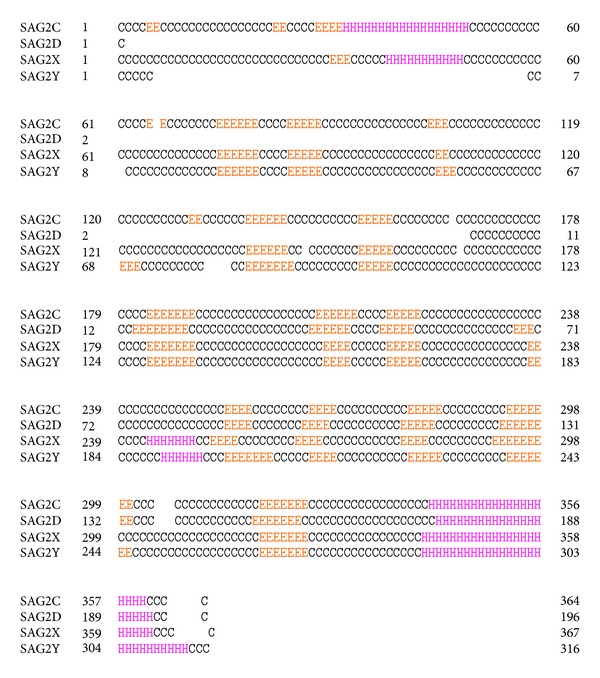
The 2D Structures of SAG2C, -2D, -2X, and -2Y proteins. PSIPREDv3.0 was used to predict the secondary structure for SAG2C, -2D, -2X, and -2Y proteins (C stands for coil, H stands for α-helix, and E stands for β-strand).
Furthermore, we used SMART to identify domains of these proteins (Figure 3). SAG2C, SAG2X, and SAG2Y all have two domains, while SAG2D only has one domain. SAG2D has an insertion of an adenosine, causing a frame shift and a premature stop codon, presumably leading to a truncated protein. SAG2C and SAG2D have transmembrane segments, while no transmembrane segments were identified on SAG2X and SAG2Y. From Figure 3, we could see that these proteins have no signal peptides, indicating that they are mature proteins. Members of the SAG2 family also differ in terms of open reading frame size, with the smaller SAG2D protein consisting of only one SAG domain, whereas SAG2C, SAG2X, and SAG2Y contain two SAG domains interrupted by a single intron. This indicates that SAG2C, SAG2X, and SAG2Y proteins have similar structure domains except SAG2D protein, which only has one domain.
Figure 3.
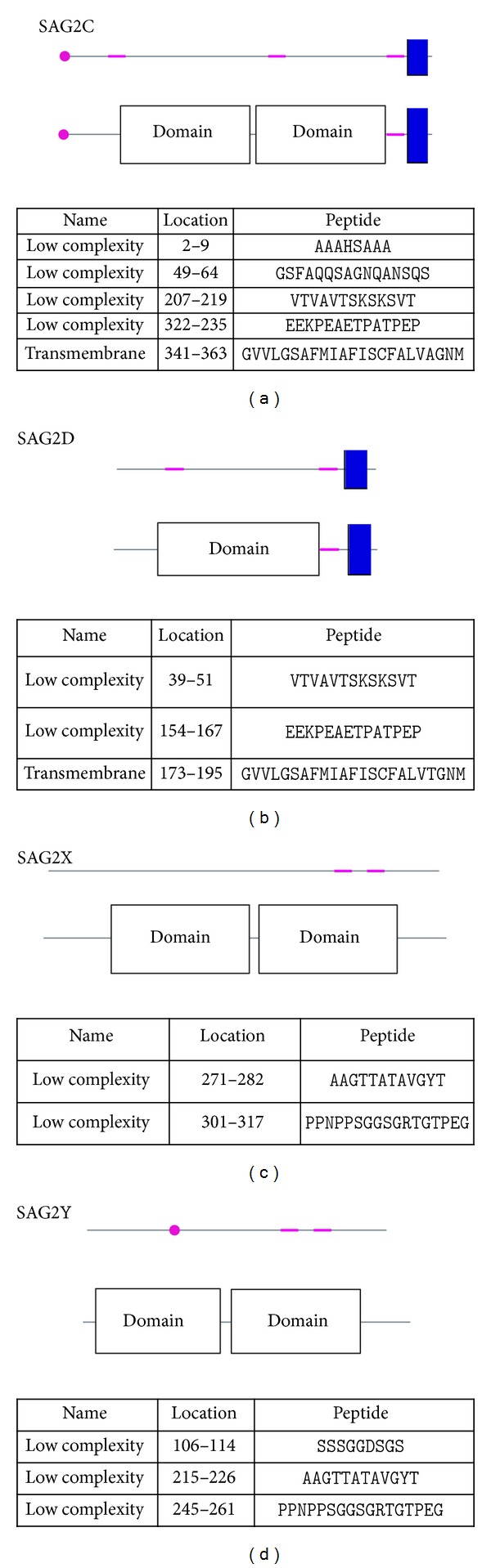
Prediction for protein domain. A web-based tool—SMARTM was used to figure out the domains of these proteins: transmembrane segments predicted by the TMHMM2 program (segments in blue color), segments of low compositional complexity determined by the SEG program (segments in purple color), signal peptides determined by the SignalP program (segments in red color), and domain (segments in gray color).
3.3. Construction of 3D Model for SAG2C, -2D, -2X, and -2Y Proteins
3D model of SAG2C, -2D, -2X, and -2Y proteins were constructed by I-TASSER server. Five models were set up for each protein by Dr. Zhang's lab [19]. We selected the model with highest confidence C-score, which estimates the quality of predicted models by I-TASSER. It was calculated based on the significance of threading template alignments and the convergence parameters of the structure assembly simulations [20]. C-score is typically in the range of [−5, 2], and model with a C-score above 2 suggested a high confidence.
Low temperature replicas (decoys) generated during the simulation were clustered by SPICKER and top five cluster centroids were selected to generate full atomic protein models. The cluster density was defined as the number of structure decoys at each unit of space in the SPICKER cluster. A higher cluster density meant that the structure occurs more often in the simulation trajectory and therefore a better quality model. Table 2 showed the parameters for construction D model of each protein.
Table 2.
Parameters for predicted best 3-D model.
| Name | C-scorea | TM-scoreb | RMSDb | No. of decoysc | Cluster densityd |
|---|---|---|---|---|---|
| SAG2C | −2.18 | 0.46 ± 0.15 | 11.8 ± 4.5 | 460 | 0.0285 |
| SAG2D | −1.45 | 0.54 ± 0.15 | 8.5 ± 4.5 | 4662 | 0.0842 |
| SAG2X | −2.22 | 0.45 ± 0.15 | 11.9 ± 4.4 | 455 | 0.0280 |
| SAG2Y | −1.59 | 0.52 ± 0.15 | 9.9 ± 4.6 | 2877 | 0.0525 |
aC-score is a confidence score for estimating the quality of predicted models by I-TASSER. C-score is typically in the range of [−5, 2], where a C-score of higher value signifies a model with a high confidence and vice versa.
bTM-score and RMSD are known standards for measuring structural similarity between two structures which are usually used to measure the accuracy of structure modeling when the native structure is known.
cNumber of decoys represents the number of structural decoys that are used in generating each model.
dCluster density represents the density of cluster.
The best model of each protein was selected and viewed via VMD program (Figure 4). SAG2C, -2X, and -2Y have obvious two domains, D1 and D2, which are formed by two β-strands separated by one α-helix; SAG2D has one domain which is formed by one β-strand separated by one α-helix. The β-strands rotate to form a sheet tube that is a common character of these proteins. Furthermore, the binding sites of residues in the model were predicted and showed in Table 3.
Figure 4.
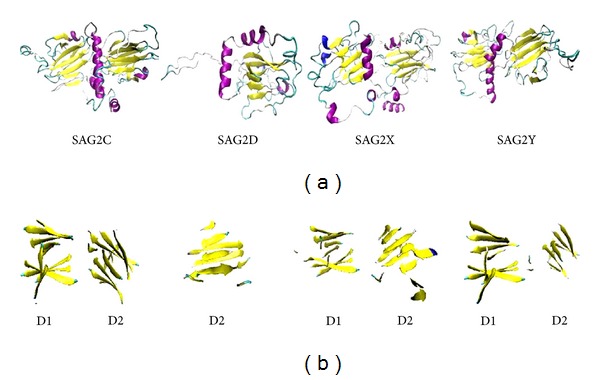
The 3D models of SAG2C, -2D, -2X, and -2Y. The sequences of proteins were sent to Dr. Zhang's lab from the website http://zhanglab.ccmb.med.umich.edu/I-TASSER/. The 3D models with the highest score for each protein were selected. The models were viewed by VMD software, color method was secondary structure (yellow: β-strands, purple: α-helix, gray: coil), and draw method was new cartoon. The domain of each model was shown out in sheet form.
Table 3.
Prediction binding site residues in the model.
| Protein model | C-scoreLB a | TM-scoreb | RMSDc | IDENd | Cov.e | BS-scoref | Lig. name | Predicted binding site residues in the model |
|---|---|---|---|---|---|---|---|---|
| SAG2C | 0.09 | 0.350 | 7.07 | 0.031 | 0.604 | 0.84 | MAL | 222, 223, 224, 225, 226, 227, 273, 279 |
| SAG2D | 0.11 | 0.435 | 4.90 | 0.043 | 0.668 | 0.75 | ANP | 96, 153, 155 |
| SAG2X | 0.06 | 0.388 | 5.94 | 0.053 | 0.597 | 0.79 | NA | 261, 292, 326 |
| SAG2Y | 0.07 | 0.412 | 6.36 | 0.057 | 0.687 | 0.77 | FES | 144, 145, 146, 147, 149, 150, 151, 153, 243 |
aC-scoreLB is the confidence score of predicted binding site. C-scoreLB values range between [0-1], where a higher score indicates a more reliable ligand-binding site prediction.
bTM-score is a measure of global structural similarity between query and template protein.
cRMSD is the RMSD between residues that are structurally aligned by TM-align.
dIDEN is the percentage sequence identity in the structurally aligned region.
eCov. represents the coverage of global structural alignment and is equal to the number of structurally aligned residues divided by length of the query protein.
fBS-score is a measure of local similarity (sequence and structure) between template binding site and predicted binding site in the query structure.
Previous analysis of SAG2C, -2D, -2X, and -2Y structures revealed that the five on three β sandwich fold of SAG2 was most similar to the T. gondii bradyzoite-expressed BSR4 with TM-scores of 0.583, 0.661, 0.672, and 0.670, respectively (Table 4). BSR4 is a prototypical bradyzoite surface antigen encoded in a cluster of SRS genes on chromosome IV, including the closely related paralogs SRS6 and SRS9 [8, 9]. Sequence alignment shows that SAG2C, -2D, -2X, and -2Y share 71% sequence identity with the tachyzoite-expressed BSR4. This observation is consistent with the prediction that stage-specific structural features might play an important role in the process of infection, dissemination, and pathogenesis in T. gondii. In BSR4, two strands are organized in an antiparallel fashion, followed by another strand on the lower face of the β sandwich. The dimeric structure of SAG1 showed a β sandwich, two parallel outside strands with an opposite one in between [29]. The overall topology of the five on three β sandwich D2 domain is conserved between SAG2C, -2D, -2X, -2Y and BSR4. A detailed comparison of SAG2C, -2D, -2X, -2Y and BSR4 reveals a similarity in topology of the D1 and D2 domain consistent with the lower Z-score from the Dali search.
Table 4.
Similarity between SAG2C, -2D, -2X, -2Y and top identified structural analogs BSR4 protein.
| Protein model | TM-scorea | RMSDb | IDENc | Cov.d |
|---|---|---|---|---|
| SAG2A | 0.583 | 1.20 | 0.244 | 0.596 |
| SAG2B | 0.661 | 2.42 | 0.167 | 0.755 |
| SAG2C | 0.673 | 1.68 | 0.179 | 0.698 |
| SAG2D | 0.670 | 2.85 | 0.194 | 0.763 |
aTM-score of the structural alignment between the query structure and known structures in the PDB library.
bRMSD is the RMSD between residues that are structurally aligned by TM-align.
cIDEN is the percentage sequence identity in the structurally aligned region.
dCov. represents the coverage of the alignment by TM-align and is equal to the number of structurally aligned residues divided by length of the query protein.
By comparison, the next most similar structure is SproSAG (surface antigen glycoprotein) with a substantially reduced TM-score [30, 31]. SporoSAG is a dominant surface coat protein expressed on the surface of sporozoites. SporoSAG crystallized as a monomer and displayed unique features of the SRS β sandwich fold compared to SAG1 and BSR4 [9]. Intriguingly, the structural diversity is localized to the upper sheets of the β sandwich fold and may have important implications for multimerization and host cell ligand recognition. By fit analysis, SAG2D fits well on the C-terminal of the protein SAG2C. SAG2X and SAG2Y fit pretty well from C-terminal to N-terminal (Figure 5).
Figure 5.

Fit analyses of SAG2C, -2D, -2X, and -2Y. Swiss-Pdb Viewer was used to show the fitness. SAG2C and SAG2X are in yellow color; SAG2D and SAG2Y are in blue color.
3.4. Conserved HLA-Restricted CD8+ T Cells Epitope Prediction
Epitope prediction algorithm consensus was used to predict peptides that could stimulate human to induce effective and protective immune response against T. gondii. We want to see if they have similar epitopes scattered on the surface of their protein. The epitopes from SAG2C, -2D, -2X, and -2Y were predicted using the software from IEDB (http://www.immuneepitope.org/) which could identify novel HLA-class I restricted CD8+ T cell epitopes derived from T. gondii. 16 peptides were selected based on a high HLA allele binding score (percentile rank < 3).
From Table 5, we can see three HLA-A*0201-restricted peptides: VVLGSAFMI, FMIAFISCF, AFISCFALV; four HLA-A*1101-restricted peptides: QVTVAVTSK, SSPQNIFYK, QVGTQTECK, KVLINIEEK; and two HLA-B*0702-restricted peptides: LPSSPQNIF, KPEAETPAT shared by SAG2C and SAG2D. From Table 6, we can see two HLA-A*0201-restricted peptides: ALVPNSSLV, VLSSSFMIV; three HLA-A*1101-restricted peptides: ALAITSTTK, SSAQTFFYK, KVLISVEKR; and two HLA-B*0702-restricted peptides: LPSSAQTFF, RPDSDATAT shared by SAG2X and SAG2Y.
Table 5.
Predicted HLA restricted CD8+ T cell epitopes for SAG2C, -2D of T. gondii.
| Allele | Sequence | Pecentile rank | Method used | Location | |
|---|---|---|---|---|---|
| SAG2C | SAG2D | ||||
| HLA-A*0201 | VVLGSAFMI | 3.4 | Consensus (ANN, SMM, CombLib Sidenev 2008) | 342–350 | 174–182 |
| HLA-A*0201 | FMIAFISCF | 1.4 | Consensus (ANN, SMM, CombLib Sidenev 2008) | 348–356 | 180–188 |
| HLA-A*0201 | AFISCFALV | 2.8 | Consensus (ANN, SMM) | 351–359 | 183–191 |
| HLA-A*1101 | QVTVAVTSK | 1.45 | Consensus (ANN, SMM) | 206–214 | 38–46 |
| HLA-A*1101 | SSPQNIFYK | 0.2 | Consensus (ANN, SMM) | 290–298 | 122–130 |
| HLA-A*1101 | QVGTQTECK | 2.35 | Consensus (ANN, SMM) | 308–316 | 140–148 |
| HLA-A*1101 | KVLINIEEK | 1 | Consensus (ANN, SMM) | 316–324 | 148–156 |
| HLA-B*0702 | LPSSPQNIF | 1.1 | Consensus (ANN, SMM, CombLib Sidenev 2008) | 288–296 | 120–128 |
| HLA-B*0702 | KPEAETPAT | 2.6 | Consensus (ANN, SMM, CombLib Sidenev 2008) | 324–332 | 156–164 |
Table 6.
Predicted HLA-restricted CD8+ T cell epitopes for SAG2X, -2Y of T. gondii.
| Allele | Sequence | Pecentile rank | Method used | Location | |
|---|---|---|---|---|---|
| SAG2C | SAG2D | ||||
| HLA-A*0201 | ALVPNSSLV | 1.6 | Consensus (ANN, SMM, CombLib Sidenev 2008) | 260–268 | 204–212 |
| HLA-A*0201 | VLSSSFMIV | 1.2 | Consensus (ANN, SMM, CombLib Sidenev 2008) | 346–354 | 290–298 |
| HLA-A*1101 | ALAITSTTK | 1.6 | Consensus (ANN, SMM) | 208–216 | 152–160 |
| HLA-A*1101 | SSAQTFFYK | 0.1 | Consensus (ANN, SMM) | 289–297 | 234–242 |
| HLA-A*1101 | KVLISVEKR | 2.75 | Consensus (ANN, SMM) | 311–319 | 263–271 |
| HLA-B*0702 | LPSSAQTFF | 3 | Consensus (ANN, SMM, CombLib Sidenev 2008) | 285–293 | 232–240 |
| HLA-B*0702 | RPDSDATAT | 2.2 | Consensus (ANN, SMM, CombLib Sidenev 2008) | 314–322 | 271–279 |
More interestingly, when we marked the HLA-restricted epitopes on the alignment sequences of the proteins, we found that the epitopes restricted by the same type of HLA allele are located at the same domains of the proteins (Figure 6). Our results indicated that the epitopes from SAG2C, -2D, -2X, and -2Y can be recognized by the proper MHC-I molecular and present on the cell surface to induce immune response in the host CD8+ T cells which might be helpful on vaccine study and diagnosis for this parasitic disease. Some identified peptides from these proteins have been proven to be recognized by PBMC cells from proper HLA-restricted T. gondii seropositive individuals and significantly induced IFN-γ production in T cells from immunized mice [32, 33] and therefore confirmed our predictions.
Figure 6.

Identifying HLA-restricted epitopes on the surface of 3D models. The predicted HLA-restricted epitopes sequences shown in Tables 2 and 3 were marked out on the surface of 3D models of SAG2C, -2D, -2X, and -2Y. The 3D structures of proteins were shown using Surf method. Red color balls stand for epitopes restricted by HLA-A*1101, green color balls stand for epitopes restricted by HLA-A*0201, and blue color balls stand for epitopes restricted by HLA-B*0702.
4. Conclusions
In this study, we have conducted a detailed bioinformatic and structural characterization analysis of the bradyzoite proteins SAG2C, -2D, -2X, and -2Y. The characterization of SAG2C, -2D, -2X, and -2Y provided structural view of the T. gondii SRS family members at chronic bradyzoite stage. Our bioinformatic analysis clearly showed that SAG2C, -2D, -2X, and -2Y are homologous protein members of the SAG2 subfamily. Consistently, our structural analysis demonstrated that SAG2C, -2D, -2X, and -2Y are similar to two other bradyzoite SAG2 members, BSR4 and SPOROSAG, rather than tachyzoite SAG1. This result indicated that SAG2 family has conserved structure at bradyzoite stage but a great difference from SAG1 at tachyzoite stage. Furthermore, the predicted conserved peptides and HLA-restricted epitopes shed interesting light on vaccine study and diagnosis for this parasitic disease.
Conflict of Interests
The authors wish to declare that there is no known conflict of interests associated with this publication and there has been no other significant financial support for this work that could have influenced its outcome.
Acknowledgments
This study was supported by a Grant from the National Natural Science Foundation Project of China (no. 81171604) and no. 49 China postdoc foundation.
References
- 1.McLeod R, Kieffer F, Sautter M, Hosten T, Pelloux H. Why prevent, diagnose and treat congenital toxoplasmosis? Memorias do Instituto Oswaldo Cruz. 2009;104(2):320–344. doi: 10.1590/s0074-02762009000200029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tenter AM, Heckeroth AR, Weiss LM. Toxoplasma gondii: from animals to humans. International Journal for Parasitology. 2000;30(12):1217–1258. doi: 10.1016/s0020-7519(00)00124-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chew WK, Wah MJ, Ambu S, Segarra I. Toxoplasma gondii: determination of the onset of chronic infection in mice and the in vitro reactivation of brain cysts. Experimental Parasitology. 2012;130(1):22–25. doi: 10.1016/j.exppara.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Soldati D, Boothroyd JC. Transient transfection and expression in the obligate intracellular parasite Toxoplasma gondii . Science. 1993;260(5106):349–352. doi: 10.1126/science.8469986. [DOI] [PubMed] [Google Scholar]
- 5.Jung C, Lee CY, Grigg ME. The SRS superfamily of Toxoplasma surface proteins. International Journal for Parasitology. 2004;34(3):285–296. doi: 10.1016/j.ijpara.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Manger ID, Hehl AB, Boothroyd JC. The surface of Toxoplasma tachyzoites is dominated by a family of glycosylphosphatidylinositol-anchored antigens related to SAG1. Infection and Immunity. 1998;66(5):2237–2244. doi: 10.1128/iai.66.5.2237-2244.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lekutis C, Ferguson DJP, Boothroyd JC. Toxoplasma gondii: identification of a developmentally regulated family of genes related to SAG2. Experimental Parasitology. 2000;96(2):89–96. doi: 10.1006/expr.2000.4556. [DOI] [PubMed] [Google Scholar]
- 8.Machado AV, Caetano BC, Barbosa RP, et al. Prime and boost immunization with influenza and adenovirus encoding the Toxoplasma gondii surface antigen 2 (SAG2) induces strong protective immunity. Vaccine. 2010;28(18):3247–3256. doi: 10.1016/j.vaccine.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Saeij JPJ, Arrizabalaga G, Boothroyd JC. A cluster of four surface antigen genes specifically expressed in bradyzoites, SAG2CDXY, plays an important role in Toxoplasma gondii persistence. Infection and Immunity. 2008;76(6):2402–2410. doi: 10.1128/IAI.01494-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SK, Karasov A, Boothroyd JC. Bradyzoite-specific surface antigen SRS9 plays a role in maintaining Toxoplasma gondii persistence in the brain and in host control of parasite replication in the intestine. Infection and Immunity. 2007;75(4):1626–1634. doi: 10.1128/IAI.01862-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He XL, Grigg ME, Boothroyd JC, Garcia KC. Structure of the immunodominant surface antigen from the Toxoplasma gondii SRS superfamily. Nature Structural Biology. 2002;9(8):606–611. doi: 10.1038/nsb819. [DOI] [PubMed] [Google Scholar]
- 12.Crawford J, Grujic O, Bruic E, Czjzek M, Grigg ME, Boulanger MJ. Structural characterization of the bradyzoite surface antigen (BSR4) from Toxoplasma gondii, a unique addition to the surface antigen glycoprotein 1-related superfamily. Journal of Biological Chemistry. 2009;284(14):9192–9198. doi: 10.1074/jbc.M808714200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crawford J, Lamb E, Wasmuth J, Grujic O, Grigg ME, Boulanger MJ. Structural and functional characterization of SporoSAG: a SAG2-related surface antigen from Toxoplasma gondii . Journal of Biological Chemistry. 2010;285(16):12063–12070. doi: 10.1074/jbc.M109.054866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Notredame C, Higgins DG, Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment. Journal of Molecular Biology. 2000;302(1):205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 15.Di Tommaso P, Moretti S, Xenarios I, et al. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Research. 2011;39(2):W13–W17. doi: 10.1093/nar/gkr245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buchan DWA, Ward SM, Lobley AE, Nugent TCO, Bryson K, Jones DT. Protein annotation and modelling servers at University College London. Nucleic Acids Research. 2010;38(2):W563–W568. doi: 10.1093/nar/gkq427.gkq427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadowski MI, Jones DT. The sequence-structure relationship and protein function prediction. Current Opinion in Structural Biology. 2009;19(3):357–362. doi: 10.1016/j.sbi.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Research. 2000;28(1):231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roy A, Xu D, Poisson J, Zhang Y. A protocol for computer-based protein structure and function prediction. Journal of Visualized Experiments. 2011;3(57):p. e3259. doi: 10.3791/3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9, article 40 doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols. 2010;5(4):725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. Journal of Molecular Graphics. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 23.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Research. 2003;31(13):3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 25.Guex N, Peitsch MC, Schwede T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;30(1):S162–S173. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 26.Moutaftsi M, Peters B, Pasquetto V, et al. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus. Nature Biotechnology. 2006;24(7):817–819. doi: 10.1038/nbt1215. [DOI] [PubMed] [Google Scholar]
- 27.Sidney J, Assarsson E, Moore C, et al. Quantitative peptide binding motifs for 19 human and mouse MHC class i molecules derived using positional scanning combinatorial peptide libraries. Immunome Research. 2008;4(1, article 2) doi: 10.1186/1745-7580-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lundegaard C, Nielsen M, Lund O. The validity of predicted T-cell epitopes. Trends in Biotechnology. 2006;24(12):537–538. doi: 10.1016/j.tibtech.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 29.Barragan A, David Sibley L. Transepithelial migration of Toxoplasma gondii is linked to parasite motility and virulence. Journal of Experimental Medicine. 2002;195(12):1625–1633. doi: 10.1084/jem.20020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wallner B, Elofsson A. Can correct protein models be identified? Protein Science. 2003;12(5):1073–1086. doi: 10.1110/ps.0236803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graille M, Stura EA, Bossus M, et al. Crystal structure of the complex between the monomeric form of Toxoplasma gondii surface antigen 1 (SAG1) and a monoclonal antibody that mimics the human immune response. Journal of Molecular Biology. 2005;354(2):447–458. doi: 10.1016/j.jmb.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 32.Cong H, Mui EJ, Witola WH, et al. Towards an immunosense vaccine to prevent toxoplasmosis: protective Toxoplasma gondii epitopes restricted by HLA-A*0201. Vaccine. 2011;29(4):754–762. doi: 10.1016/j.vaccine.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cong H, Mui EJ, Witola WH, et al. Human immunome, bioinformatic analyses using HLA supermotifs and the parasite genome, binding assays, studies of human T cell responses, and immunization of HLA-A*1101 transgenic mice including novel adjuvants provide a foundation for HLA-A03 restricted CD8+T cell epitope based, adjuvanted vaccine protective against Toxoplasma gondii . Immunome Research. 2010;6(1, article 12) doi: 10.1186/1745-7580-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]