

Abstract
Hb Baden (β18Val→Met) is a rare variant hemoglobin that has never been functionally or clinically characterized. We describe a Hb Baden heterozygote who exhibits normal growth and development, as well as age- and gender-appropriate hematological parameters. Surprisingly, in vitro analyses demonstrate that Hb Baden is relatively unstable and exhibits an abnormally high affinity for O2. These properties are likely to affect the physiologies of individuals who inherit the βBaden mutation in trans to a determinant for either a functionally relevant hemoglobin-opathy or a mild thalassemia. The data also provide insights into the function of the AB-segment/A-helix of the β-globin, supporting a structural model in which this poorly understood region serves as a scaffold that fixes the positions of other helices that directly impact β-globin function.
Introduction
Several hundred variant human hemoglobins (Hbs) have been catalogued and functionally characterized, providing critical insights into the structural determinants of Hb O2 affinity, allostery, cooperativity, and stability. The properties of one heterotetramer--Hb Baden (α2βV18M2)--have never been established, despite the likelihood that these descriptions would provide critical insights into functional roles subserved by the two structural motifs (the AB-segment and the A-helix) that flank the β18 residue. The amino-acid residues in these structures play uncertain, and largely unpredictable roles in heme-O2 binding, allosteric response to pH and phosphates, and other critical characteristics of hemoglobin heterotetramers. The most widely studied A-helix mutation, a Glu→Val at codon 6, exerts a negligible effect on Hb-O2 binding but permits the deoxygenated heterotetramer to polymerize, resulting in clinical sickle cell disease. In contrast, other A-helix mutations variably affect Hb O2-binding affinity(1, 2) and may(3) or may not(4) alter heterotetramer stability. A full accounting of the characteristics of Hb Baden would help to clarify the contribution of β-globin amino-acid residues 4-22 to normal heterotetramer function.
Hb Baden has been described on a single occasion, in two members of a family of northern European (Germanic) descent(5). While the original report identified the relevant mutation--a GTG→ATG transition at β-globin codon 18--neither the biochemical attributes of Hb Baden nor its phenotypical consequences could be determined. In this pedigree, the allelic gene carried a second mutation in cis (IVS-I-5 G→C) that activated several cryptic splice sites to produce a particularly severe β+-thalassemia(6). Consequently, both of the reported index cases expressed Hb Baden at extremely low levels (<3%), prohibiting informative hematological descriptions and precluding functional characterizations of the variant hemoglobin. Clinical and laboratory analyses were further complicated in the propositus, who carried a separate determinant for Hb Dhonburi (α2βV126G2) on his remaining β-globin gene.
We report the case of an adolescent male who is heterozygous for Hb Baden (α2βV18M2) and does not carry complicating β-globin gene defects either in cis or in trans. In this genetic context, the mutant allele does not elicit a clear hematological phenotype. Functional analyses of Hb Baden, though, demonstrate abnormalities in both O2-binding characteristics as well as constitutive heterotetramer stability. Based upon previous crystallographic studies of Hb with established coordinates of the β18Val within the β-globin subunit, we propose that the AB-segment/A-helix of the β-chain plays a critical role in maintaining the position and/or orientation of adjacent β-chain helices that directly interact with the O2-binding heme prosthesis.
Methods
Clinical and bench studies were conducted at the direction of the patient's attending hematologist, with prior consent from all subjects.
HPLC
Clarified hemolysates were resolved over a 50 mm × 4.6 mm Poly CAT A column (Poly LC, Columbia MD) using a D-7000 controller (Hitachi). Analyses were conducted with buffer A (40 mM Bis-Tris, 5 mM EDTA, pH 6.5), and buffer B (buffer A + 0.2 M sodium chloride) using a linear 25-90% buffer B gradient. The eluate was continuously monitored at 410 nm.
Hemoglobin isolation
Hemolysates were fractionated over a 2.5 × 20 cm Sephadex CM column using a linear potassium phosphate gradient (10 mM, pH 6.8 to 20 mM, pH 8.0) and the eluate analyzed at A576. Fractions corresponding to individual hemoglobins were pooled, concentrated using a Centricon 10-kDa filter (Millipore) and purity validated by HPLC analysis.
Hemoglobin oxygen affinity
Oxygen-equilibrium curves (OECs) were determined on a HEMOX analyzer (TCS, Southampton PA). Analyses of intact cells were conducted at 37°C using 15-20 mmL of fresh EDTA-anticoagulated blood resuspended in 30 mM TES buffer [134 mM NaCl, 5 mM KCl, 8 mM glucose, 0.1% BSA, and 0.01% antifoam (SAG-10, Union Carbide) adjusted to pH 7.4](7). Clarified hemolysates were studied at 25°C in potassium phosphate buffer (100 mM, pH 7.4) supplemented with 0.01% antifoam and 0.04% hexamethylphosphoramide (Fisher Scientific) as stabilizing agent(8). Each P50 value was averaged from two independent analyses. Hill plots were constructed from OEC data using proprietary HAS-200 software (TCS).
Hemoglobin stability
Chemical
Purified hemoglobins were diluted to ∼0.1 mM in buffer (0.1 mM Tris, pH 7.4), then diluted 10-fold in prewarmed buffer containing 17% (v:v) isopropanol. Aliquots were incubated at 37°C for 5 minutes(9), and denatured hemoglobins precipitated by centrifugation at 14 × g for five minutes. The supernatant A576 was subsequently determined using a SpectraMax plate reader (Molecular Devices). Mechanical: Hemoglobins were diluted to ∼13 mM with potassium phosphate buffer (10 mM, pH 8.0); aliquots (2 ml) were agitated for defined intervals at a setting of 2000 on a Maxi-Mix III type 65800 apparatus (Thermolyne)(10). Denatured products were precipitated by centrifugation, and soluble hemoglobin determined by A576 spectrophotometry.
Sequencing
Genomic DNA prepared from whole blood was amplified by polymerase chain reaction using synthetic oligomers positioned in the β-globin 5′- and 3′-flanking regions (5′gggcagagccatctattgc3′ and 5′GACCTCCCACATTCCCTTTTTAG3′, respectively). Sequencing was conducted by the NAPCore facility at The Children's Hospital of Philadelphia using β-globin gene-specific DNA primers (IDT, Coralville IA).
Mass spectrometry
Analyses were conducted by the Protein and Proteomics Core Facility at The Children's Hospital of Philadelphia. Hemolysate was resolved on a denaturing 4-12% bis-tris gel (Invitrogen). A gel slice containing the ∼16-kDa globin apoprotein was subjected to in-gel tryptic digestion(11) and resolved by reverse-phase HPLC on a ProteoPep nanocapillary column (New Objective). Tryptic fragments were separated using mobile phases A (1% methanol/0.1% formic acid) and B (phase A + 79% acetonitrile) over a 90 minute interval using a linear 5-45% phase B gradient. Peptides were eluted into a LTQ-Orbitrap XL mass spectrometer (ThermoFisher) at 300 nL/min, and repetitively scanned between m/z values of 300-1800 at an Orbitrap resolution of 60,000. Data-dependent MS/MS scans were conducted on the five most abundant ions. Additional instrument parameters are provided in Supplemental Information (online).
Globin identification
MS/MS spectra were searched using Sorcerer-SEQUEST (ver. 4.0.3, SagenResearch) against a custom sequence database comprising all globin variants in the UniProt knowledgebase (http://www.uniprot.org/uniprot/, release 15.15), concatenated with amino-acid sequences from yeast, E. coli, and other non-globin protein sources. Additional details of the search parameters are available as Supplemental Information (online).
In silico structural modeling
Due to failure to obtain either liganded or unliganded crystals of the mutant protein after several attempts, a structural model of βBaden was generated utilizing the Crystallographic Object-Oriented Toolkit (COOT)(12), using the known structures of deoxy (2HHB) and CO-liganded (1LJW) Hb A as templates.
Results
Patient description
The propositus is a 14-year old male who was lost to medical follow-up after routine perinatal screening revealed heterozygosity for an unspecified variant β-globin chain. The patient's interval history was unremarkable except for small congenital atrial- and ventricular-septal defects that spontaneously closed during childhood. There was no family history of anemia, polycythemia, hemoglobinopathy, or thalassemia. The patient's mother is of Lebanese, Irish, and German extraction, while his father claims English, Irish, and Native American ancestry. The propositus' physical examination was age-appropriate; specifically, growth and development were normal, and neither the spleen nor the liver were enlarged. Routine clinical laboratory studies (Table I) were within accepted limits, including a Hb of 16.0 g/dL (age-specific reference range 12.0-16.9 g/dL). Electrophoretic analysis revealed Hb A (56.5%), Hb A2 (2.7%), and a third Hb (40.9%) that was not definitively identified. Hematological evaluations of the mother and both of the patient's adolescent siblings were normal, including CBC analyses and hemoglobin electrophoreses. Blood from the father was not available for study.
Hemoglobin identification
The referring hospital provisionally identified the patient's abnormal hemoglobin as Hb Zoeterwoude (α2βV23A2)(13), based upon a characteristic elution profile over a Varian HPLC apparatus. This assignment was inconsistent with the patient's clinical presentation--normal values for Hb level and RBC number--as heterozygosity for Hb Zoeterwoude, a high O2-affinity hemoglobin, appears to effect frank polycythemia(13). Consequently, we conducted both genetic and protein-based analyses to directly identify the variant hemoglobin. An ∼1.5-kb DNA fragment, corresponding to the patient's full-length β-globin genes, was generated by PCR amplification of genomic DNA and subsequently subjected to automated sequencing. This analysis revealed a single-copy GTG→ATG transition at β-globin codon 18 (Fig. 1A) consistent with heterozygosity for βBaden. Importantly, no other mutations were identified; specifically, the cis-acting IVS-I-5 G→C that precluded analyses of the original index cases of Hb Baden(5). The amplicon was subsequently ligated into an expression vector and subclones sequenced (not shown), confirming the presence of both wild-type G and mutant A at position 1 of β-globin codon 18 and predicting a Val→Met missense substitution at helix position A15.
Figure 1. Identification of Hb Baden.

(A) Genomic sequencing. PCR-amplified β-globin genes were subjected to automated sequencing using site-specific DNA primers. The normal β-globin gene sequence is illustrated. An arrow indicates the position of the G→A transition that defines βBaden. (B) HPLC analysis of clarified hemolysate. Patient (top) and normal control samples (bottom) are illustrated. Positions of Hb A and Hb Baden are indicated. (C) Mass spectrometric analyses of wild-type and variant globins. Assigned MS/MS spectra of T-3 tryptic peptides from βA(top) and βBaden(bottom). Masses of corresponding b-ions (which retain the N-terminal β18 amino-acid) differ by 32 Da, while masses of corresponding y-ions (which delete the amino-acid at this position) are identical. (D) Full-scale mass spectrum. The m/z values for βA, βBaden, and βBaden-oxidized Met are illustrated.
The original pedigree that expressed Hb Baden carried several additional β-globin gene mutations, both in cis and in trans to the β18 GTG→ATG transition(5). This situation prohibited phenotypical description of an uncomplicated Hb Baden heterozygote and, in addition, precluded direct confirmation of its predicted amino-acid sequence. To remedy both issues, we assessed clarified hemolysate from the propositus using a standard HPLC method. Two peaks were observed: a prominent signal corresponding to Hb A, and a second signal--accounting for ∼53% of the total--that eluted with a shorter retention time (Fig 1B). Patient hemolysate was subsequently subjected to mass spectrometric analyses. MS/MS spectra of corresponding tryptic fragments from βA (Fig 1C, top) and βBaden (Fig 1C, bottom) demonstrate a 32 kDa mass shift in the m/z values for all of the b ions (which extend from the amino terminus) consistent with a Val→Met exchange at the first position of each peptide fragment. A full scan mass spectrum for the undissociated tryptic fragments (Fig 2D) calculates the abundance of βBaden as 0.68 (relative to βA), consistent with modest instability of Hb Baden in intact erythrocytes. Additionally, a small fraction of the variant globin peptide appeared as an oxidized product (Fig 2D), reflecting either methodological artifact and/or modest globin-chain instability. Consequently, genetic and protein studies agree on the identity of the variant hemoglobin, and indicate the likely effects of a Val→Met substitution on heterotetramer function.
Figure 2. O2-binding properties of Hb Baden.
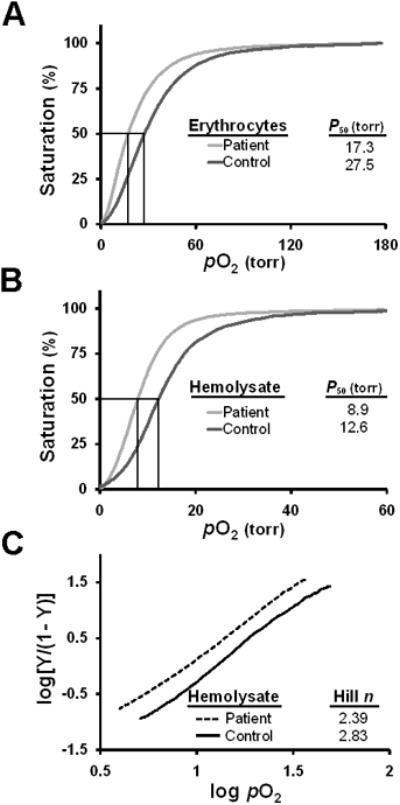
(A, B) Oxygen equilibrium curves. Tracings for intact erythrocytes (panel A) and clarified hemolysate (panel B) were generated for the propositus (black) and a normal donor control (grey). A vertical line indicates the P50 value for each sample. (C) Hill plots of clarified hemolysates. Hill plots of intact erythrocytes from the propositus (black) and a normal control donor (gray) are illustrated. Values of the best-fit lines, determined by linear regression analyses, are indicated.
O2-binding characteristics of Hb Baden
The hematological phenotype of the propositus predicted critical characteristics of Hb Baden likely to be affected by the β18Val→Met mutation. The high-normal value for Hb anticipated that the variant heterotetramer might exhibit increased affinity for O2. We assessed this possibility by conducting oxygen equilibrium curve (OEC) analyses on patient and control samples using a HEMOX analyzer. Fresh, intact red blood cells from the propositus exhibited a P50 value of 17.3 torr, significantly lower than the P50 value of 27.5 torr obtained for normal control cells (Fig 2A). Similarly, clarified hemolysate from the propositus displayed a decrease in P50 value relative to control hemolysate (8.9 and 12.6 torr, respectively; Fig 2B). Both studies concur with an increase in the affinity of Hb Baden for liganded O2. A Hill plot of data from the hemolysate study indicted a modest reduction in globin subunit cooperativity (Hill n=2.39) relative to control cells (n=2.83; normal value 2.8-3.0)(14) (Fig 2C), suggesting that the Val→Met mutation modestly alters the thermodynamic barrier to the fully liganded state. Because both the OEC and Hill analyses were conducted using cells (or hemolysate) containing a mixture of Hb Baden and normal Hb A, the data underestimate the true magnitude of the impact of a β18 Val→Met substitution on heterotetramer function.
Stability of Hb Baden
Two independent lines of evidence suggested that the β18Val→Met mutation might reduce the stability of the α2βBaden2 heterotetramer. Both Hb electrophoretic and mass spectrometric analyses observed a deficit in Hb Baden (relative to Hb A), while MS/MS studies additionally observed the presence of oxidized βBaden subunits that might be attributed to globin instability. We formally investigated this property by assessing the capacity of Hb Baden to resist denaturation under defined chemical and physical conditions. Chromatographically purified Hb Baden was highly sensitive to isopropanol denaturation, mirroring the chemical instability of control Hb S (Fig 3A). In contrast, Hb Baden--like control Hb A--was relatively resistant to mechanical stress (Fig 3B). The discordant response of Hb Baden to these denaturant stimuli, which affect different determinants of heterotetramer stability, are consistent with the anticipated effects of the β18Val→Met substitution on heterotetramer structure and, in combination with clinical laboratory and mass spectrometric analyses, predict significant instability in vivo in intact cells.
Figure 3. Stability of purified Hb Baden to defined denaturant stimuli.

(A) Chemical stability. Temporal stabilities are illustrated for Hbs A, S, and Baden when incubated in 17% isopropanol. (B) Mechanical stability. Temporal stabilities are illustrated for Hbs A, S, and Baden when mechanically agitated.
Discussion
Nearly two decades after Hb Baden was originally described(5), neither the phenotypes of heterozygotes, nor the biochemical characteristics of the variant heterotetramer have been described. Our studies address both issues, detailing the normal hematological parameters in a Hb Baden carrier (Table I), and demonstrating both an increase in O2-binding affinity (Fig 2) and a reduction in the stability (Fig 3) of heterotetramers carrying a β18Val→Met. These results elucidate the critical importance of AB-segment/A-helix residues to hemoglobin function, and implicate specific inter-helix contacts that participate in these processes.
Hb Baden exhibits compelling defects in O2-binding affinity and heterotetrameric stability that illuminate the importance of the AB-segment/A-helix residues to normal hemoglobin function. The established position of β18Val in Hb A crystal structure suggests an attractive mechanism for the increased O2 affinity observed for Hb Baden (Fig 2). Our in silico modeling studies of the relevant region indicate potential steric interactions between the side chain of the mutant Met18 and surrounding amino-acid residues (Fig 4). While βVal18 does not directly contact the protoporphyrin prosthesis, it is closely apposed to regions of the B and G helices, as well as to a third helix--the E helix--which is integral to the structure of the heme-binding pocket. Not surprisingly, E-helix residues are known to be critical determinants of both Hb stability and Hb O2-binding affinity(14). Three residues from the AB-segment/A-helix--βVal18, βTrp15, and βVal23--form a tightly packed hydrophobic pocket with βLeu114 and βPhe118 from the G helix and βLeu68 from the E helix. In βBaden subunits, the large side-chain of mutant β18Met would be expected to disrupt amino-acid packing in this multi-structure hydrophobic pocket, altering the positions and/or orientations of associating helices and, consequently, the nature of E-helix interactions with the protoporphyrin ring. Any shift in the positions of E-helix residues βHis63 (E7) or βVal67 (E11) would be anticipated to impact Hb O2 affinity because of the role that these amino acids play in regulating ligand access to heme iron(15). This model could account for the elevated O2 affinities of heterotetramers comprising embryonic β-like subunits [e.g., Hb Gower-2 (α2(x003B5)2)](16) which, like βBaden, contain a Met at position 18. Repeated attempts to obtain mutant crystals to validate this proposition have not yet been successful.
Figure 4. Ribbon model of amino-acid residues neighboring the β-globin AB-segment/A-helix.
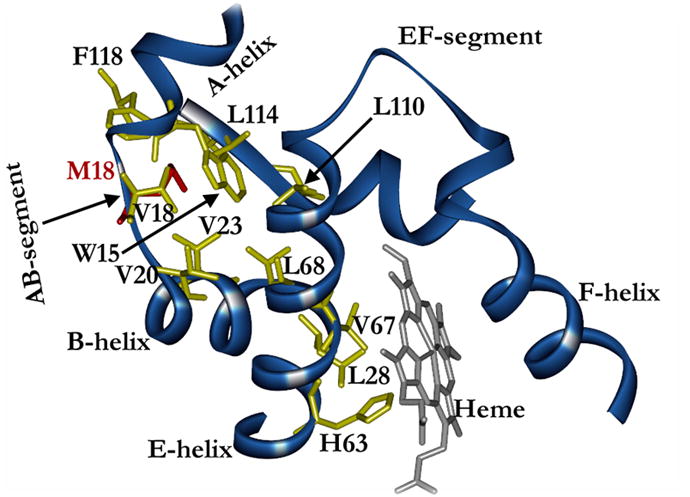
The M18 mutation (red) is encased in a nonpolar pocket formed by β-globin residues V18, W15, V23, V20, L68, L114, and F118. Structural changes in this region are transmitted through the E helix, altering its interaction with the heme prosthesis (grey).
The stability of heterotetrameric hemoglobin also appears to be critically dependent upon the integrity of the multi-helix hydrophobic pocket. A naturally occurring mutation that alters this region (Hb Belfast, β15Trp→Arg)(3) significantly reduces heterotetramer stability, while amino-acid substitution at a neighboring position that is oriented away from the pocket (Hb J Lens, β13Ala→Asp) exerts little effect on hemoglobin metabolism(4). This model is fully consistent with the specific sensitivity of Hb Baden to chemical, but not mechanical denaturants (Fig 3). Isopropanol is believed to promote denaturation by integrating into internal, nonpolar regions of native protein(17), such as the β-globin multi-helix contact region. A β18Val→Met would be predicted to disrupt the tight packing of nonpolar assemblies, permitting isopropanol access that would further compromise the structural integrity of this region. Both the abnormal O2-binding and stability attributes of Hb Baden suggest that the AB-segment/A-helix functions as a peptide ‘scaffold’ that fixes the positions and orientations of helices that contribute to the structure--and function--of the heme pocket.
The results of mass spectrometric analyses validate both the anticipated amino-acid sequence of βBaden, and also some of its critical functional characteristics. While an artifactual process cannot be fully excluded, the presence of a met-oxide form of the βBaden T-3 peptide (reflecting N-terminal sulfoxidation of the N-terminal Met; Fig 2D) appears to validate other studies of Hb Baden instability (Fig 3) and suggests that these measurements accurately describe the in vivo characteristics of βBaden.
Finally, our observations may have some importance for the management of the rare individual who carries the βBaden allele. While the high O2 affinity and instability of Hb Baden do not effect a clinically relevant phenotype in heterozygotes, they pose a potential risk to doubly heterozygous progeny who carry a functionally significant β-globin gene mutation in trans. A βBaden/β°-thalassemia genotype would likely result in clinically significant changes in both the quantity and O2-transport characteristics of the patient's hemoglobin. Genetic counseling would likely benefit selected patients with Hb Baden who plan on having children.
Supplementary Material
Acknowledgments
This work was funded through NIH awards R01-HL082754 (JER), R01-HL061399 (JER), and U54-HL070596 (OA). The authors thank Lynn A. Spruce of the CHOP Protein and Proteomics Core for expert assistance with mass spectrometry.
Footnotes
Disclosure of Conflicts of Interest: None of the five authors have actual or potential conflicts of interest.
Authorship Contributions: Osheiza Abdulmalik: Designed and conducted experiments, analyzed data, wrote manuscript
Martin K. Safo: Analyzed data
Steven M. Seeholzer: Conducted experiments, analyzed data, wrote manuscript
Nicole Hasbrouck: Analyzed data
Toshio Asakura: Analyzed data
J. Eric Russell: Analyzed data, wrote manuscript
References
- 1.Tondo CV, Bonaventura J, Bonaventura C, Brunori M, Antonini E. Functional properties of hemoglobin Porto Alegre (α2β9Ser→Cys2) and the reactivity of its extra cysteinyl residue. Biochemica et Biophysica Acta. 1974;342:15–30. doi: 10.1016/0005-2795(74)90101-9. [DOI] [PubMed] [Google Scholar]
- 2.Harano T, Harano K, Ueda S, Shibata S, Imai K, Seki M. Hemoglobin Machida (β6(A3)Glu→Gln), a new abnormal hemoglobin discovered in a Japanese family: Structure, function, and biosynthesis. Hemoglobin. 1982;6:531–535. doi: 10.3109/03630268209083766. [DOI] [PubMed] [Google Scholar]
- 3.Kennedy CC, Blundell G, Lorkin PA, Lang A, Lehmann H. Haemoglobin Belfast 15 (A12) Tryptophan→Argenine: A new unstable haemoglobin variant. British Medical Journal. 1974;4:324–326. doi: 10.1136/bmj.4.5940.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Djoumessi S, Rousseaux J, Dautrevaux M. Structural studies of a new hemoglobin: Hb J Lens, β13(A10) Ala→Asp. FEBS Letters. 1981;136:145–147. doi: 10.1016/0014-5793(81)81234-3. [DOI] [PubMed] [Google Scholar]
- 5.Divoky V, Bisse E, Wilson JB, Gu LH, Wieland H, Heinrichs I, et al. Heterozygosity for the IVS-I-5 (G→C) mutation with a G→A change at codon 18 (Val→Met; Hb Baden) in cis and a T→G mutation at codon 126 (Val→Gly; Hb Dhonburi) in trans resulting in a thalassemia intermedia. Biochemica et Biophysica Acta. 1992;1180:173–179. doi: 10.1016/0925-4439(92)90065-u. [DOI] [PubMed] [Google Scholar]
- 6.Kazazian HH, Orkin SH, Antonarakis SE, Sexton JP, Boehm CD, Goff SC, et al. Molecular characterization of seven β-thalassemia mutations in Asian Indians. EMBO J. 1984;3:593–596. doi: 10.1002/j.1460-2075.1984.tb01853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asakura T. Automated method for determination of oxygen equilibrium curves of red cell suspensions under controlled buffer conditions and its clinical applications. Crit Care Med. 1979;7:391–395. doi: 10.1097/00003246-197909000-00008. [DOI] [PubMed] [Google Scholar]
- 8.Asakura T, Adachi K, Schwartz E. Stabilizing effects of various organic solvents on protein. J Biol Chem. 1978;253:6423–6425. [PubMed] [Google Scholar]
- 9.Carrell R, Kay R. A simple method for the detection of unstable haemoglobins. British Journal of Haematology. 1972;23:615–619. doi: 10.1111/j.1365-2141.1972.tb07096.x. [DOI] [PubMed] [Google Scholar]
- 10.Asakura T, Ohnishi T, Friedman S, Schwartz E. Abnormal precipitation of oxyhemoglobin S by mechanical shaking. Proc Natl Acad Sci USA. 1974;71:1594–1598. doi: 10.1073/pnas.71.5.1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 12.Emsley P, Cowtan K. Model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 13.Harteveld CL, Groeneveld JHM, van Dam B, Delft PV, Akkerman N, Arkesteign S, et al. Hb Zoeterwoude [β23(B5)Val→Ala]: A bew β-globin variant found in association with erythrocytosis. Hemoglobin. 2005;29:11–17. [PubMed] [Google Scholar]
- 14.Bunn HF, Forget BG. Hemoglobin: Molecular, Genetic, and Clinical Aspects. Philadelphia: W. B. Saunders; 1986. [Google Scholar]
- 15.Tucker PW, Phillips SEV, Perutz MF, Houtchens R, Caughey WS. Structure of hemoglobins Zurich [His E7(63)β→Arg] and Sydney [Val E11(67)β→Ala] and role of the distal residues in ligand binding. Proc Natl Acad Sci USA. 1978;75:1076–1080. doi: 10.1073/pnas.75.3.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Z, Russell JE. Expression, purification, and characterization of human hemoglobins Gower-1 (ζ2ε2), Gower-2(a2ε2), and Portland-2 (ζ2β2) assembled in complex transgenic-knockout mice. Blood. 2001;97:1099–1105. doi: 10.1182/blood.v97.4.1099. [DOI] [PubMed] [Google Scholar]
- 17.Kim HC, Schwartz E. Unstable hemoglobins. In: Williams WJ, Beutler E, Erslev AJ, Lichtman MA, editors. Hematology. Fourth. New York: McGraw-Hill; 1990. pp. 1707–1710. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.