

Abstract
BACKGROUND & AIMS
Capillarization, characterized by loss of differentiation of liver sinusoidal endothelial cell (LSEC), precedes the onset of hepatic fibrosis. We investigated whether restoring differentiation to LSEC in liver affects their interactions with hepatic stellate cells (HSCs) and thereby promotes quiescence of HSCs and regression of fibrosis.
METHODS
Rat LSECs were cultured with inhibitors and/or agonists and examined by scanning electron microscopy for fenestrae in sieve plates. Cirrhosis was induced in rats using thioacetamide, followed by administration of BAY 60-2770, an activator of soluble guanylate cyclase (sGC). Fibrosis was assessed by Sirius red staining; expression of α-smooth muscle actin was measured by immunoblot analysis.
RESULTS
Maintenance of LSEC differentiation requires vascular endothelial growth factor-A stimulation of nitric oxide (NO)-dependent signaling (via sGC and cGMP) and NO-independent signaling. In rats with thioacetamide-induced cirrhosis, BAY 60-2770 accelerated the complete reversal of capillarization (restored differentiation of LSEC) without directly affecting activation of HSC or fibrosis. Restoration of differentiation to LSEC led to quiescence of HSC and regression of fibrosis, in the absence of further exposure to BAY 60-2770. Activation of sGC with BAY 60-2770, prevented progression of cirrhosis, despite continued administration of thioacetamide.
CONCLUSIONS
Differentiation of LSEC has an important role in activation of HSC and the fibrotic process in rats.
Keywords: VEGF, rat model, chronic liver disease, fenestration
Introduction
The liver sinusoidal endothelial cell (LSEC) undergoes loss of its highly specialized phenotype prior to activation of the hepatic stellate cell (HSC) and fibrosis 1, 2. If changes in LSEC phenotype not only precede HSC activation, but also were causally linked to it, this would significantly enhance our understanding of the development of chronic liver disease.
There is evidence that LSEC and HSC maintain each other’s differentiated phenotype. VEGF-A (henceforth referred to as VEGF) production by either HSC or hepatocytes maintains LSEC differentiation 3. Conversely, in vitro studies have shown that differentiated LSEC (i.e. fenestrated LSEC) prevent HSC activation and promote reversal of activated HSC to quiescence, but that LSEC lose this effect when they are de-differentiated or “capillarized” 4. Capillarization is defined as the in vivo loss of LSEC fenestration with development of an organized basement membrane 5. Capillarization occurs in fibrotic liver in humans 5–7 and experimental animal models 8–10 and precedes onset of fibrosis in alcoholic liver injury 1, in non-alcoholic fatty liver disease 2, and in experimental liver disease models induced by either intragastric alcohol infusion or thioacetamide (DeLeve LD et al, unpublished observations). Taken together, these findings suggest that capillarization not only precedes fibrosis, but may also be permissive for it and that changes in LSEC differentiation might be an integral part of the development of fibrosis.
LSEC fenestration is maintained by paracrine secretion of VEGF by hepatocytes or HSC and downstream autocrine production of NO by VEGF-stimulated eNOS 3, 11. Nitric oxide (NO) could act through either the soluble guanylate cyclase (sGC)/cyclic GMP (cGMP)/protein kinase G (PKG) pathway 12 or through protein S-nitrosylation 13. To examine the role of capillarization in the fibrotic process, we examined which of these signaling pathways control LSEC differentiation in vitro, using the presence of fenestration as a surrogate marker for LSEC differentiation and defenestration as a surrogate marker for capillarization. In vivo studies examined whether reversal of capillarization by activation of the relevant signaling pathway promotes HSC quiescence and regression of fibrosis, and prevents progression of cirrhosis.
Methods
Reagents
Provided in supplemental material.
Animal studies
Male Sprague-Dawley rats (body weight 220 –250 g) were obtained from Bantin and Kingman Laboratories (Fremont, CA). Thioacetamide, 200 mg/kg i.p., was given twice weekly. BAY 60-2770 14, 0.3 mg/kg i.g., was administered daily. VEGF antisense oligonucleotides, 20 mg/kg i.p., were given twice weekly for 4 weeks.
All protocols were reviewed and approved by the Animal Care and Use Committee at the University of Southern California to ensure ethical and humane treatment of animals. This study followed the guidelines outlined in the NIH “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86-23 revised 1985).
Cell isolation and culture
LSEC were isolated by collagenase perfusion, iodixanol density gradient centrifugation, and centrifugal elutriation as previously described and modified 15, 16. Yields from normal rat liver averaged 100 million cells with >95% viability and 99% purity as determined by uptake of formaldehyde-treated serum albumin, a specific marker of LSEC 17–19.
HSC were provided by the USC Non-parenchymal Liver Cell sub-core. HSC were isolated by collagenase/pronase digestion and Stractan density gradient centrifugation. Co-culture of HSC and LSEC was performed as previously described 4 (details in supplemental material).
Scanning electron microscopy and quantitative imaging
Sample preparation for scanning electron microscopy (SEM) and porosity (percentage of LSEC surface occupied by fenestrae) measurements of cells and liver tissue were performed as previously described 20 (details in supplemental material).
Evaluation of fibrosis and cirrhosis
Assessment of fibrosis and cirrhosis in Sirius red stained sections was performed blindly by G.C.K. Whole section-scanning morphometric image analysis was performed blindly by R.D.A. as previously described 2.
Statistics
All data, expressed as mean ± standard error of the mean (SE), were from at least three separate experiments. Groups were compared by analysis of variance (ANOVA) with a posteriori contrast by least significant difference; or by Student t test using the Microsoft Excel Analysis ToolPak (Microsoft, Redmond, WA). Results with p < 0.05 were considered significant.
Results
VEGF-stimulated NO/cGMP pathway is required to maintain LSEC differentiation
The key morphological features of differentiated LSEC, presence of fenestrae grouped into sieve plates, are rapidly lost in vitro 3, 21, 22. LSEC cultured for 2 days lost fenestration, but fenestration was well maintained in the presence of 40 ng/ml VEGF (Figure 1A). Conversely, knockdown of hepatic VEGF protein by antisense oligonucleotides (Figure 1B) led to marked sinusoidal defenestration (Figure 1C), confirming that VEGF maintains LSEC fenestration in vivo. VEGF acts through eNOS-catalyzed NO production 3 and NO signaling can occur through the soluble guanylate cyclase (sGC)/cGMP/protein kinase G (PKG) pathway 12 and/or through protein S-nitrosylation 13.
Figure 1. VEGF is required to maintain LSEC phenotype both in vitro and in vivo.
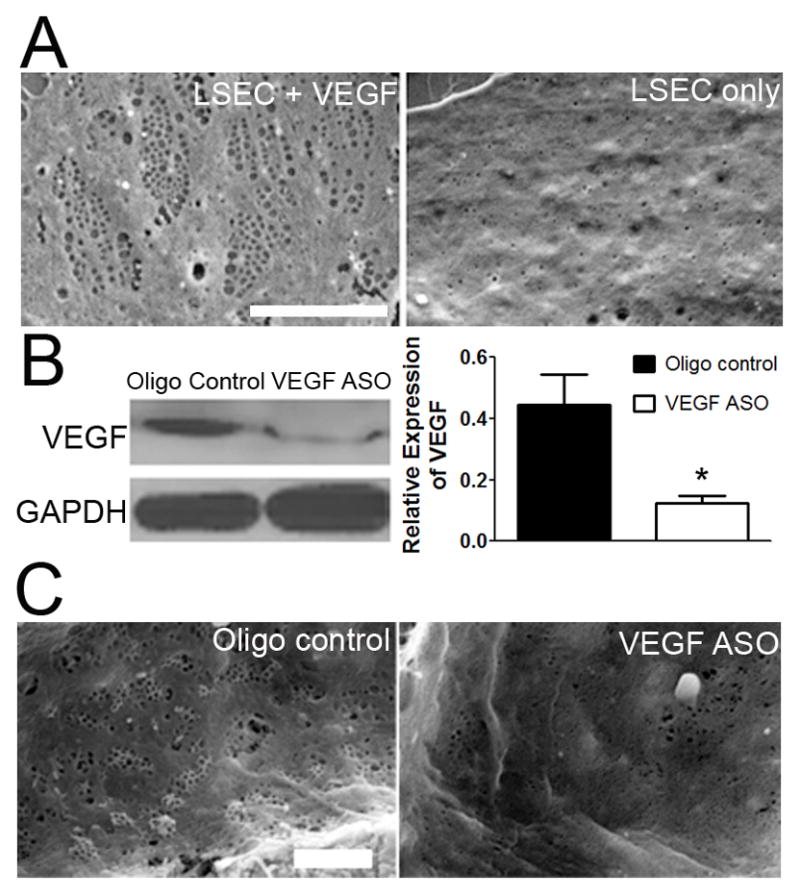
(A) Representative SEM of LSEC cultured with (left panel) and without (right panel) VEGF for 2 days show loss of fenestrae in sieve plates in vitro in the absence of VEGF. Scale bar, 5μm. (B) Hepatic expression of VEGF on immunoblot with densitometry and (C) representative SEM of hepatic sinusoids from rats treated with VEGF ASO or control oligonucleotides. * p<0.05. Scale bar, 2μm. All figures represent n ≥ 3.
Inhibition of sGC activity by 10 μM ODQ (Supplemental figure 1A) or PKG activity by 10 μM Rp-8-pCPT-PET-cGMPS (Supplemental figure 1B) completely blocked VEGF maintenance of LSEC fenestration (Figure 2A and B), confirming that the VEGF/cGMP pathway is necessary to maintain LSEC fenestration.
Figure 2. VEGF-stimulated cGMP is necessary but not sufficient to maintain LSEC phenotype in vitro.

Either ODQ (sGC inhibitor) or Rp-8-pCPT-PET-cGMPS (PKG inhibitor) completely blocks VEGF-stimulated (A) fenestration and (B) porosity in LSEC cultured for 2 days. Scale bar: 5μm. * p<0.001 versus VEGF control, n=3. (C) Either YC-1 (sGC activator) or 8-pCPT-cGMP (cGMP analog) without VEGF fails to normalize fenestration. However, VEGF+L-NAME (eNOS inhibitor) +YC-1 or VEGF+ODQ+8-pCPT-cGMP normalizes LSEC fenestration; thus both VEGF independent of NO plus the VEGF-stimulated cGMP pathway are required, whereas the experiment with VEGF + L-NAME + YC-1 also demonstrates that protein S-nitrosylation is not necessary. Scale bar: 5μm. (D) Porosity measurement of experiments described in (C). * p<0.001 versus VEGF control; NS, not significant; n = 3.
Maintenance of LSEC differentiation requires both VEGF stimulated-NO via the cGMP pathway plus VEGF via an NO-independent pathway
Primary LSEC were cultured with YC-1 (an NO-independent sGC activator) or 8-pCPT-cGMP (a cGMP analog) in the absence of VEGF to determine whether rescuing the cGMP pathway alone is sufficient to maintain LSEC differentiation. Neither 30 μM YC-1 nor 100 μM 8-pCPT-cGMP was able to maintain normal LSEC fenestration and porosity in the absence of VEGF (Figure 2C and D), though 30 μM YC-1 lead to cellular cGMP levels that were twice as high as in VEGF-stimulated LSEC. These data demonstrate that maintaining the cGMP pathway by itself fails to maintain normal LSEC fenestration. Thus the cGMP pathway is necessary, but not sufficient, to maintain LSEC differentiation.
In addition to the classical cGMP pathway, NO can also induce protein S-nitrosylation. To determine whether protein S-nitrosylation is required, LSEC were treated with VEGF, 3mM NG-nitro-L-arginine methyl ester (L-NAME) to inhibit VEGF-stimulated NO production, and YC-1 to stimulate sGC. NO production is not stimulated under these conditions, yet LSEC maintained normal fenestration (Figure 2C and D), demonstrating that protein S-nitrosylation is not necessary to maintain fenestration. Furthermore, LSEC maintained fenestration when cultured with VEGF, ODQ to block VEGF-stimulated cGMP production, and the cGMP analog, 8-pCPT-cGMP (Figure 2C and D). In conjunction with the studies described in the preceding paragraph, these two experiments demonstrate that maintenance of LSEC differentiation requires both VEGF-stimulated NO working through the cGMP pathway plus VEGF working independently of NO.
To confirm that VEGF-stimulated NO is needed, L-NAME was added to inhibit VEGF-stimulated NO production. LSEC completely defenestrated after a 2-day incubation with VEGF plus L-NAME (Figure 3). To further validate that the NO-independent pathway is required, the following experiments were performed. To determine whether NO without VEGF is sufficient, DETA-NONOate (an NO donor) was added to the culture medium. 6μM DETA-NONOate produced similar amounts of NO in the culture medium and cGMP levels in LSEC to LSEC cultured with VEGF (data not shown), yet the NO donor failed to maintain normal LSEC fenestration (Figure 3). This demonstrates that NO without VEGF is not sufficient. However, when LSEC were incubated with VEGF, L-NAME (to block VEGF-stimulated NO) plus DETA-NONOate, LSEC porosity was maintained (Figure 3). Taken together, these data confirm that both VEGF-independent of NO plus VEGF-stimulated NO are required to maintain normal LSEC fenestration.
Figure 3. Both VEGF-independent of NO plus VEGF-stimulated NO are required to maintain normal LSEC fenestration.
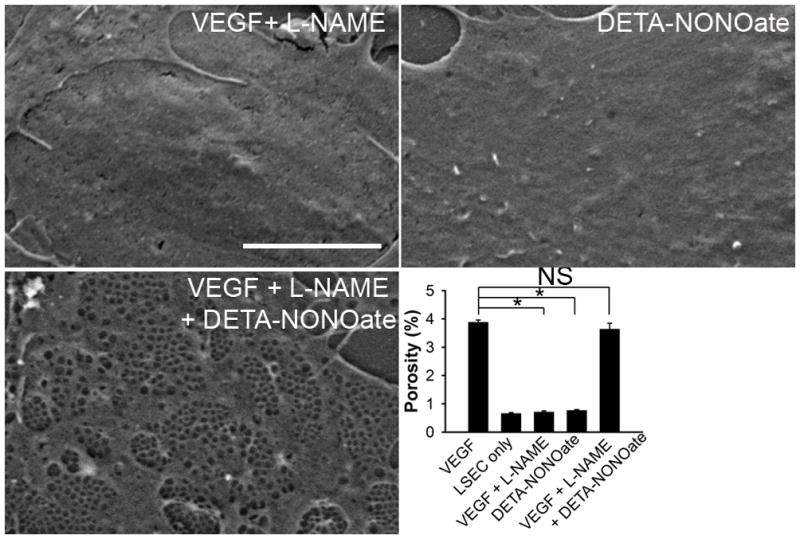
LSEC cultured with VEGF+L-NAME (top left panel) or DETA-NONOate (NO donor; top right panel) for 2 days lack fenestration, while LSEC cultured with VEGF+L-NAME+DETA-NONOate (bottom left panel) for 2 days demonstrate normal fenestration. Porosity is shown in the bottom right panel. Scale bar, 5μm. * p<0.001versus VEGF control; NS, not significant; n = 3.
Activation of sGC accelerates reversal of capillarization
Control sinusoids contain numerous fenestrae grouped into sieve plates (Figure 4A) and treatment with 3 weeks of thioacetamide (TAA) significantly defenestrated sinusoids (Figure 4A). Quantitative morphometry showed that TAA caused a 3 to 4-fold reduction of porosity, which is an indicator of both number and size of fenestrae (Figure 4B). One week after discontinuing TAA there was minimal restoration of fenestration (Figure 4A), as confirmed by porosity measurement (Figure 4B). Furthermore, LSEC cGMP levels decreased significantly after onset of capillarization and remained low 1 week after discontinuation of TAA (Figure 4C), consistent with previous reports 23. However, when rats received 3 weeks of TAA followed by 1 week of BAY 60-2770 (an NO-independent sGC activator 14), LSEC cGMP levels were normalized (Figure 4C) and fenestration (Figure 4A) and porosity (Figure 4B) were restored. Spontaneous reversal of capillarization did not occur until 2 weeks after discontinuation of TAA (data not shown). These results demonstrate that the cGMP pathway is suppressed during capillarization and that rescuing this pathway accelerates reversal of capillarization.
Figure 4. In vivo sGC activation restores LSEC phenotype in thioacetamide (TAA) -induced capillarization.

(A) Representative SEM of hepatic sinusoids. Top left panel: normal hepatic sinusoid with fenestrae grouped into sieve plates. Top right panel: capillarization after 3 weeks of TAA. Bottom left panel: minimal reversal of capillarization 1 week after discontinuing TAA. Bottom right panel: complete reversal of capillarization by 1-week of daily sGC activator after discontinuing TAA. Scale bar: 2μm. (B) Porosity from rats treated as in (A). * P<0. 001. (C) Cellular cGMP level in LSEC isolated from rats treated as indicated in (A). * P<0.001. n = 3.
Normalization of LSEC phenotype promotes HSC quiescence and regression of fibrosis
To determine in vivo whether reversal of capillarization promotes regression of fibrosis, 5 treatment groups were studied (Figure 5A). First, we investigated whether sGC activation has a direct effect on HSC. Three weeks of TAA (group 1) induced HSC activation (Figure 5B and C) and cirrhosis (Figure 5D) in rats. One week of solvent (group 2) or treatment with the sGC activator, BAY 60-2770 (group 3), following cessation of TAA did not significantly decrease HSC activation (Figure 5B and C) or fibrosis (Figure 5D and E). These data demonstrate that 1 week of the sGC activator did not directly decrease HSC activation in vivo although, as noted in the previous paragraph, LSEC fenestration had completely normalized. To examine whether restoration of LSEC differentiation would decrease HSC activation and fibrosis, rats received solvent treatment alone for an additional week. HSC activation, as assessed by α-SMA immunoblot analysis (Figure 5B) and immunofluorescence (Figure 5C), was significantly reduced in cirrhotic rats that received 1 week of the sGC activator followed by 1 week of solvent treatment (group 5). Sirius red staining and whole section-scanning morphometry (Figure 5B and C) showed a significant decline in fibrosis compared to group 4 rats that received 2 weeks of solvent after TAA (note: LSEC were capillarized after TAA and 1 week of solvent). These data demonstrate that after resolution of capillarization, the normalized LSEC promotes HSC quiescence and accelerates regression of fibrosis in the absence of the sGC activator. The decline in α-SMA with sGC activator-induced regression of fibrosis (group 5) was substantially greater than the decline in desmin (supplemental figure 2B), suggesting that the decrease in HSC activation is due to a combination of reversion to quiescence and apoptosis of activated HSC.
Figure 5. Restoration of the differentiated LSEC phenotype accelerates regression of thioacetamide (TAA)-induced early cirrhosis.

(A) Protocol for 5 treatment groups (n = 9–12). (B) Immunoblot with densitometry and (C) immunohistochemistry show elevated α-SMA expression after 3 weeks of TAA treatment. 1 week of solvent or sGC activator after discontinuing TAA does not alter α-SMA expression, but α-SMA expression is significantly reduced when the 1week of sGC activator is followed by 1 week of solvent (group 5) compared to 2 weeks of solvent after TAA (group 4). * p<0.001. Scale bar, 30 μm. (D) Representative Sirius red stain shows early cirrhosis in 3-week TAA treated rats (top middle). Cirrhosis persists after 1 week of either solvent (top right) or sGC activator (bottom left) after discontinuation of TAA, but only bridging fibrosis is observed when the 1 week of sGC activator is followed by 1 week of solvent (bottom right), compared to persistent cirrhosis in rats that received 2 weeks of solvent after TAA (bottom middle). Scale bar, 1.2 mm. (E) Quantification of fibrosis by scanning morphometry. * p<0.05.
VEGF and VEGF-receptor 2 (VEGF-R2) expression and hepatic vein serum VEGF increased with progression of fibrosis, consistent with reports in the literature 24–26, and decreased with sGC activator-induced regression of fibrosis (group 5; Supplemental figure 3B–E). eNOS expression stayed unchanged during fibrogenesis, consistent with reports by other groups 25, 26, but eNOS expression increased in the sGC activator-treated groups (groups 3 and 5), i.e. in rats with reversal of LSEC capillarization (Supplemental figure 3B).
Despite ongoing treatment with TAA, sGC activation restores LSEC differentiation and prevents progression of cirrhosis
Rats were treated with 6 weeks of TAA to induce cirrhosis and received co-treatment with BAY 60-2770 or solvent from week 4 to 6 of TAA. SEM showed that LSEC were defenestrated after both 3 and 6 weeks of TAA (group I and II), whereas rats that received 6 weeks of TAA and co-treatment with the sGC activator during the final 3 weeks (group III) had completely normal fenestration and porosity at the end of week 6 (Figure 6B). There was reduced expression of α-SMA in group III rats treated with TAA plus the sGC activator (Figure 6C and D) compared to group II rats that received 6 weeks of TAA alone. Sirius red staining and morphometric analysis showed more fibrosis after 6 weeks of TAA (group II) compared to 3 weeks of TAA (group I) treatment, but there was no progression in fibrosis in rats treated with 6 weeks of TAA plus the sGC activator from week 4 to 6 (group III) compared to group I rats treated with just 3 weeks of TAA (Figure 6E and F). These data indicate that in vivo sGC activation normalizes LSEC phenotype and completely prevents progression of fibrosis despite ongoing TAA exposure.
Figure 6. In vivo sGC activation prevents progression and accelerates regression of cirrhosis by restoration of LSEC differentiation.
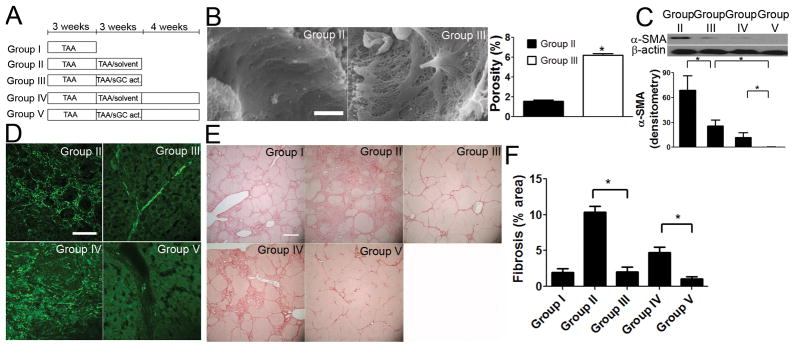
(A) Protocol for 5 treatment groups (n = 5). (B) Representative SEM (scale bar, 2μm) and porosity measurements (n=4) show that sGC activator treatment complete reverses capillarization despite ongoing TAA (group III) compared to solvent control (group II). * P<0. 001. (C) Immunoblot (n = 5) and (D) immunohistochemistry show that co-treatment of sGC with TAA from week 4 to 6 (group III) reduced α-SMA expression compared to 6-week TAA treated rats (group II). 4 weeks after discontinuation of treatment, α-SMA expression in group V was further decreased compared to 4 weeks earlier (group III) or compared to the TAA solvent control group (group IV). * P<0.05. Scale bar, 30 μm. (E) Representative Sirius red staining and (F) scanning morphometry thereof (n=5) show more fibrosis after 6 weeks TAA (group II) compared to 3 weeks TAA (group I). Extent of fibrosis is unchanged after 6 weeks TAA plus sGC activator from week 4 to 6 (group III) compared to 3 weeks TAA (group I). 4 weeks after discontinuation of treatment, regression of fibrosis is greater after sGC activator (group V) compared to the solvent group (group IV). Scale bar, 1.2 mm. * P<0.001.
VEGF and VEGF-R2 expression and hepatic vein serum VEGF increased after 6 weeks of TAA (group II), but decreased significantly when sGC activator was given along with TAA (group III vs group I and II) (Supplemental figure 4B–E). Increased eNOS expression was also observed in rats treated with TAA plus the sGC activator, compared to rats treated TAA without the sGC activator, i.e. rats with reversal of capillarization (Supplemental figure 4B).
Regression of more advanced fibrosis
Liver was assessed 4 weeks after cessation of treatment in rats that received 3 weeks of TAA alone followed by an additional 3 weeks of TAA with (group V) or without (group IV) 3 weeks of co-treatment with the sGC activator. In group V rats that received the sGC activator, α-SMA expression was markedly and significantly decreased 4 weeks after discontinuation of treatment compared to 4 weeks earlier (group III) or compared to the TAA plus solvent control group (group IV, Figure 6C and D). Sirius red staining and morphometric analysis showed that the sGC activator treatment group had significantly less fibrosis than the solvent control group 4 weeks after discontinuation of treatment (Figure 6E and F). VEGF and VEGF-R2 expression and hepatic vein serum VEGF were decreased, while eNOS expression was increased in the sGC activator treatment group comparing to the control group (Supplemental figure 4B–E). Both α-SMA and desmin decreased with regression of fibrosis, but the decrease in α-SMA was substantially greater (Supplemental figure 5B).
Mechanism of reversal of activated HSC to quiescence by sGC activation
The mechanism of action of the sGC activator on HSC was examined in vitro. HSC were allowed to activate in culture over 3 days. From day 3 to day 6, HSC were cultured under 4 different conditions: cultured alone, cultured with 5μg/ml BAY 60-2770, in co-culture with LSEC freshly isolated on day 3, or in co-culture with LSEC freshly isolated on day 3 plus BAY 60-2770 (Figure 7A). HSC cultured alone became activated and co-culture with LSEC did not decrease the number of activated HSC, presumably because these LSEC had become capillarized (Supplemental Figure 6A). HSC cultured with sGC activator showed some reduction in α-SMA positive cells compared with HSC cultured alone (Figure 7A; 47.4±2.6% versus 87.4±6.5% α-SMA positive cells; n = 4, p<0.001); doses of BAY 60-2770 greater than 5 μg provided no additional effect (data not shown). However, in the presence of both LSEC and sGC activator, there was a dramatic reduction in α-SMA positive HSC (6.3±1.0% α-SMA positive HSC) compared to culture with sGC activator alone (n=4, p < 0.001). Thus while sGC activation has a limited benefit in reversing HSC activation in vitro, near-complete reversal of the population to quiescence required co-culture with both LSEC and sGC activator. LSEC cultured with activated HSC plus the sGC activator maintained fenestration (Supplemental Figure 6B); differentiated LSEC cause reversal of activated HSC to quiescence 4, which suggests that (near-)complete reversal of activated HSC to quiescence by the sGC activator occurs through its action on LSEC differentiation. Rat cardiac microvascular endothelial cells did not prevent HSC activation, which indicates that this effect of LSEC is not a universal microvascular endothelial cell property (Supplemental figure 7).
Figure 7. Complete reversal of HSC activation by the sGC activator requires the presence of LSEC and is independent of NO.
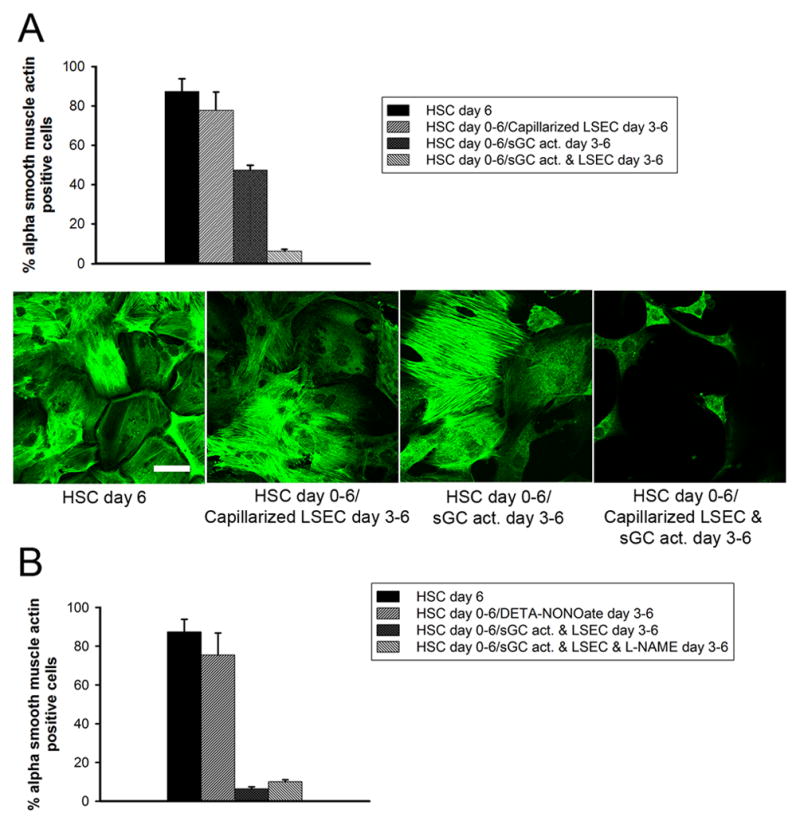
(A) Upper panel: percentage α-SMA-positive HSC determined by confocal microscopy is lower in HSC cultured with LSEC plus BAY60-2770 compared to BAY60-2770 alone (p<0.0001, n=4); lower panel: representative photomicrographs of HSC. HSC day 6: HSC cultured alone for 6 days; HSC day 0–6, LSEC day 3–6: HSC cultured alone from day 0–3, followed by co-culture from day 3–6 with LSEC isolated on day 3 (note: LSEC cultured with activated HSC for 3 days are capillarized); HSC day 0–6, sGC activator day 3–6: HSC cultured alone from day 0–6, with BAY 60-2770 from day 3–6; HSC day 0–6, sGC activator + LSEC day 3–6: HSC cultured alone for 3 days, followed by co-culture from day 3–6 with LSEC isolated on day 3 plus BAY 60-2770 (note: LSEC cultured for 3 days with BAY60-2770 remain differentiated) (Scale bar: 40μm. n = 4–6 for each group). (B) NO does not promote HSC quiescence. The percentage α-SMA-positive HSC is not significantly different in HSC cultured with or without DETA-NONOate or in HSC co-cultured with LSEC and BAY 60-2770 with or without L-NAME. HSC day 6: HSC cultured alone for 6 days; HSC day 0–6, DETA-NONOate day 3–6: HSC cultured alone from day 0–6,with DETA-NONOate added on day 3; HSC day 0–6, BAY + LSEC day 3–6: HSC cultured alone for 3 days, followed by co-culture from day 3–6 with LSEC isolated on day 3 plus BAY 60-2770; HSC day 0–6, sGC activator + LSEC + L-NAME day 3–6: HSC cultured alone for 3 days, followed by co-culture from day 3–6 with LSEC isolated on day 3 plus BAY 60-2770 with L-NAME. n = 3–6 for each group.
The studies in the previous paragraph support the concept that the sGC activator works on HSC through an LSEC-dependent mechanism. We previously reported that LSEC-mediated reversal of HSC activation was mediated by NO 4. We re-examined that in the current studies. To determine whether exogenous NO alone promotes reversal of activated HSC to quiescence, the NO donor DETA-NONOate was added to HSC that had been in culture for 3 days. DETA-NONOate did not reduce the number of α-SMA positive cells compared to HSC cultured alone (Figure 7; 75.4±11.3% versus 87.4±6.5% α-SMA positive HSC; n = 4, NS). To examine whether blocking NO production from LSEC abolishes the effect of LSEC plus sGC activator on HSC activation, L-NAME was added to HSC that had been in culture for 3 days (Figure 7B). L-NAME did not block the reversal of activated HSC to quiescence induced by LSEC plus sGC activator. These two pieces of data suggest that NO does not mediate the LSEC effect on HSC phenotype. Our previous report 4 was a misinterpretation of the data: the most likely interpretation is that L-NAME blocked VEGF-stimulated NO production, resulting in capillarized LSEC (Figure 3) that were unable to promote reversal of activated HSC to quiescence.
Discussion
The major findings reported here are as follows. Maintenance of LSEC differentiation in vitro requires VEGF-stimulated NO working through sGC activation plus VEGF working through an NO-independent pathway. In vivo studies demonstrated that sGC activation can reverse LSEC capillarization in cirrhosis. Once sGC activation normalizes the LSEC phenotype, there is subsequent accelerated reversal of HSC activation and fibrosis, even in the absence of the sGC activator. The sGC activator had a limited effect on reversal of HSC activation in vitro, but near-complete reversal of the HSC population to quiescence occurred with co-culture with LSEC plus sGC activator. The sGC activator also reversed capillarization and prevented progression of cirrhosis despite ongoing thioacetamide treatment.
These findings demonstrate that restoration of LSEC differentiation promotes HSC quiescence and thereby accelerates regression and prevents progression of fibrosis. These in vivo findings confirm previous in vitro studies 4 of the effect of LSEC on HSC activation and the lack of an effect once LSEC are capillarized. Thus the proposed paradigm is that differentiated LSEC have a gatekeeper function that promotes HSC quiescence and that this gatekeeper function is lost with capillarization. sGC activation-induced differentiation of LSEC lead to a decrease in α-SMA expression of HSC with a decline in desmin-positive cells. However as the decline in desmin-positive cells (total HSC number) was substantially less than the decline in α-SMA-positive cells (activated HSC), this suggests that there was both an element of HSC apoptosis as well as reversion of HSC to quiescence. This mix of reversion to quiescence and apoptosis is consistent with recent reports by other investigators 27, 28. TUNEL was negative (data not shown) in all groups, but this was likely a false negative: TUNEL is a freeze frame of a rapidly turning over process and may be difficult to capture.
The sGC activator prevented HSC activation in vitro (data not shown), consistent with previous reports using cGMP analogues 29. However we observed no direct effect of the sGC activator on HSC in vivo in established fibrosis and only a limited in vitro effect on activated HSC. These seemingly contradictory findings are reconciled by the observation that quiescent HSC express the α1β1 heterodimeric form of sGC required for cGMP generation and downstream events, but that expression of α1β1 heterodimeric sGC is undetectable in activated HSC 30. Thus sGC activation may work through both preservation of LSEC phenotype and prevention of HSC activation when it prevents the transition from normal to fibrotic liver. However once fibrosis is established, HSC are already activated and non-responsive to the sGC activator. Thus in established fibrosis, sGC activation induces regression of existing fibrosis and prevents progression of fibrosis by restoring LSEC differentiation with subsequent crosstalk of SEC with HSC, rather than by directly acting on the HSC.
VEGF-stimulated NO maintains LSEC differentiation 3. The current study examines the downstream signaling and demonstrates that the VEGF-stimulated cGMP pathway is necessary, but not sufficient, to maintain LSEC differentiation. Two VEGF pathways are required to maintain LSEC differentiation: VEGF independent of NO plus VEGF stimulated-NO acting through the cGMP pathway. Multiple signaling pathways are activated by VEGF, including MAPK (mitogen-activated protein kinase), FAK (focal adhesion kinase), PI3K/Akt (phosphatidylinositol 3-kinase), and PLC- γ (phospholipase C-γ) 31. Actin cytoskeletal filaments may play a role in LSEC fenestration 32–34. The MAPK pathway responsible for actin reorganization might therefore be a good candidate for the NO-independent pathway. Although VEGF is a key factor in maintaining LSEC differentiation, VEGF and VEGF-R2 are increased in fibrosis, suggesting that capillarization is due to disruption of signaling downstream of VEGF. As restoration of cGMP levels in vivo was sufficient to normalize the LSEC phenotype, this demonstrates that the limiting defect responsible for capillarization in this model of cirrhosis was in the NO/sGC/cGMP pathway. These findings are consistent with observed decreases in eNOS activity, NO production and cellular cGMP levels in LSEC from cirrhotic liver 23, 35, 36.
There are several clinical implications to the findings reported here. First, restoring LSEC phenotype in capillarized liver may be a viable approach to promoting regression of fibrosis or preventing progression of disease in the face of an ongoing insult. Second, in aging-related pseudo-capillarization 37, 38, loss of fenestration decreases clearance of chylomicron remnants. The increase in circulating chylomicron remnants is thought to play a key role in initiation of atherosclerosis 39, 40. Furthermore, the decline in LSEC scavenger receptor-mediated endocytosis with aging 41 may contribute to progression of atherosclerosis 41, 42. Thus restoring LSEC differentiation could be a therapeutic approach for atherosclerosis. Third, there is a large body of literature on endothelial cell-pericyte interactions in various vascular beds 43–47. This suggests that the current findings might also pertain to other organs, e.g. the lung or kidney, and provide a similar therapeutic strategy for fibrosis.
Supplementary Material
Acknowledgments
The authors thank Robert S. McCuskey (University of Arizona) for assistance with scanning electron microscopy, and Michelle MacVeigh-Aloni for assistance with immunostaining and confocal microscopy.
Grant Support: This work was supported by National Institutes of Health grant R01-DK66423 (L.D.D) and by the Histology and Microscopy sub-core of the USC Research Center for Liver Diseases (National Institutes of Health grant P30DK048522).
Abbreviations
- α-SMA
α-smooth muscle actin
- eNOS
endothelial nitric oxide synthase
- HSC
hepatic stellate cell
- LSEC
liver sinusoidal endothelial cell
- L-NAME
NG-nitro-L- arginine methyl ester
- NO
nitric oxide
- OsO4
osmium tetroxide
- PKG
protein kinase G
- SEM
scanning electron microscopy
- TAA
thioacetamide
- sGC
soluble guanylate cyclase
- VEGF
vascular endothelial growth factor
- VEGF-R2
vascular endothelial growth factor 2
Footnotes
Disclosures: None of the authors have a financial conflict of interest with the work in this manuscript.
Author Contributions: G.X. designed and performed experiments, analyzed data, and wrote the manuscript; X.W., L.W. and L.W. designed and performed experiments; R.D.A. performed whole section-scanning morphometric image analysis of fibrosis; G.C.K performed semi-quantitative blind scoring of liver histology; W.A.G. provided VEGF and Antisense Oligonucleotides and guidance as to their use; L.D.D designed experiments, supervised research, analyzed data, and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Horn T, Christoffersen P, Henriksen JH. Alcoholic liver injury: defenestration in noncirrhotic livers-a scanning electron microscopic study. Hepatology. 1987;7:77–82. doi: 10.1002/hep.1840070117. [DOI] [PubMed] [Google Scholar]
- 2.DeLeve LD, Wang X, Kanel GC, et al. Prevention of Hepatic Fibrosis in a Murine Model of Metabolic Syndrome with Non-Alcoholic Steatohepatitis. American Journal of Pathology. 2008;173:993–1001. doi: 10.2353/ajpath.2008.070720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeLeve LD, Wang X, Hu L, et al. Rat liver sinusoidal endothelial cell phenotype is under paracrine and autocrine control. Am J Physiol-Gastrointest Liver Physiol. 2004;287:G757–763. doi: 10.1152/ajpgi.00017.2004. [DOI] [PubMed] [Google Scholar]
- 4.DeLeve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48:920–930. doi: 10.1002/hep.22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaffner F, Popper H. Capillarization of hepatic sinusoids in man. Gastroenterology. 1963;44:239–242. [PubMed] [Google Scholar]
- 6.Xu B, Broome U, Uzunel M, et al. Capillarization of hepatic sinusoid by liver endothelial cell-reactive autoantibodies in patients with cirrhosis and chronic hepatitis. American Journal of Pathology. 2003;163:1275–89. doi: 10.1016/S0002-9440(10)63487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horn T, Junge J, Christoffersen P. Early alcoholic liver injury: changes of the Disse space in acinar zone 3. Liver. 1985;5:301–310. doi: 10.1111/j.1600-0676.1985.tb00253.x. [DOI] [PubMed] [Google Scholar]
- 8.Martinez-Hernandez A, Martinez J. The Role of Capillarization in Hepatic Failure: Studies in Carbon Tetrachloride-induced Cirrhosis. Hepatology. 1991;14:864–874. doi: 10.1002/hep.1840140519. [DOI] [PubMed] [Google Scholar]
- 9.Mori T, Okanoue T, Sawa Y, et al. Defenestration of the Sinusoidal Endothelial Cell in a Rat Model of Cirrhosis. Hepatology. 1993;17:891–897. [PubMed] [Google Scholar]
- 10.Warren A, Bertolino P, Benseler V, et al. Marked changes of the hepatic sinusoid in a transgenic mouse model of acute immune-mediated hepatitis. Journal of Hepatology. 2007;46:239–46. doi: 10.1016/j.jhep.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 11.Yamane A, Seetharam L, Yamaguchi S, et al. A new communication system between hepatocytes and sinusoidal endothelial cells in liver through vascular endothelial growth factor and Flt tyrosine kinase receptor family (Flt-1 and KDR/Flk-1) Oncogene. 1994;9:2683–2690. [PubMed] [Google Scholar]
- 12.Griffith TM. Studies of endothelium-derived relaxant factor (EDRF), its nature and mode of action. European Heart Journal. 1985;6:37–49. doi: 10.1093/oxfordjournals.eurheartj.a061753. [DOI] [PubMed] [Google Scholar]
- 13.Hess DT, Matsumoto A, Kim S-O, et al. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 14.Knorr A, Hirth-Dietrich C, Alonso-Alija C, et al. Nitric oxide-independent activation of soluble guanylate cyclase by BAY 60-2770 in experimental liver fibrosis. Arzneimittelforschung. 2008;58:71–80. doi: 10.1055/s-0031-1296471. [DOI] [PubMed] [Google Scholar]
- 15.DeLeve LD, Wang X, McCuskey MK, et al. Rat liver endothelial cells isolated by anti-CD31 immunomagnetic sorting lack fenestrae and sieve plates. American Journal of Physiology- Gastrointestinal and Liver Physiology. 2006;291:G1187–9. doi: 10.1152/ajpgi.00229.2006. [DOI] [PubMed] [Google Scholar]
- 16.Steffan AM, Gendrault JL, McCuskey RS, et al. Phagocytosis, an unrecognized property of murine endothelial liver cells. Hepatology. 1986;6:830–836. doi: 10.1002/hep.1840060505. [DOI] [PubMed] [Google Scholar]
- 17.Blomhoff R, Eskild W, Berg T. Endocytosis of formaldehyde-treated serum albumin via scavenger pathway in liver endothelial cells. Biochemical Journal. 1984;218:81–6. doi: 10.1042/bj2180081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blomhoff R, Smedsrød, et al. Preparation of isolated liver endothelial cells and Kupffer cells in high yield by means of an enterotoxin. Experimental Cell Research. 1984;150:194–204. doi: 10.1016/0014-4827(84)90714-6. [DOI] [PubMed] [Google Scholar]
- 19.Eskild W, Kindberg GM, Smedsrod B, et al. Intracellular transport of formaldehyde-treated serum albumin in liver endothelial cells after uptake via scavenger receptors. Biochemical Journal. 1989;258:511–20. doi: 10.1042/bj2580511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie G, Wang L, Wang X, et al. Isolation of periportal, midlobular, and centrilobular rat liver sinusoidal endothelial cells enables study of zonated drug toxicity. American journal of physiology Gastrointestinal and liver physiology. 2010;299:G1204–10. doi: 10.1152/ajpgi.00302.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Géraud C, Schiedzewski K, Demory A, et al. Liver Sinusoidal Endothelium: A Microenvironment-Dependent Differentiation Program in Rat Including the Novel Junctional Protein Liver Endothelial Differentiation-Associated Protein-1. Hepatology. 2010;52:313–326. doi: 10.1002/hep.23618. [DOI] [PubMed] [Google Scholar]
- 22.Sellaro TL, Ravindra AK, Stolz DB, et al. Maintenance of hepatic sinusoidal endothelial cell phenotype in vitro using organ-specific extracellular matrix scaffolds. Tissue Engineering. 2007;13:2301–2310. doi: 10.1089/ten.2006.0437. [DOI] [PubMed] [Google Scholar]
- 23.Rockey DC, Chung JJ. Reduced nitric oxide production by endothelial cells in cirrhotic rat liver: endothelial dysfunction in portal hypertension. Gastroenterology. 1998;114:344–351. doi: 10.1016/s0016-5085(98)70487-1. [DOI] [PubMed] [Google Scholar]
- 24.Mejias M, Garcia-Pras E, Tiani C, et al. Beneficial effects of sorafenib on splanchnic, intrahepatic, and portocollateral circulations in portal hypertensive and cirrhotic rats. Hepatology. 2009;49:1245–56. doi: 10.1002/hep.22758. [DOI] [PubMed] [Google Scholar]
- 25.Gracia-Sancho J, Russo L, Garcia-Caldero H, et al. Endothelial expression of transcription factor Kruppel-like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut. 2011;60:517–24. doi: 10.1136/gut.2010.220913. [DOI] [PubMed] [Google Scholar]
- 26.Trebicka J, Hennenberg M, Laleman W, et al. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology. 2007;46:242–53. doi: 10.1002/hep.21673. [DOI] [PubMed] [Google Scholar]
- 27.Tröger J, Gwak GY, Schwabe R. Hepatic stellate cells revert to a quiescent phenotype during liver fibrosis regression and do not undergo mesenchymal-epithelial transition. Hepatology. 2011;54:S1. [Google Scholar]
- 28.Kisseleva T, Paik Y, Scholten D, et al. Hepatic stellate cells revert to an inactive phenotype during regression of fibrosis. Hepatology. 2010;52:S1. [Google Scholar]
- 29.Kawada N, Kuroki T, Uoya M, et al. Smooth Muscle [alpha]-Actin Expression in Rat Hepatic Stellate Cell Is Regulated by Nitric Oxide and cGMP Production. Biochemical and Biophysical Research Communications. 1996;229:238–242. doi: 10.1006/bbrc.1996.1786. [DOI] [PubMed] [Google Scholar]
- 30.Failli P, Defranco RMS, Caligiuri A, et al. Nitrovasodilators Inhibit Platelet-Derived Growth Factor-Induced Proliferation and Migration of Activated Human Hepatic Stellate Cells. Gastroenterology. 2000;119:479–492. doi: 10.1053/gast.2000.9354. [DOI] [PubMed] [Google Scholar]
- 31.Olsson A-K, Dimberg A, Kreuger J, et al. VEGF receptor signalling ? in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 32.Braet F, DeZanger R, Baekeland M, et al. Structure and dynamics of the fenestrae-associated cytoskeleton of rat sinusoidal endothelial cells. Hepatology. 1995;21:180–189. [PubMed] [Google Scholar]
- 33.Braet F, Spector I, Zanger RD, et al. A novel structure involved in the formation of liver endothelial cell fenestrae revealed by using the actin inhibitor misakinolide. Proc Natl Acad Sci U S A. 1998;95:13635–13640. doi: 10.1073/pnas.95.23.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tazaki T, Sasaki T, Uto K, et al. p130Cas, Crk-associated substrate plays essential roles in liver development by regulating sinusoidal endothelial cell fenestration. Hepatology. 2010;52:1089–1099. doi: 10.1002/hep.23767. [DOI] [PubMed] [Google Scholar]
- 35.Gupta TK, Toruner M, Chung MK, et al. Endothelial dysfunction and decreased production of nitric oxide in the intrahepatic microcirculation of cirrhotic rats. Hepatology. 1998;28:926–31. doi: 10.1002/hep.510280405. [DOI] [PubMed] [Google Scholar]
- 36.Shah V, Toruner M, Haddad F, et al. Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental cirrhosis in the rat. Gastroenterology. 1999;117:1222–8. doi: 10.1016/s0016-5085(99)70408-7. [DOI] [PubMed] [Google Scholar]
- 37.Cogger VC, Warren A, Fraser R, et al. Hepatic sinusoidal pseudocapillarization with aging in the non-human primate. Experimental Gerontology. 2003;38:1101–7. doi: 10.1016/j.exger.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 38.McLean AJ, Cogger VC, Chong GC, et al. Age-related pseudocapillarization of the human liver. Journal of Pathology. 2003;200:112–117. doi: 10.1002/path.1328. [DOI] [PubMed] [Google Scholar]
- 39.Le Couteur DG, Warren A, Cogger VC, et al. Old age and the hepatic sinusoid. Anatomical Record. 2008;291:672–83. doi: 10.1002/ar.20661. [DOI] [PubMed] [Google Scholar]
- 40.Botham KM, Wheeler-Jones CPD. Introduction to the Biochemical Society Focused Meeting on Diet and Cardiovascular Health: chylomicron remnants and their emerging roles in vascular dysfunction in atherosclerosis. Biochemical Society Transactions. 2007;35:437–9. doi: 10.1042/BST0350437. [DOI] [PubMed] [Google Scholar]
- 41.Simon-Santamaria J, Malovic I, Warren A, et al. Age-related changes in scavenger receptor-mediated endocytosis in rat liver sinusoidal endothelial cells. The journals of gerontology. Series A, Biological sciences and medical sciences. 2010:65. doi: 10.1093/gerona/glq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li R, Oteiza A, Sorensen KK, et al. Role of liver sinusoidal endothelial cells and stabilins in elimination of oxidized low-density lipoproteins. American journal of physiology Gastrointestinal and liver physiology. 2011;300:G71–81. doi: 10.1152/ajpgi.00215.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circulation Research. 2005;97:512–23. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 44.Armulik A, Genové G, Mäe M, et al. Pericytes regulate the blood-brain barrier. Nature. 2010 doi: 10.1038/nature09522. in press. [DOI] [PubMed] [Google Scholar]
- 45.Daneman R, Zhou L, Kebede AA, et al. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature. 2010 doi: 10.1038/nature09513. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hirschi KK, Rohovsky SA, D’Amore PA. Cell-cell interactions in vessel assembly: a model for the fundamentals of vascular remodelling. Transplant Immunology. 1997;5:177–178. doi: 10.1016/s0966-3274(97)80034-2. [DOI] [PubMed] [Google Scholar]
- 47.Siemerink MJ, Augustin AJ, Schlingemann RO. Mechanisms of ocular angiogenesis and its molecular mediators. Developments in opthalmology. 2010;46:4–20. doi: 10.1159/000320006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.