

Abstract
Purpose
Prospectively identifying who will benefit from adjuvant chemotherapy (ACT) would improve clinical decisions for non-small-cell lung cancer (NSCLC) patients. In this study, we aim to develop and validate a functional gene set that predicts the clinical benefits of ACT in NSCLC.
Experimental Design
An 18-hub-gene prognosis signature was developed through a systems biology approach, and its prognostic value was evaluated in six independent cohorts. The 18-hub-gene set was then integrated with genome-wide functional (RNAi) data and genetic aberration data to derive a 12-gene predictive signature for ACT benefits in NSCLC.
Results
Using a cohort of 442 Stage I–III NSCLC patients who underwent surgical resection, we identified an 18-hub-gene set which robustly predicted the prognosis of patients with adenocarcinoma in all validation datasets across four microarray platforms. The hub genes, identified through a purely data-driven approach, have significant biological implications in tumor pathogenesis, including NKX2-1, Aurora Kinase A, PRC1, CDKN3, MBIP, RRM2. The 12-gene predictive signature was successfully validated in two independent datasets (N=90 and N=176). The predicted benefit group showed significant improvement in survival after ACT (UT Lung SPORE data: hazard ratio=0.34, p=0.017; JBR.10 clinical trial data: hazard ratio=0.36, p=0.038), while the predicted non-benefit group showed no survival benefit for two datasets (hazard ratio=0.80, p=0.70; hazard ratio= 0.91, p=0.82).
Conclusions
This is the first study to integrate genetic aberration, genome-wide RNAi data, and mRNA expression data to identify a functional gene set that predicts which resectable patients with non-small-cell lung cancer will have a survival benefit with ACT.
Keywords: non-small-cell lung cancer, predictive gene signature, adjuvant chemotherapy, integrative analysis, hub genes
Introduction
Lung cancer is the leading cause of cancer-related mortality worldwide (1). Even after an apparent complete resection of non-small-cell lung cancer (NSCLC), 33% of patients with pathologic stage IA and 77% with stage IIIA disease die within 5 years of diagnosis. Several randomized trials have demonstrated that there is a survival benefit with adjuvant chemotherapy (ACT) in resected NSCLC (2–6). However, the effect is modest - only 4–15% improvement in 5-year survival, while at the same time, such treatment may cause serious adverse effects (5, 7). Because the response to standard chemotherapy in lung cancer varies, it would be very helpful to prospectively identify the subgroup(s) of patients who are unlikely to benefit from ACT, and therefore, can be spared the side effects of unnecessary treatment.
Recently, several groups have developed gene expression signatures aiming to classify lung cancer patients into groups with distinct clinical outcomes (8–21). However, most current molecular signatures for lung cancer are prognostic only, and do not provide any estimation as to whether a patient would benefit from ACT. In addition, the signatures often contain large numbers of genes, with limited information about the functional importance of the genes. All of these problems limit the clinical application of those signatures. In this study, we used a systems biology approach to construct a survival-related gene network in NSCLC and identified 18 “hub” genes, which consistently co-expressed with many survival-related genes and hence play important roles in multiple biological processes. Here we show that the 18-hub-gene set is functionally important and predicts the overall prognosis of NSCLC patients with Stage I–III disease. Our previous RNAi screening study (22) identified “synthetic lethal” genes; knockdown of these genes enhanced the cancer-killing effects of paclitaxel, which implies that these genes modulate chemotherapy drugs’ effects in cancer cells. Recently, genetic aberration data have been successfully used to identify several key lung cancer driver genes in tumorigenesis(23, 24). By integrating synthetic lethal genes and genetic aberration information with the hub genes, we identified a 12-gene set that predicts ACT benefits in patients with Stage I – IIIA NSCLC. This 12-gene set was validated in two independent datasets, including the University of Texas Lung Specialized Program of Research Excellence (UT Lung SPORE) cohort (n=176) and the National Cancer Institute of Canada Clinical Trials Group JBR.10 clinical trial cohort (n=90).
Materials and Methods
Patients and samples
UT Lung SPORE cohort
Patients were eligible to enter the study if they underwent curative resection for NSCLC at MD Anderson Cancer Center between December 1996 and June 2007. Those with radiation therapy were excluded from the study. All tissue samples were obtained by surgical resection from patients who had provided written informed consent. Tissues were stored at −140°C after being snap frozen in liquid nitrogen. Serial sectioning of each sample was used to histologically evaluate tumor and malignant cells content before RNA extraction(25). The primary tumor tissues from 176 patients were randomly selected from the UT Lung SPORE tumor collection based on stringent, predefined quality control procedures, including the presence of ≥70% tumor tissue and ≥50% malignant cells in the frozen tissue used for RNA extraction. In this cohort, 133 patients are adenocarcinomas (ADCs) and 43 patients are squamous cell carcinomas (SCCs); 49 patients received ACT (mainly carboplatin plus taxanes) and 127 patients did not receive ACT. The clinical information and gene expression data for the UT Lung SPORE cohort were deposited in GEO database (GSE42127).
Samples from other groups
In addition to the UT Lung SPORE data, 7 public NSCLC microarray datasets (10, 13, 17, 26–29) were used in this study. The National Cancer Institute Director’s Challenge Consortium study (Consortium dataset)(13), which is the largest independent public available lung cancer microarray dataset and involves 442 resected ADCs, was used as the training set. Six datasets were used to validate the prognosis signature: UT lung SPORE data, GSE3141 (ADC n=58, SCC n=53), GSE8894 (ADC n=62, SCC n=76), GSE11969 (ADC n=90, and SCC n=35), GSE13213 (ADC n=117), GSE4573 (SCC n=129). Among these 6 datasets, three (GSE 13213, GSE8894 and GSE11969) are Asian cohorts. Two datasets were used to validate the predictive signature: UT lung SPORE data and GSE14814 that includes 90 samples (49 patients with vinorelbine plus cisplatin ACT and 41 patients without ACT) collected from the JBR.10 trial. Table 1 provides detailed information on these datasets. Since 43 out of 133 samples in the original JBR.10 dataset (GSE14814) were also included in the Consortium data (training set), these 43 samples were excluded from the JBR.10 dataset to ensure the independence between the training and validation sets.
Table 1.
Clinical characteristics of patients in the validation datasets.
| SPORE New data | GSE13213 Tomida2009 | GSE11969 Matsuyama2011 | GSE8894 Lee2008 | GSE3141 Bild2006 | GSE4573 Raponi2006 | GSE14814 Zhu2010 | |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Total Patients | n = 176 | n = 117 | n = 149 | n = 138 | n = 111 | n = 129 | n = 90 |
| Gender | |||||||
| Female | 83 (47.2%) | 57 (48.7%) | 48 (32.2%) | 34 (24.6%) | - | 47 (36.4%) | 23 (25.6%) |
| Male | 93 (52.8%) | 60 (51.3%) | 101 (67.8%) | 104 (75.4%) | - | 82 (63.6%) | 67 (74.4%) |
|
| |||||||
| Stage | |||||||
| I | 112 (63.6%) | 79 (67.5%) | 78 (52.3%) | - | 62 (55.9%) | 73 (56.6%) | 45 (50.0%) |
| II | 32 (18.2%) | 13 (11.1%) | 26 (17.4%) | - | - | 33 (25.6%) | 45 (50.0%) |
| III | 30 (17.0%) | 25 (21.4%) | 45 (30.2%) | - | - | 23 (17.8%) | - |
| IV | 1 (0.6%) | - | - | - | - | - | - |
| Unknown | 1 (0.6%) | - | - | 138 (100%) | 49 (44.1%) | - | - |
|
| |||||||
| Histology | |||||||
| ADCs | 133 (75.6%) | 117 (100%) | 90 (60.4%) | 62 (44.9%) | 58 (52.3%) | - | 28 (31.1%) |
| SCCs | 43 (24.4%) | - | 35 (23.5%) | 76 (55.1%) | 53 (47.7%) | 129 (100%) | 52 (57.8%) |
| Others | - | - | 24 (16.1%) | - | - | - | 10 (11.1%) |
|
| |||||||
| Median Follow-up (Months) | 47.4 | 68 | 78 | 41.8 | 31.1 | 34.2 | 64.8 |
|
| |||||||
| Platform | Illumina Human-WG6 V3 | Agilent 44K | Agilent 21.6K custom array | Affy U133 Plus_2 | Affy. U133 Plus_2 | Affy. U133A | Affy. U133A |
RNA extraction and microarray profiling
The frozen tissues specimens were processed on the cryostat to generate multiple 5-micron thick sections for subsequent homogenization using an electric homogenizer. Before RNA extraction, histology sections were stained and reviewed to assess the percentage of tumor. Total RNA was extracted using TRIREAGENT (Life Technologies, NY, USA) according to manufacturer’s protocol. The nanodrop spectrophotometer (Thermo Fisher, Wilmington, DE, USA) was used to estimate the concentration of RNA while the quality of the RNA was assessed on Nano Series II RNA LAB-chips using Agilent Bioanalyzer 2100 (Agilent Technologies, Inc., Santa Clara, CA, USA). All samples selected for RNA profiling have an RNA integrity number (RIN) ≥5. Total RNA was processed for analysis on the Illumina Human-6 V3 arrays according to Illumina protocols for first- and second-strand synthesis, biotin labeling and fragmentation.
Microarray data preprocessing
The UT Lung SPORE Illumina beadarray data were processed using Model-Based Background Correction (MBCB) method (30). For the Consortium and GSE14814 datasets, the raw Affymetrix. cel data was downloaded from caArray database and Gene Expression Omnibus (GEO), respectively. Both datasets were then preprocessed by the Robust Multiarray Average (RMA) algorithm and quantile-quantile normalization (31). For datasets that did not provide raw data file (GSE3141, GSE4573, and GSE8894) or used the Agilent platforms (GSE11969 and GSE13213), we downloaded the author processed data from GEO. All gene expression values were log2 transformed. The Entrez IDs were used to map genes across microarray platforms.
Survival analysis
Overall survival time was calculated from the date of surgery until death or last follow-up contact. Survival curves were estimated using the Kaplan-Meier method (32) and were compared using log-rank test. Univariate and multivariate survival analyses were performed using Cox proportional-hazards model(33). Meta-analysis was used to combine the results across different test sets. It was performed using the R package metagen (34). The overall combined estimate of the hazard ratio was estimated based on their values and standard errors in individual validation set.
Gene network analysis
The lung cancer survival-related gene network was constructed using the Consortium dataset. The association between the expression level of each probeset and survival time was evaluated using multivariate Cox model adjusted for age, cancer stage, and sample processing sites. The false discovery rate (FDR) was calculated from a beta-uniform mixture model (35). All probesets that passed the FDR criteria (FDR<10%) were included in gene network analysis. When there are multiple probesets corresponding to a single gene, the expression levels from the probesets were averaged to derive the gene level expression. The Sparse PArtial Correlation Estimation (SPACE) algorithm(36) was used to construct the network of survival-associated genes using their expression values in the Consortium dataset. From the constructed gene network, genes with at least 7 connections to other genes were identified as “hub” genes.
Prediction methods
Supervised principal component analysis (37, 38) was applied to construct the prediction model, which is based on the linear combinations of gene expressions of the provided gene set in the training data set. Then we apply the risk prediction model to the test set, and derive a risk score for each samples based on their gene expressions. The test set samples are divided in to two equal-sized risk groups based on the median of the predicted risk scores. For the prediction model we used the first 3 principal components, which the default parameter of the program with prediction (superPC R package). The training and validation strategy is illustrated in Figure 1. Please see the Supplementary SWEAVE report for all analysis details including the models, parameters and procedures for this study.
Figure 1.

Schematic of the study design for: (A) the development and validation of the 18-hub-gene prognosis signature; and (B) the development and validation of the 12-gene predictive signature.
Results
Identification of an 18-hub-gene set
From the Consortium dataset, we identified 797 genes (Figure 1) whose expression levels were associated with patients’ overall survival time (FDR < 10%). Next, we constructed a lung cancer survival-related gene network (see Method Section) based on expression changes of these 797 genes across 442 lung cancer samples in the Consortium dataset (Figure 2A). We identified 18 hub genes that are connected with at least 7 other genes in the constructed network. Among these 18 genes (summarized in Figure 2B), RRM2, AURKA, PRC1, and CDKN3 are associated with poor prognosis, while the remaining 14 genes are associated with good prognosis.
Figure 2.

(A) The topology of the constructed survival related gene network in NSCLC. The gene expression of 797 survival-related genes (false discovery rate< 10%) from 442 ADC samples in the Consortium dataset was used to construct the gene network based on the Sparse PArtial Correlation Estimation (SPACE) algorithm. Each node represents one gene (only nodes with at least one connection are shown). The genes with at least 7 connections with other genes were identified as hub genes and labeled in red. (B) Composition of the 18 survival related hub genes. Network Connectivity refers to the number of genes that the hub gene has direct connection with based on the constructed gene network. The hazard ratios (HR) and P-values for each gene were derived from Cox models adjusted for age, cancer stage, and sample processing sites. P-values for synthetic lethal were from our previous study(22), and P-values less than 0.05 were highlighted in yellow. Genetic alteration information was from the Tumorscape program (http://www.broadinstitute.org/tumorscape): “+” indicates the genes with significant amplification and “−” indicates significant deletion in lung cancer. The gene symbols of the 12 genes with either synthetic lethal or genetic alteration were highlighted in red.
Prognosis performance of the 18-hub-gene set
Robustness of the prognostic signature
A prognostic signature was developed using the expression of the 18-hub-gene set and patients’ survival outcomes from the Consortium dataset (training set) based on the superPC method. The prognostic signature was evaluated in ADC patients from 5 independent validation sets across 4 different microarray platforms, including: UT Lung SPORE (Illumina-6 V3), GSE3141 and GSE8894 (Affymetrix U133Plus2), GSE11969 (Agilent 21.6K custom array) and GSE13213 (Agilent 44K). Patients receiving adjuvant chemotherapy were excluded from the validation sets. Remarkably, the prognostic signature consistently predicted overall survival in all 5 validation sets. The predicted high-risk group has significantly worse survival outcomes than the predicted low-risk group: GSE3141 (n=58, HR=2.06 [1.01–4.2], p=0.042), UT Lung SPORE (n=94, HR=2.85 [1.36–5.97], p=0.0038), GSE8894 (n=62, HR=3.73 [1.45–9.59], p=0.0034), GSE11969(n=90, HR=1.87 [0.99–3.53], p=0.049), GSE13213 (n=117, HR=2.74 [1.51–4.98], p=0.00058) (Figure 3A). Since most of the public datasets did not provide complete demographic information, we performed multivariate survival analysis in UT Lung SPORE data. The predicted high-risk group has significantly worse survival outcomes than the predicted low-risk group (HR=2.93 [1.25–6.88], p=0.0137) after adjusting for stage, age and gender (Table S2). Furthermore, the 18-hub-gene signature consistently predicted the prognosis of patients with stage I disease: GSE3141 (n=30, HR=3.88 [1.18–12.8], p=0.016), UT Lung SPORE (n=67, HR=3.18 [1.14–8.84], p=0.019), GSE11969 (n=52, HR=2.85 [0.99–8.23], p=0.043), GSE13213 (n=79, HR=5.31 [1.99–14.2], p=0.00020) (Figure 3B).
Figure 3.

Validation of the 18-hub-gene signature in six independent data sets. (A) Lung ADC patients. (B) Stage I Lung ADC patients. (C) Lung SCC patients. The high- and low-risk groups were defined based on the 18-hub-gene signature which was derived from the Consortium data. The median of the estimated risk scores was used as the cut off to partition the patients into high-risk and low-risk groups. Dashed Grey and solid black lines indicate predicted high- and low-risk groups. Grey and black filled circles represent censored samples. Hazard ratio (HR) compares the overall survival of the high-risk group and the low-risk group. P values were obtained by the log-rank test. The patients with chemotherapy were excluded from the validation sets.
The 18-hub-gene prognostic signature is ADC-specific
ADC and SCC are two major NSCLC histology subtypes with fundamentally different molecular makeup (39). Since the 18-hub-gene prognostic signature was derived from a cohort of ADC patients only, we wanted to determine whether it was specific for ADC or could also predict prognosis for SCC patients. We tested the 18-hub-gene prognostic signature in SCC patients from GSE3141 (n=53), UT Lung SPORE (n=33), GSE8894 (n=76), GSE11969 (n=35), GSE4573 (n=129). The results (Figure 3C) show that the signature does not predict survival in any of the 5 datasets. Note that 4 datasets (GSE3141, SPORE, GSE8894 and GSE13213) have both ADC and SCC patients, and the 18-hub-gene signature has significant prognostic values in all ADC sub-cohorts, but not in any SCC sub-cohorts. These results show that the 18-hub-gene prognostic signature is ADC-specific (p=0.00047 for interaction between histology and signature). In addition, 15 out of the 18 hub genes express differently between ADC and SCC patients, and unsupervised clustering analysis based on the expression of the 18 hub genes divided the patients into an ADC dominated group and a SCC dominated group (Figure S1).
The 18-hub-gene set has better performance than top-ranked genes
Selecting an optimal small set of genes from a large candidate gene list is a critical step for developing clinically practical molecular assays. The most widely used ranking based approaches (10) select genes with the most prominent p values obtained from individual gene-based testing. We derived an 18-top-ranked-gene set containing 18 genes with the most significant association with the survival outcome based on the multivariate Cox model adjusted for age, cancer stage, and sample processing sites using the Consortium dataset. Here, we compared the performance of the 18-hub–gene set with the 18-top-ranked-gene set and the whole 797 survival related gene set (797-SR-gene set), all derived from the Consortium dataset.
Comparing the prognostic performances
Using the Consortium dataset as the training set, the prognosis performances of the 18-hub-gene set, 18-top-ranked-gene set and 797-SR-gene set were compared in 5 independent validation sets for ADC patients. Figure 4A shows that the 18-hub-gene signature consistently predicted prognosis in all 5 validation sets (HR=2.46, p=1.74E-08 from meta-analysis), and outperformed the 18-top-ranked-gene signature (HR=1.88, p=4.45E-05 from meta-analysis), which predicted prognosis (with p value < 0.05) in only 2 out of 5 datasets. Furthermore, the 18-hub-gene signature has similar or even better prognostic performance than the 797-SR-gene signature (HR=2.24, p=2.72E-07) (Figure 4A). It suggests that the hub-gene approach can effectively reduce the number of genes in the signature without sacrificing the prediction performance.
Figure 4.

Comparison of the 18-hub-gene set, 18-top-ranked-gene set and 797-SR-gene signature: (A) Summary of the prognostic performance for 18 hub-gene set, 18 top-ranked-gene set and 797-SR-gene set. The training data is the Consortium data; the validation sets include five different datasets. *Overall HR and p-values were calculated from meta-analysis. (B) Expression variation across the population in the Consortium dataset based on principal component analysis. (C) Pair-wise mutual information distance based on expression values in the Consortium dataset. (D) Entropy of expression values in the Consortium dataset.
Comparing the information content
We used information theory approach (see supplementary methods) to study the reason why the hub-gene approach works well. The 18-hub-gene set has significantly higher pair-wise mutual information distance (a measure for independency) than the 18-top-ranked-gene set (p=1E-9, Figure 4B), indicating that the hub-gene set has lower information redundancy than the top-ranked-gene set. As a result, the 18-hub-gene set has much higher entropy (a measure for information content, Figure 4D) and captures more variation across patient population (Figure 4C) than the 18-top-ranked-gene set. In summary, the hub-gene approach can effectively retain information while largely reducing the number of genes in the signature, which is important for developing clinically practical assays.
Derivation of a 12-gene set
Figure 1B illustrates the procedures for deriving and validating the 12-gene signature. First, we found that 7 out of the 18 hub genes have significant genetic aberration in lung cancer using the Tumorscape program (http://www.broadinstitute.org/tumorscape) (Figure 2B), including a key lung cancer driver gene (NKX2-1) (23). Furthermore, 9 out of the 18 hub genes were “synthetic lethal” with paclitaxel for NSCLC (i.e. siRNA gene-specific knockdowns which killed NSCLC cells only in the presence of paclitaxel) based on our previous study (22) (Figure 2B). In total, 12 out of 18 hub genes either have genetic aberration or are ‘synthetic lethal’ for paclitaxel in lung cancer. These genes are DOCK9, RRM2, AURKA, HOPX, NKX2-1, TTC37, COL4A3, IFT57, C1orf116, HSD17B6, MBIP, and ATP8A1. We developed a prediction model (12-gene signature) using the expression of these 12 genes and patients’ survival outcomes in the Consortium dataset (training set) based on the superPC model and tested its prognostic effects on five independent ADC cohorts. The predicted high-risk group has significantly worse survival outcomes than the predicted low-risk group in the testing cohorts (Figure S2), so this 12-gene signature can predict prognosis in early stage NSCLC.
The 12-gene signature predicts survival benefits from ACT in NSCLC
Because these 12 genes are “hubs” of the survival related genes, and play roles in cell response to chemotherapy drugs or have genetic aberrations in lung cancer, we hypothesize that this 12-gene set can predict survival benefits from ACT in NSCLC. To test this hypothesis, we tested whether the 12-gene signature can predict which patients would benefit from ACT using two independent validation sets: (1) 90 NSCLC samples from JBR.10 clinical trial (17) in which 49 patients received vinorelbine plus cisplatin ACT treatment and 41 patients did not receive ACT; (2) 176 NSCLC samples from UT Lung SPORE in which 49 patients received ACT (mainly Carboplatin plus Taxanes) and 127 patients did not receive ACT. Each patient in the validation sets was classified into a high- or low-risk group based on the 12-gene signature. Different from the prognosis biomarkers, no study has shown that the predictive biomarkers for chemotherapy are ADC- or SCC- specific. Therefore, we tested the 12-gene signature in all NSCLC patients as other predictive biomarker studies (8, 17, 40). For the JBR.10 dataset, the ACT-treated patients showed longer survival than those without ACT (HR 0.36 [0.13–0.97], p=0.038; Figure 5A) in the high-risk group; while patients with ACT treatment had no significant survival benefits (HR, 0.91[0.391–2.11], p=0.823; Figure 5A) in the low-risk group. Furthermore, the patients with ACT treatment even have worse survival outcomes in the first 21 months for the low-risk group. The signature has a similar predictive effect in the UT Lung SPORE data: the patients who received ACT had better overall survival in the high-risk group (HR=0.34 [0.13–0.86], p=0.017, Figure 5B), but not in the low-risk group (HR=0.80 [0.266–2.42], p=0.70, Figure 5B).
Figure 5.
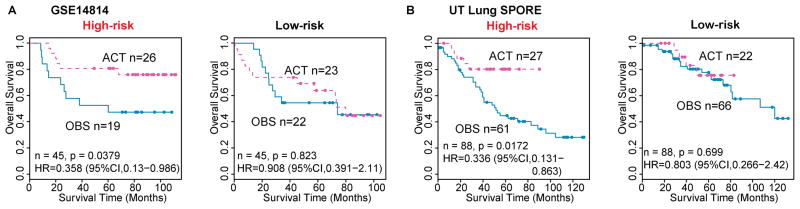
Validation of the 12-gene predictive signature in two independent data sets. (A) JBR.10 clinical trial dataset. The high- and low-risk groups were defined by the 12-gene signature. The patients were divided into two equal-sized risk groups based on their estimated risk scores. In the high-risk group, patients with ACT (pink line) have significantly longer survival time than patients without ACT (Observation group, blue line). In the low-risk group, patients with ACT (pink line) do not have significantly longer survival time than patients without ACT (Observation group, blue line). (B) UT Lung SPORE dataset. The risk groups were defined by the same 12-gene signature. In the high-risk group, patients with ACT (pink line) have significantly longer survival time than patients without ACT (Observation group, blue line). In the low-risk group, patients with ACT (pink line) do not have significantly longer survival time than patients without ACT.
Discussion
This is the first study to use systems biology approaches to identify hub genes for prognostic and predictive signatures in lung cancer. Feature selection, which selects the most predictive genes while excluding the redundant genes to reduce the cost, is a critical step in developing a clinically practical molecular assay. A commonly used selection approach is based on ranking the performance of individual features (genes), and selecting the top ranked features. However, the combination of top ranked individual genes may not be optimal, because it does not consider relationship and potential information redundancy among genes. In this study, we applied a systems biology approach to identify hub genes which have 7–30 connections with other genes in the constructed survival-related network (Figure 2), so the expression changes of these hub genes will affect many other genes and lead to substantial changes at the system level. This 18-hub-gene set has higher information content (Figure 4B–D) than the 18-top-ranked-gene set and has remarkably robust prognosis performances across different datasets and microarray platforms. From the Molecular Signatures Database (MsigDB), we identified four lung cancer prognosis signatures derived from the same training dataset (the Consortium dataset). In addition, we identified another four NSCLC prognosis signatures with similar number of genes from the literatures (9, 17, 41, 42). We compared the prediction performances of the 18-hub-gene signature and the eight prognosis signatures in GSE13213 (n=117 for ADC) which has the most ADC patients in our testing datasets, and the 18-hub-gene signature clearly outperforms all other eight signatures (Table S3). These results indicate that the hub genes capture the key mRNA expression information related to NSCLC patients’ survival.
The 18 hub genes, identified through a purely data-driven approach, have important biological implications in tumor development, including seven cancer metastasis genes and one key lung cancer driver gene (NKX2-1), demonstrating the biological relevance of this approach. To understand the potential biological and therapeutic relevance of the identified hub gene signature, we downloaded all the gene lists from the MSigDB C2 gene sets database, and evaluated the overlap between our signatures and the gene lists (Table S4). Most notably, all of the hub genes have been identified in at least one gene list concerning cancer or carcinoma, while 7 genes are associated with cancer metastasis gene lists, and 6 genes are related to proliferation. The large overlap with cancer-associated gene lists implies that our prognostic gene signature is biologically relevant, and it is likely that the prognostic power is originated from their association with cancer metastasis or tumor cell proliferation. In particular, NKX2-1 and HOPX are important for the activation of p53 pathways and potentially helpful in repressing lung ADC development (43) (44), and could be promising candidates for lung cancer therapy.
This is also the first study to integrate RNAi functional screening data(22) with mRNA expression and genetic aberration data(23, 24) to identify a gene signature that predicting the benefits of ACT in lung cancer. This 12-gene signature is predictive for ACT benefits in NSCLC for both paclitaxel or vinorelbine plus cisplatin (JBR.10 clinical trial cohort), and commonly used combinations such as carboplatin plus taxanes (UT Lung SPORE cohort). The 12-gene signature is both prognostic for ADC patients and predictive for adjuvant chemotherapy, so this signature has the potential to facilitate clinical decisions on using adjuvant chemotherapy for early stage NSCLC patients. In addition, the 18-gene set is a stronger prognostic signature for early stage ADC patients, so the 18-gene signature could be helpful if the goal is to predict patients’ prognosis only. In addition, the EGFR mutation and ALK rearrangement could be important to patient response to chemotherapy, and further studies are need to test how these mutations could affect the usage of the 12-gene signature.
Although the current study shows the promising results and interesting functional relevance of the 12-gene signature, one limitation of this study is that the sample size is not big enough (45) to test the interaction between the signature and the treatment groups. Since the long-term survival outcome may be confounded by other non-treatment factors, we tested the interaction between signature groups and treatment using the survival in first three years after treatment. For JBR10 data, the interaction between the 12-gene signature and the treatment groups is significant (p=0.0005). The SPORE testing data is from a retrospective study. This dataset has limited sample size with treatments and the follow-up time is short, so the number of observed events is too small to reach the significant p value for the interaction term. Therefore, a further prospective study with large sample size is needed to valid the 12-gene signature as a predictive signature.
In this study, the 18-hub-gene prognosis signature was validated in 6 independent datasets across five different microarray platforms (including Affymetrix U133Plus2, Affymetrix U133A, Illumina Human-6 V3, Agilent 21.6K custom arrays, and Agilent 44K), and the validation cohorts include three studies conducted in western countries and three studies conducted in Asia. The prognosis performances are consistent across these heterogeneous populations and experimental techniques. We tested the 12-gene predictive signature in two independent cohorts: the JBR.10 clinical trial and the UT Lung SPORE cohort. To our knowledge, this is the first study to include two validation datasets for predictive signatures in lung cancer. Zhu et al (17) and Chen et al (8) developed a predictive signature for ACT lung cancer, but it was only tested on the JBR 10 trial data. The UT Lung SPORE cohort used carboplatin plus taxanes based ACT treatments and the microarray experiment platform is different. All these results show the robustness of the prognosis and predictive signatures developed from this study. To facilitate other researchers to reproduce the results in this study, we have provided a literate programming R package (SWEAVE report) in the supplementary material.
In summary, through systems biology approaches we have identified a robust 18-hub-gene signature for prognosis of resected NSCLC patients. Furthermore, we developed a 12-gene prognostic and predictive signature for ACT benefit in NSCLC patients using integrative analysis approaches. A prospective clinical study is needed to further validate the clinical value of the prognosis and predictive signatures in the decision-making process of ACT for resected NSCLC patients.
Supplementary Material
Statement of Translational Relevance.
Randomized clinical trials have demonstrated the survival benefit of adjuvant chemotherapy (ACT) in resected non-small-cell lung cancer (NSCLC). Because the response to standard chemotherapy in lung cancer varies, it would be very helpful to prospectively identify patients who will benefit from ACT to guide the treatment plan. In this study, we used a systems biology approach to identify an 18-hub-gene signature that can robustly predict the prognosis of patients with early stage adenocarcinoma of the lung. Furthermore, we integrated these hub genes with genetic aberration and genome-wide RNAi functional data to derive a 12-gene set that is predictive for survival benefit with ACT. This 12-gene predictive signature has been validated in two independent NSCLC cohorts. As this predictive signature contains a small set of genes and has shown robust predictive power across platforms and studies, it may have therapeutic utility in determining which early stage NSCLC patients would benefit from ACT.
Acknowledgments
Grant support
This work was supported by NIH grants 5R01CA152301 (to H.T., Y.X and I.I.W), University of Texas SPORE in Lung Cancer (P50CA70907 to J.D.M, Y.X and I.I.W), P30CA142543 (to Y.X), 4R33DA027592( to G.X), NSF grant DMS-0907562(to G.X), NASA grant NNJ05HD36G(to Y.X), and CPRIT RP101251(to Y.X).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
No authors have any competing interests.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Douillard JY, Rosell R, De Lena M, Carpagnano F, Ramlau R, Gonzales-Larriba JL, et al. Adjuvant vinorelbine plus cisplatin versus observation in patients with completely resected stage ib-iiia non-small-cell lung cancer (adjuvant navelbine international trialist association [anita]): A randomised controlled trial. Lancet Oncol. 2006;7:719–27. doi: 10.1016/S1470-2045(06)70804-X. [DOI] [PubMed] [Google Scholar]
- 3.Kato H, Ichinose Y, Ohta M, Hata E, Tsubota N, Tada H, et al. A randomized trial of adjuvant chemotherapy with uracil-tegafur for adenocarcinoma of the lung. N Engl J Med. 2004;350:1713–21. doi: 10.1056/NEJMoa032792. [DOI] [PubMed] [Google Scholar]
- 4.The International Adjuvant Lung Cancer Trial Collaborative Group. Cisplatin-based adjuvant chemotherapy in patients with completely resected non-small-cell lung cancer. N Engl J Med. 2004;350:351–60. doi: 10.1056/NEJMoa031644. [DOI] [PubMed] [Google Scholar]
- 5.Winton T, Livingston R, Johnson D, Rigas J, Johnston M, Butts C, et al. Vinorelbine plus cisplatin vs. Observation in resected non-small-cell lung cancer. N Engl J Med. 2005;352:2589–97. doi: 10.1056/NEJMoa043623. [DOI] [PubMed] [Google Scholar]
- 6.Strauss GM, Herndon JE, II, Maddaus MA, Johnstone DW, Johnson EA, Harpole DH, et al. Adjuvant paclitaxel plus carboplatin compared with observation in stage ib non-small-cell lung cancer: Calgb 9633 with the cancer and leukemia group b, radiation therapy oncology group, and north central cancer treatment group study groups. J Clin Oncol. 2008;26:5043–51. doi: 10.1200/JCO.2008.16.4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olaussen KA, Mountzios G, Soria JC. Ercc1 as a risk stratifier in platinum-based chemotherapy for nonsmall-cell lung cancer. Curr Opin Pulm Med. 2007;13:284–9. doi: 10.1097/MCP.0b013e32816b5c63. [DOI] [PubMed] [Google Scholar]
- 8.Chen DT, Hsu YL, Fulp WJ, Coppola D, Haura EB, Yeatman TJ, et al. Prognostic and predictive value of a malignancy-risk gene signature in early-stage non-small cell lung cancer. J Natl Cancer Inst. 2011;103:1859–70. doi: 10.1093/jnci/djr420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen HY, Yu SL, Chen CH, Chang GC, Chen CY, Yuan A, et al. A five-gene signature and clinical outcome in non-small-cell lung cancer. N Engl J Med. 2007;356:11–20. doi: 10.1056/NEJMoa060096. [DOI] [PubMed] [Google Scholar]
- 10.Lee ES, Son DS, Kim SH, Lee J, Jo J, Han J, et al. Prediction of recurrence-free survival in postoperative non-small cell lung cancer patients by using an integrated model of clinical information and gene expression. Clin Cancer Res. 2008;14:7397–404. doi: 10.1158/1078-0432.CCR-07-4937. [DOI] [PubMed] [Google Scholar]
- 11.Lu Y, Lemon W, Liu PY, Yi Y, Morrison C, Yang P, et al. A gene expression signature predicts survival of patients with stage i non-small cell lung cancer. PLoS Med. 2006;3:e467. doi: 10.1371/journal.pmed.0030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Navab R, Strumpf D, Bandarchi B, Zhu CQ, Pintilie M, Ramnarine VR, et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc Natl Acad Sci U S A. 2011;108:7160–5. doi: 10.1073/pnas.1014506108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nature Medicine. 2008;14:822–7. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomida S, Koshikawa K, Yatabe Y, Harano T, Ogura N, Mitsudomi T, et al. Gene expression-based, individualized outcome prediction for surgically treated lung cancer patients. Oncogene. 2004;23:5360–70. doi: 10.1038/sj.onc.1207697. [DOI] [PubMed] [Google Scholar]
- 15.Wigle DA, Jurisica I, Radulovich N, Pintilie M, Rossant J, Liu N, et al. Molecular profiling of non-small cell lung cancer and correlation with disease-free survival. Cancer Res. 2002;62:3005–8. [PubMed] [Google Scholar]
- 16.Xie Y, Xiao G, Coombes KR, Behrens C, Solis LM, Raso G, et al. Robust gene expression signature from formalin-fixed paraffin-embedded samples predicts prognosis of non-small-cell lung cancer patients. Clin Cancer Res. 2011;17:5705–14. doi: 10.1158/1078-0432.CCR-11-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu CQ, Ding K, Strumpf D, Weir BA, Meyerson M, Pennell N, et al. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non-small-cell lung cancer. J Clin Oncol. 2010;28:4417–24. doi: 10.1200/JCO.2009.26.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeong Y, Xie Y, Xiao G, Behrens C, Girard L, Wistuba II, et al. Nuclear receptor expression defines a set of prognostic biomarkers for lung cancer. PLoS Med. 2010;7:e1000378. doi: 10.1371/journal.pmed.1000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kratz JR, He J, Van Den Eeden SK, Zhu ZH, Gao W, Pham PT, et al. A practical molecular assay to predict survival in resected non-squamous, non-small-cell lung cancer: Development and international validation studies. Lancet. 2012;379:823–32. doi: 10.1016/S0140-6736(11)61941-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boutros PC, Lau SK, Pintilie M, Liu N, Shepherd FA, Der SD, et al. Prognostic gene signatures for non-small-cell lung cancer. Proc Natl Acad Sci U S A. 2009;106:2824–8. doi: 10.1073/pnas.0809444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roepman P, Jassem J, Smit EF, Muley T, Niklinski J, van de Velde T, et al. An immune response enriched 72-gene prognostic profile for early-stage non-small-cell lung cancer. Clin Cancer Res. 2009;15:284–90. doi: 10.1158/1078-0432.CCR-08-1258. [DOI] [PubMed] [Google Scholar]
- 22.Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007;446:815–9. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- 23.Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–8. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maitra A, Wistuba II, Gazdar AF. Microdissection and the study of cancer pathways. Curr Mol Med. 2001;1:153–62. doi: 10.2174/1566524013364121. [DOI] [PubMed] [Google Scholar]
- 26.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–7. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 27.Matsuyama Y, Suzuki M, Arima C, Huang QM, Tomida S, Takeuchi T, et al. Proteasomal non-catalytic subunit psmd2 as a potential therapeutic target in association with various clinicopathologic features in lung adenocarcinomas. Mol Carcinog. 2011;50:301–9. doi: 10.1002/mc.20632. [DOI] [PubMed] [Google Scholar]
- 28.Raponi M, Zhang Y, Yu J, Chen G, Lee G, Taylor JM, et al. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res. 2006;66:7466–72. doi: 10.1158/0008-5472.CAN-06-1191. [DOI] [PubMed] [Google Scholar]
- 29.Tomida S, Takeuchi T, Shimada Y, Arima C, Matsuo K, Mitsudomi T, et al. Relapse-related molecular signature in lung adenocarcinomas identifies patients with dismal prognosis. J Clin Oncol. 2009;27:2793–9. doi: 10.1200/JCO.2008.19.7053. [DOI] [PubMed] [Google Scholar]
- 30.Xie Y, Wang X, Story M. Statistical methods of background correction for illumina beadarray data. Bioinformatics. 2009;25:751–7. doi: 10.1093/bioinformatics/btp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of affymetrix genechip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. Journal of the American Statistical Association. 1958:457–81. [Google Scholar]
- 33.Collett D. Modelling survival data in medical research. Chapman & Hall/CRC; 2003. [Google Scholar]
- 34.Schwarzer G. Meta: Meta-analysis with r. 2012. [Google Scholar]
- 35.Pounds S, Morris SW. Estimating the occurrence of false positives and false negatives in microarray studies by approximating and partitioning the empirical distribution of p-values. Bioinformatics. 2003;19:1236–42. doi: 10.1093/bioinformatics/btg148. [DOI] [PubMed] [Google Scholar]
- 36.Peng J, Wang P, Zhou N, Zhu J. Partial correlation estimation by joint sparse regression models. Journal of the American Statistical Association. 2009;104:735–46. doi: 10.1198/jasa.2009.0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bair E, Tibshirani R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2004;2:E108. doi: 10.1371/journal.pbio.0020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Breiman L, Friedman J, Stone JC, Olshen RA. Classification and regression trees. Chapman & Hall/CRC; 1984. [Google Scholar]
- 39.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–80. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olaussen KA, Dunant A, Fouret P, Brambilla E, Andre F, Haddad V, et al. DNA repair by ercc1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med. 2006;355:983–91. doi: 10.1056/NEJMoa060570. [DOI] [PubMed] [Google Scholar]
- 41.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 42.Bianchi F, Nuciforo P, Vecchi M, Bernard L, Tizzoni L, Marchetti A, et al. Survival prediction of stage i lung adenocarcinomas by expression of 10 genes. J Clin Invest. 2007;117:3436–44. doi: 10.1172/JCI32007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sweet-Cordero A, Mukherjee S, Subramanian A, You H, Roix JJ, Ladd-Acosta C, et al. An oncogenic kras2 expression signature identified by cross-species gene-expression analysis. Nat Genet. 2005;37:48–55. doi: 10.1038/ng1490. [DOI] [PubMed] [Google Scholar]
- 44.Winslow MM, Dayton TL, Verhaak RG, Kim-Kiselak C, Snyder EL, Feldser DM, et al. Suppression of lung adenocarcinoma progression by nkx2–1. Nature. 2011;473:101–4. doi: 10.1038/nature09881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sargent DJ, Conley BA, Allegra C, Collette L. Clinical trial designs for predictive marker validation in cancer treatment trials. J Clin Oncol. 2005;23:2020–7. doi: 10.1200/JCO.2005.01.112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.