

Abstract
This brief review analyzes the underlying physical principles of enzyme catalysis, with an emphasis on the role of equilibrium enzyme motions and conformational sampling. The concepts are developed in the context of three representative systems, namely dihydrofolate reductase, ketosteroid isomerase, and soybean lipoxygenase. All of these reactions involve hydrogen transfer, but many of the concepts discussed are more generally applicable. The factors that are analyzed in this review include hydrogen tunneling, proton donor-acceptor motion, hydrogen bonding, pKa shifting, electrostatics, preorganization, reorganization, and conformational motions. The rate constant for the chemical step is determined primarily by the free energy barrier, which is related to the probability of sampling configurations conducive to the chemical reaction. According to this perspective, stochastic thermal motions lead to equilibrium conformational changes in the enzyme and ligands that result in configurations favorable to the breaking and forming of chemical bonds. For proton, hydride, and proton-coupled electron transfer reactions, typically the donor and acceptor become closer to facilitate the transfer. The impact of mutations on the catalytic rate constants can be explained in terms of the factors enumerated above. In particular, distal mutations can alter the conformational motions of the enzyme and therefore the probability of sampling configurations conducive to the chemical reaction. Methods such as vibrational Stark spectroscopy, in which environmentally sensitive probes are introduced site-specifically in the enzyme, provide further insight into these aspects of enzyme catalysis through a combination of experiments and theoretical calculations.
INTRODUCTION
Enzymes are complex biological molecules that catalyze a wide range of chemical transformations. Although enzymes achieve these chemical transformations through many different types of mechanisms, they exhibit a common set of underlying physical principles. Identifying these underlying physical principles is a challenge for both experiment and theory. This review does not attempt to cover the entire field of enzyme catalysis. Instead, it focuses on the role that computation has played and will continue to play in helping to unravel the complexities of enzyme catalysis. Even within this more focused goal, we are only able to cover a small sampling of the theoretical contributions to this field and concentrate on the work from our own group to illustrate the fundamental concepts underlying enzyme catalysis.
The emphasis of this review is the role of enzyme motions and conformational sampling, particularly with respect to the catalyzed chemical reaction.1–11 We start with a brief overview of computational methods that have been utilized to investigate these aspects of enzyme reactions. Then we introduce three representative systems, namely dihydrofolate reductase (DHFR), ketosteroid isomerase (KSI), and soybean lipoxygenase (SLO), that are used to illustrate the concepts discussed in this review. Although all three of these reactions involve hydrogen transfer, many of the concepts discussed are more generally applicable to any enzymatic reaction. The following topics are covered in this review: hydrogen tunneling, proton donor-acceptor motion, hydrogen bonding and pKa shifting, electrostatics, conformational motions, preorganization and reorganization, the impact of mutations, and the vibrational Stark effect. Each topic is covered only briefly with references given to more comprehensive treatments.
COMPUTATIONAL METHODS
A variety of computational methods have been used to shed light on the factors influencing enzyme catalysis. Typically the entire enzyme, which is solvated in a bath of explicit water molecules, must be considered in order to obtain meaningful results, and a crystal structure is required to provide the initial coordinates. Classical molecular dynamics (MD) simulations of such systems provide information about the equilibrium properties, such as the stability of specific hydrogen-bonding interactions and the relative flexibility of different parts of the enzyme. Classical MD simulations of different states of the enzyme along the reaction pathway can provide this type of equilibrium information for each state, but these types of simulations do not provide any information about the process of converting from one state to another. For this purpose, quantum mechanical methods may be utilized to allow bonds to break and form.
Due to the large size of enzymatic systems, mixed quantum mechanical/molecular mechanical (QM/MM) methods are often used to study the chemical reactions catalyzed by enzymes. In some cases, the active site is treated with a QM method such as density functional theory, and the remainder of the system is treated with a standard MM forcefield. In the alternative empirical valence bond (EVB) method,12 the reaction is described as a linear combination of resonance states corresponding to the different bonding patterns of the relevant states in the chemical reaction. In the simplest case involving a single hydrogen transfer reaction, two resonance states corresponding to the proton bonded to its donor and acceptor are defined. In many implementations, the matrix elements of the EVB Hamiltonian in the basis of resonance states are represented by MM forcefield terms.
For hydrogen transfer reactions, nuclear quantum effects, such as zero point energy and hydrogen tunneling, may be important. Hybrid quantum/classical molecular dynamics methods have been developed to incorporate the nuclear quantum effects of the transferring hydrogen nucleus and, in some cases, the other vibrational modes of the active site.13,14 In the simplest case, only the transferring hydrogen nucleus is treated quantum mechanically. The hydrogen vibrational wavefunction may be calculated on a three-dimensional grid at each molecular dynamics time step, with the appropriate feedback between the quantum hydrogen nucleus and the classical nuclei. Alternatively, Feynman path integral methods, where the nucleus is represented by a set of beads, may be utilized to describe nuclear quantum effects.15,16 Another viable option is variational transition state theory with semiclassical tunneling calculations.14
Many proton and hydride transfer reactions catalyzed by enzymes are adiabatic, where the reaction occurs in the ground electronic and vibrational states. (The differentiation between adiabatic and nonadiabatic proton transfer reactions is discussed in Ref. 17.) For adiabatic reactions, the rate constant can be expressed as the product of a transmission coefficient, κ, and a transition state theory rate constant, kTST, which is often expressed in terms of the free energy barrier ΔG‡:
where kB is the Boltzmann constant. The free energy barrier can be calculated by generating the free energy profile along a collective reaction coordinate, as depicted in Figure 1. When the free energy barrier is much greater than the thermal energy, an approach such as umbrella sampling may be used to sample the entire range of the reaction coordinate.18,19 In this approach, a series of independent trajectories with different biasing potentials is propagated to generate segments of the free energy curve. Subsequently, the results are unbiased using a statistical procedure to produce the free energy curve for the original potential.20,21 To fully investigate equilibrium motions occurring on relatively long timescales, enhanced sampling techniques may be required.
Figure 1.

Schematic depiction of an adiabatic hydrogen transfer reaction. The free energy is plotted along the collective reaction coordinate, with the free energy barrier denoted by ΔG‡. The dashed red curve and red arrow indicate the decrease in the free energy barrier when the nuclear quantum effects of the transferring hydrogen are included. DA denotes the hydrogen donor-acceptor distance, which is typically larger at the reactant and product (i.e., minima) and smaller at the transition state (i.e., top of the barrier) because the donor and acceptor must get closer for the hydrogen to transfer.
In the context of an EVB potential for proton or hydride transfer, the collective reaction coordinate is often chosen to be an energy gap reaction coordinate, defined as the difference in energy between the two EVB states.12,13 In this case, the “transition state” corresponds to configurations for which the energy gap reaction coordinate is approximately zero, namely configurations at which the proton or hydride is moving in a nearly symmetric environment. In the remainder of this paper, the “transition state” will refer to these types of configurations rather than to saddle points on a potential energy surface. Note that the top of the barrier may not occur at exactly zero energy gap reaction coordinate for asymmetric systems. Moreover, alternative reaction coordinates could also be utilized.
The transmission coefficient κ accounts for dynamical recrossings of the dividing surface, which is typically chosen to pass through the top of the free energy barrier. This factor can be calculated using a reactive flux approach,15,16 in which a large ensemble of trajectories is initiated at the top of the free energy barrier and propagated backward and forward in time.13 A standard algorithm22 is used to calculate the transmission coefficient, where κ is unity if there are no recrossings of the dividing surface and decreases as the number of recrossings increases. Note that the overall rate constant, defined as the product of the transition state theory rate constant and the transmission coefficient, is independent of the position of the dividing surface.
A variety of other methods have also been used to calculate the rate constants of enzyme reactions. For example, the variational transition state theory method with semiclassical tunneling calculations has also been applied to numerous enzymatic reactions.14 Furthermore, to avoid the imposition of a specific reaction coordinate, methods such as transition path sampling23 can be used. Other reviews have discussed these types of approaches.24–26
Proton-coupled electron transfer (PCET) reactions are defined as the simultaneous transfer of an electron and a proton, often between different donors and acceptors.27–29 In general, PCET reactions may be sequential or concerted, where sequential reactions are defined to have a stable intermediate. The subtleties of these differences are discussed elsewhere,17 but here we will consider only concerted PCET reactions, which are often nonadiabatic and involve excited electronic and vibrational states.17 In this case, the rate constant can be expressed as the product of the square of the vibronic coupling, which is the product of the electronic coupling and the overlap between the reactant and product proton vibrational wavefunctions, and a term that depends exponentially on the free energy barrier. The overlap and free energy barrier relevant to nonadiabatic reactions are depicted schematically in Figure 2. Details of this nonadiabatic treatment are provided elsewhere.27
Figure 2.

Schematic depiction of a nonadiabatic PCET reaction. The free energies of the two diabatic states corresponding to the electron on its donor (De−Ae, blue) and acceptor (DeAe−, red) are plotted along the collective reaction coordinate, and the free energy barrier is denoted by ΔG‡. In the upper figures surrounded by dotted lines, the proton potential energy curves for the two diabatic states at the crossing point are plotted as functions of the hydrogen coordinate using the same color scheme as used to distinguish the two diabatic states. The ground state reactant and product proton vibrational wavefunctions are depicted, and the overlap is shaded in purple. When the proton donor-acceptor distance, DpAp, is large, the overlap is small, and when this distance decreases, the overlap increases.
THREE REPRESENTATIVE ENZYMES
DHFR catalyzes the conversion of dihydrofolate (DHF) to tetrahydrofolate (THF). The mechanism involves hydride transfer from NADPH to protonated DHF, producing NADP+ and THF,2 as depicted in Figure 3A. Crystal structures have been solved for intermediates along the entire catalytic reaction pathway, indicating significant conformational changes, particularly in the Met20 loop region prior to ligand binding.30 NMR has also been used to identify dynamic regions of the protein that change along the reaction pathway.5,31 Furthermore, mutations far from the active site have been shown to significantly impact the hydride transfer rate constant, and double and triple mutants have exhibited nonadditivity effects.32,33
Figure 3.

Chemical reactions catalyzed by each of the three representative enzymes. (A) DHFR catalyzes a process involving hydride transfer from NADPH to protonated DHF. (B) KSI catalyzes a process involving two sequential proton transfer steps: (1) proton transfer from a carbon of the steroid to Asp40, resulting in a dienolate intermediate, and (2) proton transfer from Asp40 to another carbon of the substrate, leading to an overall isomerization reaction. (C) SLO catalyzes the hydrogen abstraction from the linoleic acid substrate to the iron cofactor. This process is thought to occur by a PCET mechanism. Part (C) reproduced with permission from Ref. 46.
KSI catalyzes the isomerization of certain steroids via two sequential proton transfer reactions,34 as depicted in Figure 3B. The reaction is thought to proceed through a dienolate intermediate that is stabilized by hydrogen-bonding interactions with Tyr16 and Asp103. (Throughout this review, the KSI residues are numbered according to Pseudomonas putida KSI, although some of the results were obtained for Commamonas testosteroni KSI.) As for DHFR, crystal structures have been solved for wild-type (WT) KSI35 and a wide range of mutants.36 In addition, NMR experiments have been used to characterize the hydrogen-bonding interactions with a variety of bound inhibitors.37 More recently, vibrational Stark spectroscopy has been used to investigate the role of electrostatics in KSI, particularly upon binding of equilenin, which is viewed as an analog of the dienolate intermediate.38
SLO catalyzes the oxidation of unsaturated fatty acids, as depicted in Figure 3C with the substrate linoleic acid.39 This reaction is thought to occur by a PCET mechanism, in which the electron transfers from the π-system of the substrate to the iron of the cofactor, while the proton transfers from the C11 carbon of the substrate to the hydroxyl ligand of the cofactor.40 The kinetic isotope effect (KIE), defined as the ratio of the rate constants for hydrogen and deuterium transfer, was found to be unusually high, with a value of 81 at room temperature, while the temperature dependences of the rate constants and KIEs were found to be relatively weak.39,41 The impact of mutations on the magnitude and temperature dependence of the KIE has also been examined.42
GENERAL CONCEPTS
Hydrogen tunneling
Theoretical studies suggest that hydrogen tunneling is prevalent in enzymes, but it is also expected to occur to a similar extent in solution.43 The proton and hydride transfer reactions catalyzed by KSI and DHFR are predominantly adiabatic and hence can be described in terms of the rate constant expression given in Eq. (1). Typically the transmission coefficient κ is nearly unity for these types of reactions, and the main impact of nuclear quantum effects such as zero point energy and hydrogen tunneling is to lower the free energy barrier by 2–3 kcal/mol, as depicted in Figure 1. Our calculations of the hydride transfer reaction catalyzed by DHFR indicated that this enzyme reaction exhibits these general characteristics.44 In particular, we generated the free energy curve along the collective reaction coordinate including nuclear quantum effects, which resulted in a free energy barrier decrease of ~2 kcal/mol relative to classical MD simulations, and performed reactive flux calculations to determine the transmission coefficient, which was nearly unity. Moreover, the calculations reproduced the experimentally measured KIE of ~3 for DHFR.44 This enzymatic reaction was also studied using other methods that include nuclear quantum effects.45
In contrast, the PCET reaction catalyzed by SLO is nonadiabatic and is described by a rate constant expression with a prefactor including the square of the overlap between the reactant and product proton vibrational wavefunctions (see Figure 2).27 In this case, the nuclear quantum effects of the transferring hydrogen nucleus impact the prefactor as well as the free energy barrier. The overlap factor can lead to significant deuterium KIEs in these types of reactions. For example, the KIE for SLO has been found to be ~80 at room temperature.41 Our nonadiabatic treatment qualitatively reproduced the experimentally measured magnitude and temperature dependence of the KIE for this enzyme.40,46 On the other hand, nonadiabatic PCET reactions can also exhibit more moderate KIEs of ~3.27,28 Thus, typically a large KIE indicates that the reaction is nonadiabatic, but a moderate KIE cannot be used to distinguish between an adiabatic and nonadiabatic reaction. Note that other methods have also been used to simulate the large KIEs of this enzymatic reaction.47
Proton donor-acceptor motion
The motion of the hydrogen donor and acceptor plays a critical role in hydrogen transfer reactions. In general, the barrier to proton transfer decreases as the proton donor-acceptor distance decreases and vanishes completely at extremely short distances.43 In the context of adiabatic proton and hydride transfer reactions, the average proton donor-acceptor distance decreases as the reaction evolves along the collective reaction coordinate from reactant to transition state configurations, as depicted in Figure 1. We calculated the thermally averaged hydride donor-acceptor distance along the energy gap reaction coordinate for DHFR and found that it decreased from ~3.4 Å in the crystal structure to ~2.7 Å at the transition state.44 We observed a similar decrease in the proton donor-acceptor distances for the two sequential proton transfer reactions catalyzed by KSI.48
In the context of nonadiabatic PCET reactions, the proton donor-acceptor distance significantly influences the overlap between the reactant and product proton vibrational wavefunctions, as depicted in Figure 2. We have derived rate constant expressions that include the effects of this proton donor-acceptor motion.27,49 The rate constant tends to increase as the proton donor-acceptor distance decreases because of the larger overlap between the reactant and product proton vibrational wavefunctions, which contributes to the prefactor in the nonadiabatic rate constant expression. On the other hand, the probability of sampling these relatively short distances is smaller compared to sampling distances near the equilibrium value based on the Boltzmann distribution for the proton donor-acceptor vibrational motion. Due to the competition between these two effects, typically the dominant proton donor-acceptor distance (i.e., the distance that contributes the most to the overall rate constant) is substantially smaller than the equilibrium proton donor-acceptor distance. Our calculations on SLO indicated that the equilibrium proton donor-acceptor distance is ~2.9 Å, whereas the dominant distance is ~2.7 Å.40,46
Hydrogen bonding and pKa shifting
The roles of hydrogen bonding and pKa shifting have been explored extensively in enzyme catalysis with various computational methods. For example, hybrid quantum/classical molecular dynamics simulations have been used to examine hydrogen bonding along the collective reaction coordinate in KSI.48,50 As mentioned above, this enzymatic reaction is thought to proceed through a dienolate intermediate that is stabilized by hydrogen-bonding interactions with Tyr16 and Asp103. Our calculations illustrated that the Asp103 hydrogen bond remains predominantly constant during both proton transfer reactions, while the Tyr16 hydrogen bond strengthens during the first proton transfer reaction.48 These calculations also indicated that the hydrogen bond interaction energies were significantly greater for the intermediate state than for the reactant and product states. Thus, minor structural changes must occur during the chemical reactions to accommodate these changes in the hydrogen-bonding interactions. To better understand these interactions, a series of substituted phenolate inhibitors bound in the active site of KSI was investigated with NMR and electronic absorption experiments,37 as well as with QM/MM calculations.51
In many enzymes, the local protein environment shifts the pKa of specific residues in a manner that influences catalysis.52,53 For example, this type of pKa shifting has been shown to be significant in the active site of KSI. In aqueous solution, aspartic acid (pKa ~ 4) is more acidic than tyrosine (pKa ~ 10).52 However, QM/MM calculations on the series of substituted phenolate inhibitors bound in the active site of KSI51 suggested that electronic inductive effects along a hydrogen-bonding network of tyrosine residues, including Tyr16, Tyr57, and Tyr32 (see Figure 3B), causes the Tyr16 hydroxyl to be more acidic than the Asp103 carboxylic acid moiety, which is immersed in a relatively nonpolar local environment. The catalytic importance of these distal tyrosine residues is illustrated by the experimental observation36 that the double mutant Y32F/Y57F reduces the catalytic rate constant by a factor of ~2, a relatively small yet non-negligible effect. The strength of the hydrogen-bonding interactions between the dienolate intermediate and the enzyme residues strongly influences the free energy barriers for the proton transfer reactions.48,54
Electrostatics
Electrostatic interactions play a vital role in enzyme reactions. Electrostatics can be studied using a variety of computational methods, and many theoretical studies have illuminated the importance of electrostatics in enzymes.55–57 For example, we examined the electrostatic potential along the collective reaction coordinate by solution of the linearized Poisson-Boltzmann equation for DHFR.58 Our calculations indicated significant changes in the electrostatic potential throughout the entire enzyme as the reaction evolves from reactant to product. We also utilized a charge deletion scheme to determine the electrostatic contributions of each residue along the collective reaction coordinate and to identify the key residues that influence the changes in the electrostatic energy during the chemical reaction.58 These analyses implied that changes in the electrostatic charge distribution significantly impact the energetics of the hydride transfer reaction. Moreover, in our studies of the proton transfer reactions catalyzed by KSI,48 we found that the electrostatic interaction between the ligand and the enzyme is substantially more favorable at the intermediate than at the reactant or product, again signifying changes in the electrostatics during the chemical reaction. Such electrostatic changes are accompanied by minor conformational changes throughout the enzyme, but the degree of cause and effect is difficult to determine.
Conformational motions
Both experiments and simulations have implicated the importance of equilibrium conformational sampling in enzyme catalysis. As mentioned above, the rate constant is determined predominantly by the free energy barrier. From a thermodynamic perspective, the free energy barrier is related to the relative probabilities of sampling transition state configurations (i.e., those at the top of the barrier) and reactant configurations (i.e., those at the reactant minimum). Thus, the millisecond timescale of the chemical step for most enzyme reactions is determined by the amount of time required for the enzyme to sample phase space to obtain configurations that are conducive to the chemical reaction.3 For the case of proton and hydride transfer reactions, the conformational sampling brings the donor and acceptor closer to each other, orients them properly, and provides an appropriate electrostatic environment. Once a favorable configuration has been achieved, the breaking and forming of chemical bonds, as well as hydrogen tunneling for hydrogen transfer reactions, occur virtually instantaneously relative to the millisecond timescale of the overall reaction. Note that these concepts apply to any enzymatic reaction, not just to hydrogen transfer reactions. In general, the chemical bond rearrangement may be viewed as a rare event that occurs only when the enzymatic system is in a favorable configuration. These concepts are depicted schematically in Figure 4.
Figure 4.

Schematic depiction of the role of conformational sampling in the chemical step of an enzyme reaction. The free energy is plotted along the collective reaction coordinate. The evolution from reactant (left minimum, blue) to transition state (top of barrier, red) is caused by stochastic thermal motions, which lead to equilibrium conformational changes (purple arrow) to produce configurations that are conducive to the chemical reaction. The bond rearrangement (i.e., the breaking and forming of chemical bonds) occurs near the top of the barrier (red arrow). For hydrogen transfer reactions, hydrogen tunneling also occurs near the top of the barrier. The reacting entities (i.e., donor and acceptor) are depicted by a triangle and a square, which are further apart in the reactant and product but closer at the transition state. The changes in orientation of the shapes represent changes in orientation of the reacting entities, as well as changes in hydrogen bonding interactions and electrostatics along the collective reaction coordinate. The effects of breaking and forming chemical bonds are not explicitly shown in these shapes.
We emphasize that these conformational motions are stochastic and are simply the typical equilibrium thermal motions that occur in any condensed phase system, except that they occur in the confines of the protein structure.3 Enzymatic motions occur on a wide range of timescales, spanning femtoseconds to milliseconds. According to this perspective,3 the fast thermal fluctuations result in slower, conformational changes of the enzymatic system that eventually lead to configurations that are conducive to the chemical reaction (see the purple arrow and the resulting red configuration in Figure 4). All of these equilibrium motions contribute to the free energy barrier, which can be viewed in terms of the relative probability of achieving the favorable configurations.59 The conformational changes depicted in Figure 4 correspond to the relatively small, typically sub-Angstrom changes occurring during the chemical reaction step. Global conformational changes of the protein that occur before or after this chemical step are not considered here. Note that these concepts have been well-accepted in statistical mechanics in the context of electron transfer and other chemical reactions in solution for many years.43,60–63
We point out that these equilibrium conformational motions are distinct from the fast “promoting modes” that have been proposed to be dynamically coupled to the chemical reaction.64 We have seen no convincing evidence of the catalytic relevance of such promoting modes. These modes apparently occur on the femtosecond timescale and influence the dynamics at the top of the barrier. Typically the experimentally measured rate constant for the chemical step in enzymatic reactions is on the millisecond timescale.3 Thus, these promoting modes could be slowed down by more than ten orders of magnitude and still would not influence the rate of the enzyme-catalyzed chemical reaction. Often the chemical step is not rate limiting in the enzyme turnover cycle (i.e., product release may be rate limiting), but here we focus only on the rate constant measured for the chemical step. For example, in DHFR, the hydride transfer reaction occurring after the substrate and cofactor are bound, and before either the cofactor or product is released, has been measured to be on the millisecond timescale.3,65 As discussed above, the rate constant of the chemical step is determined mainly by the free energy barrier, which in turn is influenced by equilibrium conformational sampling.
A more complete view of enzyme catalysis, including the substrate binding and product release steps, as well as the intervening chemical step, is illustrated in Figure 5. In this depiction of the multidimensional free energy landscape for an enzyme reaction, stochastic thermal motions lead to conformational changes along both axes. The conformational changes occurring along the reaction coordinate axis facilitate the catalytic reaction. Figure 4 focuses on only the chemical reaction step (i.e., the middle free energy barrier in Figure 5) along the collective reaction coordinate, averaging over all other thermal motions.
Figure 5.
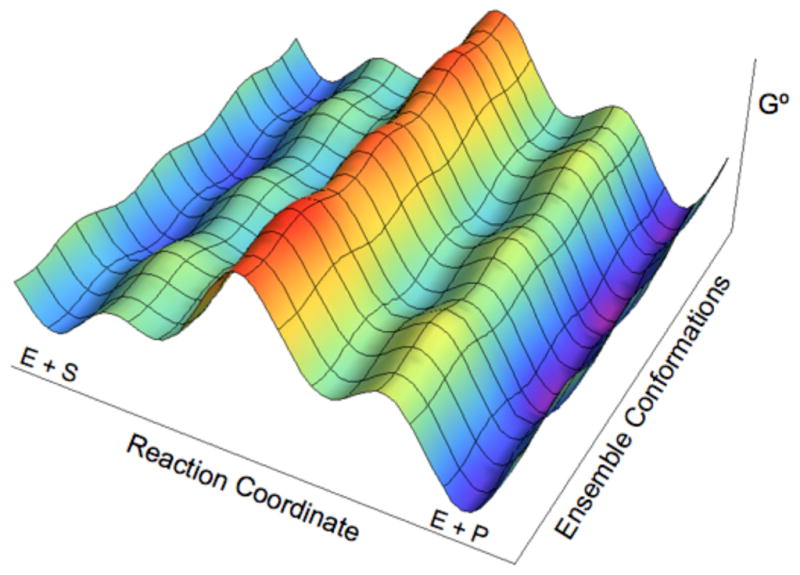
Schematic representation of the standard free energy landscape for the catalytic network of an enzyme reaction. Conformational changes occur along both axes. The conformational changes occurring along the Reaction Coordinate axis correspond to the environmental reorganization that facilitates the chemical reaction. In contrast, the conformational changes occurring along the Ensemble Conformations axis represent the ensembles of configurations existing at all stages along the reaction coordinate, leading to a large number of parallel catalytic pathways. The reaction paths can slide along and between both coordinates. For real enzymes, the number of maxima and minima along the coordinates is expected to be greater than shown. The free energy landscape and dominant catalytic pathways will be altered by external conditions and protein mutations. Figure and portions of caption reproduced from Ref. 74.
This discussion of conformational motions is applicable to both adiabatic and nonadiabatic reactions. In both cases, equilibrium conformational sampling is required to reach configurations that are conducive to the chemical reaction (i.e., to the top of the barrier in Figure 1 and the crossing point in Figure 2). As discussed earlier in this paper, the hydrogen donoracceptor motion is also important for proton, hydride, and PCET reactions. In the context of adiabatic reactions, the thermally averaged donor-acceptor distance is found to decrease along the collective reaction coordinate as the system evolves from the reactant to the transition state (see Figure 1). This decrease can be viewed as an overall conformational change that occurs on the slower timescale. In the context of nonadiabatic PCET reactions, the proton donor-acceptor motion also influences the overlap between the proton vibrational wavefunctions in the prefactor of the rate constant (see Figure 2). Even in this case, however, the proton donor-acceptor motion can still be viewed as an equilibrium thermal fluctuation. For SLO, a comparison between calculations that included the dynamical effects of this mode and calculations that treated this mode as an equilibrium distribution indicated that the dynamical effects are negligible.46
Preorganization and reorganization
Preorganization and reorganization represent important components of enzyme catalysis.8 Both of these processes involve conformational motions to some extent during the catalytic cycle. Typically the active site of the enzyme is preorganized so that the reorganization of the environment required to facilitate the chemical reaction is lower in the enzyme than in solution. Achieving a preorganized active site may require rate-limiting conformational changes, such as the opening or closing of a loop, within the overall turnover cycle. In contrast, reorganization usually involves relatively small conformational changes during the chemical step.
The perspective discussed in this review is consistent with the significance of preorganization. We emphasize, however, that even preorganized active sites require small conformational changes to enable the chemical reaction. Thus, preorganization does not eliminate the need for motion completely: the degree of reorganization may be decreased by preorganization, but reorganization of the environment is still necessary. For example, KSI has been shown to form a preorganized active site,55,66 but our calculations provided evidence of relatively small conformational changes that facilitate the proton transfer reactions by bringing the donor and acceptor closer to each other with the proper orientation for proton transfer and strengthening the hydrogen bonds between the substrate and active site residues.48 If the proton donor and acceptor remained at the distance of ~3.2 Å in the reactant state, the proton transfer would not occur. Clearly the decrease to ~2.7 Å is required to allow the proton to transfer, and this decrease in distance requires conformational changes.
Impact of mutations
In principle, mutations can influence all of the factors discussed above, particularly hydrogen bonding, electrostatics, and conformational sampling. In our studies of various DHFR mutants, we found that these mutations do not significantly alter the nuclear quantum effects (i.e., the associated decrease in the free energy barrier) or the transmission coefficient.67,68 Note that this situation could be different for other enzymes or even for other mutants of DHFR, but we have not seen any evidence supporting this possibility. Thus, we have focused on the impact of mutations on other factors.
We studied several different mutants of DHFR, focusing particularly on mutations distal to the active site.39,40 Consistent with experiments, we found that the G121V mutation in Escherichia coli DHFR decreased the rate of hydride transfer by a factor of 163, even though the mutation site was ~12 Å from the transferring hydride.67 We found that the decrease in the hydride transfer rate constant is due to an increase in the free energy barrier, which in turn results from alterations of the equilibrium conformational motions of the entire enzyme upon mutation. We identified specific differences in thermally averaged distances and angles evolving along the collective reaction coordinate for the G121V mutant compared to WT DHFR. Similar behavior was observed for other distal mutations.68 Within this conceptual framework, distal mutations can impact the catalytic rate constant by changing the equilibrium conformational sampling and therefore the probability of sampling configurations conducive to the chemical reaction. Altering this probability leads to changes in the free energy barrier and the associated rate constant. Other computational methods have also been used to study mutants of DHFR.69,70
In KSI, we studied the impact of mutations that disrupt the hydrogen bonding between the dienolate intermediate and Tyr16 and Asp103.54 Consistent with experimental measurements, the calculated rate constants for the mutants in which Tyr16 was replaced by Leu and Asp103 was replaced by Phe, as well as the associated double mutant, were significantly reduced relative to WT KSI. Our calculations indicated that the electrostatic stabilization of the dienolate intermediate relative to the reactant was greater for WT KSI than for the mutants, providing a qualitative explanation for the reduced rate constants of the mutants. In addition, these calculations suggested that the mutations alter the structure of the preorganized active site of KSI somewhat, possibly necessitating a greater degree of reorganization during the chemical step. As discussed above, this reorganization is manifested by small conformational changes due to stochastic thermal motions occurring within this preorganized active site to facilitate the two proton transfer reactions. Thus, these KSI mutants exemplify changes in hydrogen bonding, electrostatics, and possibly conformational motions.
We also investigated the impact of mutations on the nonadiabatic PCET reaction catalyzed by SLO. Specifically, we modeled experimental data42 indicating that mutation of Ile553 to less bulky residues increases the magnitude and temperature dependence of the KIE for this reaction.71 This residue borders the linoleic acid substrate but is ~15 Å from the active site iron. Based on our nonadiabatic PCET theory,27,49 the proton donor-acceptor equilibrium distance and effective frequency strongly influence the rate constants and KIEs. For these types of nonadiabatic reactions, the proton donor-acceptor distance significantly impacts the proton vibrational wavefunction overlap prefactor in the rate constant expression (see Figure 2). Our calculations on SLO illustrated that the equilibrium proton donor-acceptor distance increases and the associated frequency decreases as residue 553 becomes less bulky,71 thereby leading to the experimentally observed trends in the magnitude and temperature dependence of the KIE.
Vibrational Stark effect
The vibrational Stark effect can provide additional insights into the roles of electrostatics, hydrogen bonding, and conformational motions in enzyme catalysis. In recent experiments, thiocyanate (~SCN) probes were introduced at site-specific positions in the active site of KSI, and changes to the CN frequency were measured under various conditions.38 We utilized computational methods to calculate the vibrational frequency shifts of nitrile probes in KSI for comparison to the experimental data and to elucidate the molecular basis for the observed shifts.72 Starting with crystal structures of the enzyme containing the nitrile probe, classical MD trajectories were propagated to sample conformational space. For each configuration, a series of QM/MM calculations was performed to generate the potential energy curve associated with the CN mode. In these QM/MM calculations, the probe and nearby residues were included in the QM region, which was described by a reparameterized semiempirical QM method.72,73 The CN frequency was obtained from the energy splitting between the ground and first excited vibrational states. The spectral line shapes were computed from these calculated frequencies using the fluctuating frequency approximation.
In our initial application, we examined the frequency shifts upon equilenin binding to the active site of KSI for nitrile probes positioned at residues Met116 and Phe86. Our simulations provided atomic-level insight into the roles that key residues play in determining the electrostatic environment and hydrogen-bonding interactions experienced by the nitrile probe. For both systems, our calculations indicated that equilenin binding reorients an active site water molecule or a residue that is specifically hydrogen bonded to the nitrile probe. This reorientation alters the angle and distance at the hydrogen-bonding interface, thereby increasing the CN frequency. In these cases, the vibrational frequency shift is not due to a “classical” Stark effect, where the changes in vibrational frequency arise only from classical electrostatics and are linearly related to changes in the local electrostatic field, but rather is due to changes in specific hydrogenbonding interactions. Moreover, this initial application focused on the effects of ligand binding on the CN frequency and therefore was not directly relevant to catalysis.
Future work will center on nitrile probes inserted at various positions within the active site of DHFR for the five well-defined intermediates along the catalytic reaction pathway. These studies should provide information about hydrogen bonding, electrostatics, and conformational changes along the catalytic pathway. Time-resolved Stark effect experiments may even provide information along the collective reaction coordinate for the hydride transfer reaction itself. These types of vibrational Stark effect studies will enable a direct comparison between a calculated quantity and an experimental observable along the reaction pathway. Moreover, the simulations should provide atomic-level insight into significant aspects of enzyme catalysis.
CONCLUDING REMARKS
This review summarized the important factors that contribute to enzyme catalysis. The connection of each of these factors to enzyme motions and conformational sampling was emphasized. Overall, a combination of factors, including hydrogen bonding, pKa shifting, electrostatics, preorganization, reorganization, and conformational motions contribute to enzyme catalysis. The impact of mutations on the catalytic rate constants can be explained in terms of these various factors. Vibrational Stark spectroscopy and Stokes shift measurements will provide further insights into these aspects of enzyme catalysis. The methods discussed will allow a direct comparison between theoretical calculations and experimental data. The interplay between theory and experiment will lead to a deeper and broader understanding of enzyme catalysis.
Acknowledgments
The author is grateful to Dr. Alexander Soudackov for creating Figures 1, 2, and 4 and Phil Hanoian for creating Figures 3A and 3B. The author also thanks Phil Hanoian, Alexander Soudackov, Josh Layfield, Abir Ganguly, and Sixue Zhang for helpful discussions. This work was funded by NIH Grant No. GM56207.
Abbreviations
- DHFR
dihydrofolate reductase
- EVB
empirical valence bond
- KIE
kinetic isotope effect
- KSI
ketosteroid isomerase
- MD
molecular dynamics
- MM
molecular mechanical
- PCET
proton-coupled electron transfer
- QM
quantum mechanical
- QM/MM
quantum mechanical/molecular mechanical
- SLO
soybean lipoxygenase
- WT
wild type
References
- 1.Hammes GG. Multiple Conformational Changes in Enzyme Catalysis. Biochemistry. 2002;41:8221–8228. doi: 10.1021/bi0260839. [DOI] [PubMed] [Google Scholar]
- 2.Benkovic SJ, Hammes-Schiffer S. A Perspective on Enzyme Catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 3.Hammes-Schiffer S, Benkovic SJ. Relating Protein Motion to Catalysis. Annu Rev Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- 4.Nagel Z, Klinman JP. Tunneling and Dynamics in Enzymatic Hydride Transfer. Chem Rev. 2006;106:3095–3118. doi: 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- 5.Boehr DD, McElheny D, Dyson HJ, Wright PE. The Dynamic Energy Landscape of Dihydrofolate Reductase Catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 6.Henzler-Wildman K, Kern D. Dynamic Personalities of Proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 7.Watt ED, Shimada H, Kovrigin EL, Loria JP. The Mechanism of Rate-Limiting Motions in Enzyme Function. Proc Nat Acad Sci USA. 2007;104:11981–11986. doi: 10.1073/pnas.0702551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamerlin SC, Warshel A. At the Dawn of the 21st Century: Is Dynamics the Missing Link for Understanding Enzyme Catalysis? Proteins. 2010;78:1339–1375. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hay S, Scrutton NS. Good Vibrations in Enzyme-Catalysed Reactions. Nature Chemistry. 2012;4:161–168. doi: 10.1038/nchem.1223. [DOI] [PubMed] [Google Scholar]
- 10.Glowacki DR, Harvey JN, Mulholland AJ. Taking Ockham’s Razor to Enzyme Dynamics and Catalysis. Nature Chemistry. 2012;4:169–176. doi: 10.1038/nchem.1244. [DOI] [PubMed] [Google Scholar]
- 11.Doshi U, McGowan LC, Ladani ST, Hamelberg D. Resolving the Complex Role of Enzyme Conformational Dynamics in Catalytic Function. Proc Natl Acad Sci USA. 2012;109:5699–5704. doi: 10.1073/pnas.1117060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warshel A. Computer Modeling of Chemical Reactions in Enzymes and Solutions. John Wiley & Sons, Inc; New York: 1991. [Google Scholar]
- 13.Billeter SR, Webb SP, Iordanov T, Agarwal PK, Hammes-Schiffer S. Hybrid Approach for Including Electronic and Nuclear Quantum Effects in Molecular Dynamics Simulations of Hydrogen Transfer Reactions in Enzymes. J Chem Phys. 2001;114:6925–6936. [Google Scholar]
- 14.Truhlar DG, Gao J, Alhambra C, Garcia-Viloca M, Corchado JC, Sanchez ML, Villa J. The Incorporation of Quantum Effects in Enzyme Kinetics Modeling. Acc Chem Res. 2002;35:341–349. doi: 10.1021/ar0100226. [DOI] [PubMed] [Google Scholar]
- 15.Li D, Voth GA. Feynman Path Integral Approach for Studying Intramolecular Effects in Proton-Transfer Reactions. J Phys Chem. 1991;95:10425–10431. [Google Scholar]
- 16.Hwang JK, Chu ZT, Yadav A, Warshel A. Simulations of Quantum Mechanical Corrections for Rate Constants of Hydride-Transfer Reactions in Enzymes and Solutions. J Phys Chem. 1991;95:8445–8448. [Google Scholar]
- 17.Hammes-Schiffer S. Proton-Coupled Electron Transfer: Classification Scheme and Guide to Theoretical Methods. Energy and Environmental Science. 2012;5:7696–7703. [Google Scholar]
- 18.Allen MP, Tildesley DJ. Computer Simulation of Liquids. Clarendon Press; Oxford: 1989. [Google Scholar]
- 19.Torrie GM, Valleau JP. Monte Carlo Free Energy Estimates Using Non-Boltzmann Sampling: Application to the Sub-Critical Lennard-Jones Fluid. Chem Phys Lett. 1974;28:578–581. [Google Scholar]
- 20.Roux B. The Calculation of the Potential of Mean Force Using Computer Simulations. Comput Phys Commun. 1995;91:275–282. [Google Scholar]
- 21.Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I The Method. J Comput Chem. 1992;13:1011–1021. [Google Scholar]
- 22.Keck JC. Variational Theory of Chemical Reaction Rates Applied to Three-Body Recombinations. J Chem Phys. 1960;32:1035–1050. [Google Scholar]
- 23.Dellago C, Bolhuis PG, Csajka FS, Chandler D. Transition Path Sampling and the Calculation of Rate Constants. J Chem Phys. 1998;108:1964–1977. [Google Scholar]
- 24.Pu J, Gao J, Truhlar DG. Multidimensional Tunneling, Recrossing, and the Transmission Coefficient for Enzymatic Reactions. Chem Rev. 2006;106:3140–3169. doi: 10.1021/cr050308e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia-Viloca M, Gao J, Karplus M, Truhlar DG. How Enzymes Work: Analysis by Modern Rate Theory and Computer Simulations. Science. 2004;303:186–195. doi: 10.1126/science.1088172. [DOI] [PubMed] [Google Scholar]
- 26.Bolhuis P, Chandler D, Dellago C, Geissler P. Transition Path Sampling: Throwing Ropes over Rough Mountain Passes, in the Dark. Annu Rev Phys Chem. 2002;53:291–318. doi: 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- 27.Hammes-Schiffer S, Soudackov AV. Proton-Coupled Electron Transfer in Solution, Proteins, and Electrochemistry. J Phys Chem B. 2008;112:14108–14123. doi: 10.1021/jp805876e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huynh MH, Meyer TJ. Proton-Coupled Electron Transfer. Chem Rev. 2007;107:5004–5064. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayer JM. Proton-Coupled Electron Transfer: A Reaction Chemist’s View. Annu Rev Phys Chem. 2004;55:363–390. doi: 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]
- 30.Sawaya MR, Kraut J. Loop and Subdomain Movements in the Mechanism of Escherichia Coli Dihydrofolate Reductase: Crystallographic Evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 31.Boehr DD, McElheny D, Dyson HJ, Wright PE. Millisecond Timescale Fluctuations in Dihydrofolate Reductase Are Exquisitely Sensitive to the Bound Ligands. Proc Natl Acad Sci USA. 2010;107:1373–1378. doi: 10.1073/pnas.0914163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Goodey NM, Benkovic SJ, Kohen A. Coordinated Effects of Distal Mutations on Environmentally Coupled Tunneling in Dihydrofolate Reductase. Proc Natl Acad Sci USA. 2006;103:15753–15758. doi: 10.1073/pnas.0606976103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajagopalan PTR, Lutz S, Benkovic SJ. Coupling Interactions of Distal Residues Enhance Dihydrofolate Reductase Catalysis: Mutational Effects on Hydride Transfer Rates. Biochemistry. 2002;41:12618–12628. doi: 10.1021/bi026369d. [DOI] [PubMed] [Google Scholar]
- 34.Pollack RM. Enzymatic Mechanisms for Catalysis of Enolization: Ketosteroid Isomerase. Bioorg Chem. 2004;32:341–353. doi: 10.1016/j.bioorg.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 35.Cho HS, Ha NC, Choi G, Kim HJ, Lee D, Oh KS, Kim KS, Lee W, Choi KY, Oh BH. Crystal Structure of Δ5-3-Ketosteroid Isomerase from Pseudomonas Testosteroni in Complex with Equilenin Settles the Correct Hydrogen Bonding Scheme for Transition State Stabilization. J Biol Chem. 1999;274:32863–32868. doi: 10.1074/jbc.274.46.32863. [DOI] [PubMed] [Google Scholar]
- 36.Kim D-H, Jang DS, Nam GH, Choi G, Kim J-S, Ha N-C, Kim M-S, Oh B-H, Choi KY. Contribution of the Hydrogen-Bond Network Involving a Tyrosine Triad in the Active Site to the Structure and Function of a Highly Proficient Ketosteroid Isomerase from Pseudomonas Putida Biotype B. Biochemistry. 2000;39:4581–4589. doi: 10.1021/bi992119u. [DOI] [PubMed] [Google Scholar]
- 37.Kraut DA, Sigala PA, Pybus B, Liu CW, Ringe D, Petsko GA, Herschlag D. Testing Electrostatic Complementarity in Enzymatic Catalysis: Hydrogen Bonding in the Ketosteroid Isomerase Oxyanion Hole. PLoS Biology. 2006;4:501–519. doi: 10.1371/journal.pbio.0040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sigala PA, Fafarman AT, Bogard PE, Boxer SG, Herschlag D. Do Ligand Binding and Solvent Exclusion Alter the Electrostatic Character within the Oxyanion Hole of an Enzymatic Active Site? J Am Chem Soc. 2007;129:12104–12105. doi: 10.1021/ja075605a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rickert KW, Klinman JP. Nature of Hydrogen Transfer in Soybean Lipoxygenase 1: Separation of Primary and Secondary Isotope Effects. Biochemistry. 1999;38:12218–12228. doi: 10.1021/bi990834y. [DOI] [PubMed] [Google Scholar]
- 40.Hatcher E, Soudackov AV, Hammes-Schiffer S. Proton-Coupled Electron Transfer in Soybean Lipoxygenase. J Am Chem Soc. 2004;126:5763–5775. doi: 10.1021/ja039606o. [DOI] [PubMed] [Google Scholar]
- 41.Knapp MJ, Rickert KW, Klinman JP. Temperature Dependent Isotope Effects in Soybean Lipoxygenase-1: Correlating Hydrogen Tunneling with Protein Dynamics. J Am Chem Soc. 2002;124:3865–3874. doi: 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]
- 42.Meyer MP, Tomchick DR, Klinman JP. Enzyme Structure and Dynamics Affect Hydrogen Tunneling: The Impact of a Remote Side Chain (I553) in Soybean Lipoxygenase-1. Proc Natl Acad Sci USA. 2008;105:1146–1151. doi: 10.1073/pnas.0710643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borgis D, Hynes JT. Dynamical Theory of Proton Tunneling Transfer Rates in Solution: General Formulation. Chem Phys. 1993;170:315–346. [Google Scholar]
- 44.Agarwal PK, Billeter SR, Hammes-Schiffer S. Nuclear Quantum Effects and Enzyme Dynamics in Dihydrofolate Reductase Catalysis. J Phys Chem B. 2002;106:3283–3293. [Google Scholar]
- 45.Garcia-Viloca M, Truhlar DG, Gao J. Reaction-Path Energetics and Kinetics of the Hydride Transfer Reaction Catalyzed by Dihydrofolate Reductase. Biochemistry. 2003;42:13558–13575. doi: 10.1021/bi034824f. [DOI] [PubMed] [Google Scholar]
- 46.Hatcher E, Soudackov AV, Hammes-Schiffer S. Proton-Coupled Electron Transfer in Soybean Lipoxygenase: Dynamical Behavior and Temperature Dependence of Kinetic Isotope Effects. J Am Chem Soc. 2007;129:187–196. doi: 10.1021/ja0667211. [DOI] [PubMed] [Google Scholar]
- 47.Olsson MHM, Siegbahn PEM, Warshel A. Simulations of the Large Kinetic Isotope Effect and the Temperature Dependence of the Hydrogen Atom Transfer in Lipoxygenase. J Am Chem Soc. 2004;126:2820–2828. doi: 10.1021/ja037233l. [DOI] [PubMed] [Google Scholar]
- 48.Chakravorty DK, Soudackov AV, Hammes-Schiffer S. Hybrid Quantum/Classical Molecular Dynamics Simulations of the Proton Transfer Reactions Catalyzed by Ketosteroid Isomerase: Analysis of Hydrogen Bonding, Conformational Motions, and Electrostatics. Biochemistry. 2009;48:10608–10619. doi: 10.1021/bi901353v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soudackov A, Hatcher E, Hammes-Schiffer S. Quantum and Dynamical Effects of Proton Donor-Acceptor Vibrational Motion in Nonadiabatic Proton-Coupled Electron Transfer Reactions. J Chem Phys. 2005;122:014505. doi: 10.1063/1.1814635. [DOI] [PubMed] [Google Scholar]
- 50.Feierberg I, Aqvist J. The Catalytic Power of Ketosteroid Isomerase Investigated by Computer Simulation. Biochemistry. 2002;41:15728–15735. doi: 10.1021/bi026873i. [DOI] [PubMed] [Google Scholar]
- 51.Hanoian P, Sigala PA, Herschlag D, Hammes-Schiffer S. Hydrogen Bonding in the Active Site of Ketosteroid Isomerase: Electronic Inductive Effects and Hydrogen Bond Coupling. Biochemistry. 2010;49:10339–10348. doi: 10.1021/bi101428e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harris TK, Turner GJ. Structural Basis of Perturbed Pka Values of Catalytic Groups in Enzyme Active Sites. IUBMB Life. 2002;53:85–98. doi: 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- 53.Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. Formation and Stability of a Vinyl Carbanion at the Active Site of Orotidine 5′-Monophosphate Decarboxylase: Pka of the C-6 Proton of Enzyme-Bound Ump. J Am Chem Soc. 2008;130:1574–1575. doi: 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chakravorty DK, Hammes-Schiffer S. Impact of Mutation on Proton Transfer Reactions in Ketosteroid Isomerase: Insights from Molecular Dynamics Simulations. J Am Chem Soc. 2010;132:7549–7555. doi: 10.1021/ja102714u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kamerlin SCL, Sharma PK, Chu ZT, Warshel A. Ketosteroid Isomerase Provides Further Support for the Idea That Enzymes Work by Electrostatic Preorganization. Proc Natl Acad Sci USA. 2010;107:4075–4080. doi: 10.1073/pnas.0914579107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kamerlin SCL, Haranczyk M, Warshel A. Progress in Ab Initio Qm/Mm Free-Energy Simulations of Electrostatic Energies in Proteins: Accelerated Qm/Mm Studies of Pka, Redox Reactions and Solvation Free Energies. Journal of Physical Chemistry B. 2009;113:1253–1272. doi: 10.1021/jp8071712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Riccardi D, Konig P, Guo H, Cui Q. Proton Transfer in Carbonic Anhydrase Is Controlled by Electrostatics Rather Than the Orientation of the Acceptor. Biochemistry. 2008;47:2369–2378. doi: 10.1021/bi701950j. [DOI] [PubMed] [Google Scholar]
- 58.Wong KF, Watney JB, Hammes-Schiffer S. Analysis of Electrostatics and Correlated Motions for Hydride Transfer in Dihydrofolate Reductase. J Phys Chem B. 2004;108:12231–12241. [Google Scholar]
- 59.Note that the protein motions, and therefore this relative probability, depend on the potential energy surface, which in turn reflects the energetic interactions within the system. Thus, this perspective is not inconsistent with concepts of transition state stabilization due to substituents on the substrate or mutations within the protein. In particular, modifications that energetically stabilize the transition state relative to the reactant state tend to increase the relative probability of sampling the transition state configurations, thereby lowering the free energy barrier. An analogous argument may be made for reactant state destabilization.
- 60.Newton MD, Sutin N. Electron Transfer Reactions in Condensed Phases. Annu Rev Phys Chem. 1984;35:437–480. [Google Scholar]
- 61.Marcus RA, Sutin N. Electron Transfers in Chemistry and Biology. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 62.Staib A, Borgis D, Hynes JT. Proton Transfer in Hydrogen-Bonded Acid-Base Complexes in Polar Solvents. J Chem Phys. 1995;102:2487–2505. [Google Scholar]
- 63.Beratan DN, Skourtis SS. Electron Transfer Mechanisms. Curr Opin Chem Biol. 1998;2:235–243. doi: 10.1016/s1367-5931(98)80065-3. [DOI] [PubMed] [Google Scholar]
- 64.Schwartz SD, Schramm VL. Enzymatic Transition States and Dynamic Motion in Barrier Crossing. Nature Chemical Biology. 2009;5:551–558. doi: 10.1038/nchembio.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fierke CA, Johnson KA, Benkovic SJ. Construction and Evaluation of the Kinetic Scheme Associated with Dihydrofolate Reductase from Escherichia Coli. Biochemistry. 1987;26:4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- 66.Childs W, Boxer SG. Solvation Response Along the Reaction Coordinate in the Active Site of Ketosteroid Isomerase. J Am Chem Soc. 2010;132:6474–6480. doi: 10.1021/ja1007849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Watney JB, Agarwal PK, Hammes-Schiffer S. Effect of Mutation on Enzyme Motion in Dihydrofolate Reductase. J Am Chem Soc. 2003;125:3745–3750. doi: 10.1021/ja028487u. [DOI] [PubMed] [Google Scholar]
- 68.Wong KF, Selzer T, Benkovic SJ, Hammes-Schiffer S. Impact of Distal Mutations on the Network of Coupled Motions Correlated to Hydride Transfer in Dihydrofolate Reductase. Proc Natl Acad Sci USA. 2005;102:6807–6812. doi: 10.1073/pnas.0408343102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thorpe IF, Brooks CL., III Barriers to Hydride Transfer in Wild Type and Mutant Dihydrofolate Reductase from E. Coli. J Phys Chem B. 2003;107:14042–14051. [Google Scholar]
- 70.Rod TH, Radkiewicz JL, Brooks CL., III Correlated Motion and the Effect of Distal Mutations in Dihydrofolate Reductase. Proc Natl Acad Sci USA. 2003;100:6980–6985. doi: 10.1073/pnas.1230801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Edwards SJ, Soudackov AV, Hammes-Schiffer S. Impact of Distal Mutation on Hydrogen Transfer Interface and Substrate Conformation in Soybean Lipoxygenase. J Phys Chem B. 2010;144:6653–6660. doi: 10.1021/jp100133p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Layfield J, Hammes-Schiffer S. Calculation of Vibrational Shifts of Nitrile Probes in the Active Site of Ketosteroid Isomerase Upon Ligand Binding. Journal of Physical Chemistry B. doi: 10.1021/ja3084384. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lindquist BA, Haws RT, Corcelli SA. Optimized Quantum Mechanics/Molecular Mechanics Strategies for Nitrile Vibrational Probes: Acetonitrile and Para-Tolunitrile in Water and Tetrahydrofuran. Journal of Physical Chemistry B. 2008;112:13991–14001. doi: 10.1021/jp804900u. [DOI] [PubMed] [Google Scholar]
- 74.Benkovic SJ, Hammes GG, Hammes-Schiffer S. Free-Energy Landscape of Enzyme Catalysis. Biochemistry. 2008;47:3317–3321. doi: 10.1021/bi800049z. [DOI] [PubMed] [Google Scholar]