

Abstract
One main issue with peptide-based molecular imaging probes is their relatively low tumor affinity and short retention time. To improve peptide binding affinity, multivalency approach has been introduced. Traditionally, this approach involves the use of peptide homodimers or homomultimers in which peptide ligands of the same type are constructed with suitable linkers. Recently, a new approach using peptide heterodimers has emerged as a promising method for targeting multi-receptor over-expressed tumor cells. Significant affinity enhancements have been observed with peptide heterodimers compared with their parent peptide monomers. In a peptide hetero-dimer, two different peptide ligands capable of targeting two different receptors are covalently linked. The binding modes of peptide heterodimers can be monovalent or bivalent depending on whether simultaneous binding of two ligands can be achieved. Increased local ligand concentration and improved binding kinetics contribute to enhanced binding in both monovalent- and bivalent binding modes, while multivalency effect also plays an important role in bivalent binding mode. As many tumors overexpress multiple receptors, more peptide heterodimer-based molecular imaging probes are expected to be developed in future. This review article will discuss the peptide homodimers and heterodimers for molecular imaging with special emphasis on peptide heterodimers.
Keywords: Peptide, Heterodimer, Molecular imaging
Introduction
Ligands that bind to characteristic receptors that are over-expressed on particular tumor cell surface can be used for diagnostic and therapeutic applications after being labeled with imaging or cytotoxic agents. Peptides and antibodies are two major types of ligands for these applications. Antibodies are generally of high specificity and high affinity for tumor targeting. However, their wide application is severely limited due to the slow blood clearance, poor tissue penetration, high immunogenicity as well as high costs. When compared with antibodies, peptides have fast clearance, excellent tissue penetration and low immunogenicity (Benedetti et al. 2004). Using automatic peptide synthesizers, peptides can be produced easily and inexpensively at a relatively large scale. Synthetic peptides are also much easier to be modified when such modifications are required for increased stability, more specific targeting, better tumor affinity and pharmacokinetics. In the last decade, receptor binding peptides have attracted intense research attention and have become valuable tools in cancer imaging and therapy (Aloj and Morelli 2004; Kwekkeboom et al. 2000; Zaccaro et al. 2009).
Although peptides have many advantages over antibodies in molecular targeting, they have their own drawbacks when compared with antibodies as targeting agents. One main issue is their relatively low tumor affinity and short retention time, which makes peptides not as specific as antibodies in tumor targeting. The high binding affinity of antibodies is achieved largely through multivalent interactions. Although each individual interaction is weak, these interactions work in a cooperative way and the overall binding affinity is much higher than the sum of each individual interaction. Multivalent interactions also significantly reduce the off rate of the binding interaction.
To design peptide-based high affinity molecular imaging probes, various methods have been used including the multivalency approach (Mammen et al. 1998). Peptide homomultimers and heteromultimers have been designed and successfully applied in molecular imaging with significant affinity enhancement compared with their parent peptide monomers. Peptide homomultimer approach has been well studied and a variety of peptide homomultimers have been reported, while peptide heterodimers were not designed and applied in molecular imaging until very recently. In this review article, we will discuss briefly the peptide homodimers and heterodimers for molecular imaging with special emphasis on peptide heterodimers.
Peptide homodimer as molecular imaging agents with improved tumor affinity
One traditional way of multivalency approach is the creation of peptide homodimers or homomultimers. Peptide homodimers or multimers are constructs with two or more receptor ligands of the same type coupled through a linker system. Peptide dimers or multimers have been well documented to have much higher avidity for targeting tumor cells than peptide monomers (Zhang et al. 2006; Li et al. 2007, 2008; Liu et al. 2001; Ye et al. 2006; Sharma et al. 1996; Wu et al. 2005; Wu et al. 2007; Liu 2006). The improved affinity is believed to come from cooperative receptor–ligand interactions and receptor shielding from endogenous competition. Depending on the positions of peptide ligands, the improved receptor–ligand interactions can be true multivalent interaction when peptide ligands are well separated and can concomitantly bind to cell-surface receptors. When peptide ligands are too close for simultaneous binding, receptor–ligand interaction can also be enhanced from improved binding kinetics resulting from increased local ligand concentration (Fig. 1).
Fig. 1.
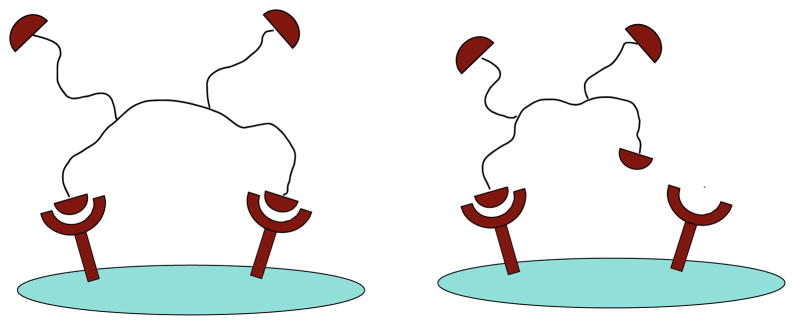
Schematic illustration of receptor–peptide multimer interaction
Previously, we have used multivalency approach to synthesize RGD peptide dimer, tetramer and octamer targeting integrin αvβ3. These peptides were radiolabeled with 18F, 64Cu and 68Ga and evaluated in vitro and in vivo. 18F-labeled RGD tetramer had much higher tumor uptake than the monomeric and dimeric RGD peptide analogs (Wu et al. 2007). Comparison of 64Cu-labeled RGD tetramer and RGD octamer indicated that the RGD octamer had significantly higher integrin binding affinity and specificity, higher tumor uptake and longer tumor retention than RGD tetramer (Li et al. 2007). The results with 68Ga-labeled RGD monomer, dimer and tetramer showed that RGD tetramer had the highest integrin affinity in vitro and highest tumor uptake in vivo (Li et al. 2008a). Overall, these studies showed integrin binding affinity in the order of octamer > tetramer > dimer > monomer. However, our studies also indicated increased non-specific targeting and imaging background with the increased size of the tracer, i.e., the no-specific targeting also follows the order of octamer > tetramer > dimer > monomer. Therefore, even though the absolute tumor uptake increased with increasing size of the tracer, the tumor/background ratio did not necessarily follow the same trend, particularly for tetramers and octamers. When considering the improved tumor uptake, prolonged retention as well as the relatively easy scale-up for commercial applications, the peptide dimer seems to be the best compound to image αvβ3-integrin expression. Recently, we also demonstrated that when two RGD peptides are connected by linkers of suitable length, further improvement of binding affinity can be achieved due to simultaneous integrin αvβ3 binding (Liu et al. 2009a, b).
Peptide heterodimers as molecular imaging agents with improved tumor affinity
Recently, a new approach using peptide heterodimers has emerged as a promising method to improve peptide affinity. In a peptide heterodimer, two different peptide ligands targeting different receptors are covalently linked by either a flexible or rigid linker with adjustable length. The molecular basis of heterodimer for tumor target is the fact that cancer cells co-express multiple peptide receptors (Handl et al. 2004; Reubi and Waser 2003). The prerequisite to design a peptide heterodimer for in vivo multireceptor molecular imaging is the knowledge that which peptide receptors are over-expressed in which tumor types. A number of peptide heterodimers have been designed and some of them have been successfully applied to molecular imaging.
Because androgen-independent prostate cancer cells overexpress gastrin-releasing peptide receptor (GRPR), various approaches have been explored to target GRPR for prostate cancer diagnosis and therapy. BBN(7-14), a truncated sequence of bombesin, an analog to GRP, has been identified as a promising ligand for this purpose. However, due to the low tumor accumulation and retention as well as unfavorable hepatobiliary excretion, further modification of the ligand structure is required for better tumor targeting effect. As androgen-independent prostate cancer cells also express moderate level of integrin αvβ3, we envisioned that a radiolabeled-BBN-RGD peptide heterodimer with dual GRPR and integrin αvβ3-targeting (BBN peptide motif for GRPR targeting and RGD peptide motif for integrin αvβ3 targeting) may significantly improve the imaging results over BBN or RGD monopeptide which recognize only one receptor type. We thus developed a BBN-RGD peptide heterodimer in which cyclic RGD was conjugated through a glutamate linker with BBN(7-14) (Fig. 2) (Li et al. 2008b). We first investigated the 18F-labeled BBN-RGD heterodimer. 18F with almost 100% positron efficiency and relatively short physical half life (t1/2 = 109.7 min) is ideally suited for peptide labeling and PET imaging. BBN-RGD peptide heterodimer was labeled with 18F-SFB (N-succin-imidyl-4-[18F]fluorobenzoate). The resulting 18F-labeled heterodimer, 18F-FB-BBN-RGD, was then used for in vitro and in vivo evaluation (Fig. 3). Receptor binding assay demonstrated that the binding affinities of this dual-receptor-targeting BBN-RGD heterodimer to GRPR and integrin αvβ3 was similar to Aca-BBN(7-14) (for GRPR binding) and c(RGDyK) (for integrin αvβ3 binding). It also had a slower washout than monomeric parent peptides. When compared with 18F-radiolabeled BBN and RGD peptides, 18F-FB-BBN-RGD had much lower liver, kidney and intestinal accumulation than the monomeric counterparts. In vitro and in vivo blocking experiments with RGD or BBN alone could only partially block the tumor cell uptake of the tracer, indicating that the tracer could still bind to the unblocked receptors even one receptor was blocked. On the other hand, RGD + BBN could completely inhibit cell uptake. This observation confirmed the dual-receptor-targeting ability of the BBN-RGD heterodimer.
Fig. 2.

18F-labeled BBN-RGD heterodimer
Fig. 3.

microPET images of mice bearing PC-3 tumor using 18F-labeled BBN-RGD, RGD and BBN peptides (Reprinted by permission of the Society of Nuclear Medicine from Li et al. 2008b)
We proposed the following binding mechanism for this BBN-RGD heterodimer (Fig. 4): because the heterodimer is linked by a short glutamate linker, simultaneous GRPR and integrin αvβ3 binding is unlikely. However, due to the dual-receptor-targeting ability of the tracer, the total number of binding sites for BBN-RGD heterodimer is the sum of GRPR and integrin αvβ3, higher than that for RGD peptide or BBN alone. The improved behavior of BBN-RGD over BBN or RGD is not only due to the increased number of receptors for signal amplification, but also improved binding kinetics. Assuming that the ligand initially binds to GRPR through the BBN moiety, the remaining RGD moiety will then be in the close vicinity of integrin αvβ3. The dissociation of BBN-RGD from GRPR will lead to rapid recomplexation of the same ligand with integrin αvβ3. If the heterodimer is initially bound to integrin instead, the dissociation of the RGD motif from integrin will reorient the BBN-RGD to bind to GRPR, resulting in an apparent low off rate of the ligand binding. Both the increased number of binding sites and the apparent low off rate of the dual-receptor-targeting ligand are expected to have enhanced tumor uptake and retention as compared to those parent single-receptor-recognizing ligands. The added molecular size and change in overall molecular charge and hydrophilicity will also have certain effects on the in vivo kinetics of the resulting probes.
Fig. 4.

Binding mechanism for BBN-RGD heterodimer (Reprinted by permission of the Society of Nuclear Medicine from Li et al. 2008b)
Our first generation of BBN-RGD heterodimer is actually a mixture of Glu-BBN-RGD (where RGD is on the side-chain δ-position) and Glu-RGD-BBN (where BBN is on the side-chain δ-position) which are unseparable by HPLC purification. Recently, we synthesized a BBN-RGD heterodimer with definite structure through solid-phase synthesis method using orthogonally protected Fmoc-Glu-OAll (Liu et al. 2009c). The synthesis strategy is described in Scheme 1. Loading of Fmoc-Met-Rink amide MBHA resin and synthesis of the bombesin peptide followed standard peptide synthesis protocols. After attaching Fmoc-Glu-OAll onto Aca, the α-allyl ester was removed by treating with Pd(Ph3P)4/CHCl3/AcOH/NMM. The α-carboxylate was activated and coupled with cyclic RGD peptide via the lysine side-chain ε-amine group. After removing the Fmoc protecting group from Glu, Glu-BBN-RGD heterodimer was cleaved and deprotected with TFA/ethandithiol (EDT)/triisopropylsilane (TIS). We then attached a mini-PEG spacer onto the amino group of the glutamate in the Glu-BBN-RGD heterodimer. The attachment of mini-PEG spacer improved 18F-labeling yield, but did not have negative effect on the biological activities of RGD and BBN as evidenced by receptor binding assay. The resulting 18F-FB-PEG3-BBN-RGD (Fig. 5) was evaluated again in PC-3 tumor model for microPET and biodistribution studies. The PEGylated BBN-RGD heterodimer demonstrated comparable tumor uptake and nonspecific tissue uptake as the first-generation BBN-RGD heterodimer. The inclusion of a hydrophilic PEG motif also resulted in rapid renal clearance as indicated by lower kidney uptake.
Scheme 1.

Synthesis of BBN-RGD heterodimer
Fig. 5.

Schematic structure of 18F-FB-PEG3-BBN-RGD
64Cu is a useful radioisotope for PET imaging. Owing to the relatively long half life (12.7 h) of 64Cu, more late time point information may be obtained from 64Cu imaging than 18F (half life, 109.7 min). 64Cu can be produced in high yield and high-specific activity from a small biomedical cyclotron. Peptides after being conjugated with macrocyclic chelators can be easily radiolabeled with 64Cu in high yield, making kit formulation of 64Cu radiotracer highly possible. We conjugated the BBN-RGD heterodimer with DOTA and NOTA (Fig. 6), two common chelators for radiometals and evaluated the behaviors of 64Cu-labeled heterodimer conjugates (Liu et al. 2009d). NOTA-BBN-RGD can be more easily labeled with 64Cu than DOTA-BBN-RGD. 64Cu-NOTA-BBN-RGD had much higher tumor uptake and tumor to non-tumor ratios and lower liver uptake than 64-Cu-DOTA-BBN-RGD. Similar to 18F-labeled BBN-RGD heterodimer, the tumor uptake of 64Cu-NOTA-BBN-RGD was also significantly higher than that of 64Cu-NOTA-RGD, 64Cu-NOTA-BBN and 64Cu-NOTA-RGD plus 64Cu-NOTA-RGD. 64Cu-NOTA-BBN-RGD also showed improved pharmacokinetics over its monomeric counterparts, as evidenced by much lower intestinal accumulation and predominant renal clearance. As a comparison, 64Cu-NOTA-BBN-RGD allowed good imaging quality and low background at late time points after injection owing to its long half life, while 18F-labeled BBN-RGD is better for tumor imaging within 2 h after injection due to its relatively high tumor uptake and low liver uptake.
Fig. 6.

DOTA and NOTA-BBN-RGD heterodimers
We also radiolabeled NOTA-BBN-RGD with 68Ga (Liu et al. 2009e). 68Ga has the physical property of high positron yield reaching 89% of all disintegrations and a short physical half life of 68 min which matches the biological half lives of many peptides and other small molecules. 68Ga can be easily obtained from an in-house 68Ge/68Ga generator (68Ge, t1/2 270.8 days) instead of a medical cyclotron. All these characteristics make 68Ga a promising radioisotope for clinical PET imaging application using an instant kit radiolabeling technique. In vitro and in vivo experiments showed that 68Ga-labeled BBN-RGD heterodimer has a significantly lower efflux ratio and higher tumor uptake when compared with 18F-labeled heterodimer, indicating that the former has more effective internalization and trapping of radiometal inside the tumor cells.
Apart from prostate cancer imaging, we also investigated the radiolabeled BBN-RGD heterodimers for breast cancer imaging (Liu et al. 2009f). Two breast cancel models, T47D and MDAMB-435, were employed in this study as representatives of ER+ and ER− breast cancers, respectively. Imaging properties of three radiolabeled (18F, 64Cu and 68Ga) heterodimeric probes were investigated. The 18F-labeled RGD-BBN showed a more rapid washout than the 64Cu- and 68Ga-labeled peptides. Although tumor uptake of 18F-FB-PEG3- BBN-RGD was lower than that of 64Cu and 68Ga labeled BBN-RGD radiotracers, 18F-FB-PEG3-BBN-RGD had the highest tumor to non-tumor organ ratio. 64Cu- and 68Ga-labeled radiotracers showed higher background than 18F-labeled tracers. In addition, the liver uptake of 64Cu-NOTA-BBN-RGD was higher than 68Ga-NOTA-BBN-RGD, probably due to the lower chelating ability of NOTA to 64Cu when compared with 68Ga. The in vitro cell uptake studies demonstrated that the cell uptake values of the radiotracers on T47D tumor cells were higher than those on MDA-MB-435 tumor cells, which may be caused by the much higher GRPR expression on the T47D cells, leading to efficient internalization of the radiotracers.
The above-described heterodimeric peptide-based radio-tracers function as a monoligand, i.e., the increased tumor affinity mainly comes from the increased local ligand concentration and the availability of two different receptors for binding. This is because the glutamate linker that connects two peptide ligands is too short, simultaneous binding of two peptide ligands to two separated different receptors are not likely. Another logical approach is to design peptide heterodimers in which two different peptide ligands may simultaneously bind to two different receptors. Molecular imaging probes based on this design have not been applied in vivo yet; however, several in vitro “proof of principle” experiments have been successfully conducted.
Peptide heterodimers were designed to simultaneously bind two G protein-coupled receptors (GPCRs): melano-cortin-4 (hMC4R) and δ-opioid (δ-OR) receptors (Vagner et al. 2008). The heterodimers were prepared by connecting melanocyte stimulating hormone [MSH(7)] ligand (H-Ser-Nle-Glu-His-dPhe-Arg-Trp-NH2) and Deltorphin-II sequence (H-Tyr-dAla-Phe-Glu-Val-Val-Gly), with a hybrid linker composed of semirigid Pro-Gly (PEGO-[Pro-Gly]x-PEGO) and/or flexible ethylene glycol moieties. The length and rigidity of the linker can be adjusted by changing the number of (Pro-Gly) repeats. The synthetic strategy is depicted in Scheme 2.
Scheme 2.

Synthesis of Delt-II-MSH(7) heterodimers
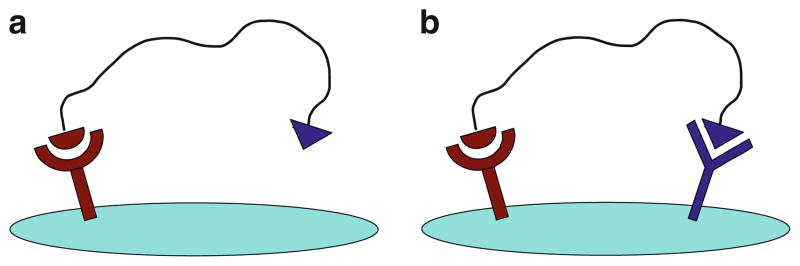
Dual-receptor binding affinities of these peptide hetero-dimers were tested in a cell system expressing both hMC4R and δ-OR receptors, by evaluating competitive binding of each heterodimer against receptor-specific labeled ligand (Eu-NDPaMSH for the hMC4R and Eu-DPLCE for the δ-OR). It was found that binding affinities to hMC4R were significant better in dual-receptor binding mode where both hMC4R and δ-OR receptors were available for binding (Fig. 7b), as compared to hMC4R monovalent binding mode, where the δ-OR receptor was blocked (by adding saturating concentrations of δ-OR blocking agent) and only the hMC4R receptor was available for binding (Fig. 7a). In dual-receptor binding mode, peptide heterodimers with optimal length demonstrated the highest binding affinity enhancement, where a maximum of approximately 48 times binding affinity enhancement was observed (binding affinity in the range of 2.5–3.5 nM IC50), when compared with monovalent mode. On the other hand, peptide heterodimer with short linkers displayed no significant enhancements of binding affinity. In dual-receptor binding mode (absence of blocking agent for the two receptors), binding affinity increases with increased linker length. A maximum of 55-fold binding affinity increase was observed when the linker reached optimized length. The experimental data support a mathematical modeling study that an optimal linker length of 20–50 Å is required to crosslink two different GPCRs by peptide heterodimer ligand.
Fig. 7.

Receptor–ligand interaction on cells under blocking and nonblocking conditions
Interestingly, with these peptide heterodimers, no significant enhancement in binding affinity to the δ-OR was observed, which is in sharp contrast with what was observed for hMC4R binding. One explanation of this observation is that hMC4R is lower expressed in cell surface compared to δ-OR. Once the less abundant hMC4R receptors were saturated by bivalent interactions of the peptide heterodimers, the remaining peptide heterodimers can only bind to the remaining more abundant δ-OR receptors in a monovalent way, which is much weaker than bivalent interaction.
Similarly, a library of peptide heterodimers MSH(7)-CCK-6 targeting hMC4R and the cholecystokinin-2 receptor (CCK-2R) were prepared using a modular solid-phase synthetic strategy (Scheme 3) (Josan et al. 2008). The strategy involves three steps: (1) assembly of the first peptide on the resin, (2) build of an appropriate linker, (3) assembly of the second peptide. Either a highly flexible PEGO or a semi-rigid Pro-Gly linker was use as linkers. Functional agents, such as fluorescent labels can be incorporated at any step in synthesis.
Scheme 3.

Synthesis of CCK(6)-MSH(7) heterodimers
Using lanthanide-based time-resolved fluorescence (TRF), evaluation of binding affinities of peptide heterodimers was done via a competitive binding assay using Hek293 cells that expressed either or both melanocortin- 4 and CCK-2 receptors. Again, the bioevaluation results demonstrated that with suitable length of linker, peptide heterodimer could simultaneously crosslink two different cell-surface receptors, resulting in increases in binding affinity. For CCK-2R binding, peptide heterodimer with a semi-rigid Pro-Gly 2 linker exhibited 20-fold enhancement in binding affinity when binding to cells expressing both hMC4R and CCK-2R (IC50 of 2.3 nM) (Fig. 8b) when compared with cells expressing only one receptor (IC50 of 46 nM) (Fig. 8a), while peptide heterodimer with a flexible PEGO3 linker exhibited 22-fold enhancement in binding affinity.
Fig. 8.
Receptor–ligand interaction on cells expressing one and two receptors
As the same in the case of heterodimer MSH(7)-deltor-phin-II targeting hMC4R and δ-OR where binding affinity increases more for low expressed receptor binding, significant enhancement of binding affinity for peptide heterodimer MSH(7)-CCK-6 was only observed at the CCK-2R, the lower abundant receptor in Hek293 cells and little for higher expressed hMC4R. It should be noted that, as compared to peptide monomers which target only one receptor, peptide heterodimers which simultaneously targeting two different receptors obtain an overall gain in binding affinity when binding to cells expressing both receptors, even though the enhancement in binding affinity occurs at only one receptor.
Imaging of peptide heterodimer binding to the dual CCK2R/hMC4R expressing cell line and ligand-induced receptor internalization was studied using MSH(7)-PEGO-[PG]6-PEGO-CCK-6 (76 atoms linker) after labeling with the fluorophore Cy5 (Xu et al. 2009). Significant binding to the cell surface was observed immediately following the incubation of Cy5-labeled MSH(7)-PEGO-[PG]6-PEGO-CCK-6. The unbounded ligand was washed away with ligand-free media after 3 min. By 10 min, the receptor–ligand complex had been substantially internalized as no significant loss of ligand was observed by adding competitive binding CCK ligand.
Recently a new peptide heterodimer based on erythropoietin mimetic peptide (EMP) and erythropoietin receptor peptide (ERP) was synthesized using chemoselective oxime chemistry (Vadas and Rose 2007). The strategy of the EMP-ERP heterodimer synthesis is shown in Scheme 4. The ERP was first prepared by solid-phase peptide synthesis. Then, five flexible ‘PEG-succ’ units were added on its N-terminus. After coupling a serine residue, the intermediate, Ser-[PEG-succ]5-ERP, was cleaved from the resin. Prior to the oxidation of terminal serine, the free cysteine of the ERP was protected by alkylation with iodoacetamide to avoid the formation of sulfonic acid. Aldehyde functionality for oxime ligation was obtained by oxidation of terminal serine with periodate. Reaction with AoA-EMP or EMPK-AoA (aminooxyacetyl-derived EMP peptides, synthesized by SPPS) afforded the N-EMP-ERP and C-EMPK-ERP heterodimers. Both EMP and ERP activate erythropoietin EPOR. EMP bind to EPOR at the same site as erythropoietin, whereas ERP interacting with the EPOR at a distant site. In vitro study has shown that ERP and EPO act in a synergic way. Thus, it is expected that the heterodimer will have strong binding affinity and better biological activity.
Scheme 4.

Synthesis of EMP-ERP heterodimer
Vascular endothelial growth factor (VEGF) ligands are main mediators of angiogenesis. VEGF receptor 2 (VEG-FR-2) presenting on both the luminal and abluminal sides of tumor are restricted to proliferating endothelial cells. Recently, high affinity heterodimers targeting VEGFR-2 were developed (Shrivastava et al. 2005; Tweedle 2006; Shrivastava et al. 2009; Tweedle 2009). The whole development process involves three critical steps: (1) phage display selection of peptide, (2) identification of peptide pairs suitable for heterodimerization and (3) chemical synthesis of heterodimer. The peptide monomers targeting VEGFR-2, with KD values as low as 3 nM, were identified using standard peptide phase display. An in vitro peptide/neutravidin-HRP (horseradish peroxidase) assay based on the avidin–biotin interaction was used to identify suitable peptide pairs which non-competitively bind to two different epitopes of a single VEGFR-2 and show synergistic binding property. Peptide heterodimer was prepared by covalently linking peptide pairs with a glutamic linker. The heterodimers were found to bind to VEGFR-2 with much higher affinity (over 100-fold increase) than either of their corresponding monomers or homodimers. They also have greater inhibition ability to block VEGF binding and VEGF-induced activation of VEGFR-2, probably due to slower off rates and/or larger sterically hinderance. Further more, the binding of peptide heterodimers are less sensitive to serum than their component peptides.
Activation of c-MET, also known as hepatocyte growth factor receptor (HGFR), is often associated with cancer metastasis. Peptide heterodimer targeting c-MET was prepared with similar strategy. A 250-fold increase in binding affinity to c-MET was obtained with the new peptide heterodimer.
It is proposed that the peptide heterodimer binds to target molecule (VEGFR-2 or c-MET) by interacting with two different epitopes of a single monomeric unit of the target molecule (Fig. 9a). Another binding mode (Fig. 9b) which a single peptide heterodimer cross-links two different monomeric units located on two different target molecules is unlikely due to the two facts: (1) the affinities of peptide heterodimers are far more superior to both mixtures of the two peptide monomers and the peptide homodimers, (2) peptide homodimers do not show significant affinity improvement over peptide monomers.
Fig. 9.

Receptor–ligand interaction through two epitopes
A general method for the preparation of peptide heterodimer targeting VEGFR-2 and c-MET was described. The peptide monomers were prepared by standard solid-phase peptide synthesis. One peptide monomer was then reacted with the excess of glutaric acid bis-N-hydroxy-succinimidyl ester to form monoacylation intermediate. After removal of excess bis-NHS ester, the monoacylation intermediate was coupled to the second peptide monomer to produce the peptide heterodimer.
A practical synthesis method to prepare a library of peptide heterodimers containing functional groups such as affinity probes, chelators and latent conjugating groups was reported (Scheme 5) (Pillai et al. 2006). In this method, a bifunctional linker (disuccinimidyl glutarate, DSG) was used to link peptide monomers bearing or not bearing spacer groups. Interestingly, the use of water soluble Adoa-Adoa spacer only lead to a limited increase in hydrophilicity of the resulting peptide heterodimer, probably due to the much shorter poly-oxyethylene moiety of the spacer, as compared to generally employed high molecular weight PEG which have been widely used to increase the water solubility of hydrophobic peptides and proteins. To ensure fully control of peptide heterodimer assembly and position of functional groups, multiple lysine Nε-amino groups other than those for tethering peptide monomers, are protected with the (4,4-dimethyl-2,6-dioxo-cyclohex-1-ylidene)-3-methylbutyl (ivDde). Deprotection was carried out in 2% hydrazine in DMF for 5 min at room temperature). Other amino acids can be used for heterodimer preparation without protection. Functional molecules can be easily incorporated into the peptide heterodimer by this approach. Attaching functional groups in general involves three steps: incorporation of an additional Nε-ivDde protected lysine into one peptide monomer before heterodimer formation, removal of ivDde of lysine after heterodimer formation and reaction of ε-amino group of lysine with the NHS ester of the required functional molecule. These peptide heterodimers demonstrate significantly higher affinity for the receptor tyrosine kinases KDR (kinase domain receptor) and c-Met than any of the constituent monomer peptides. Despite the unsymmetrical nature of the linker, almost no effect on binding was observed by switching the peptides at the two different ends of the linker. Varying linker length between 20 and 40 atoms also had not much effect.
Scheme 5.

Synthesis of heterodimers bearing functional groups
Conclusion
Many tumors overexpress multiple receptors, which can be targeted with peptide heterodimers with increased binding affinity and specificity. Peptide heterodimers can be easily prepared by simply linking two peptide monomers. The binding modes of peptide heterodimers can be monovalent or bivalent depending on whether simultaneous binding of two ligands can be achieved. Nevertheless, both monovalent and bivalent binding improve the binding efficacy. Increased local ligand concentration and improved binding kinetics contribute to enhanced binding in both monovalent and bivalent binding modes, while multivalency effect also plays an important role in bivalent binding mode. The improved affinity, the ease of large scale synthesis of peptide heterodimer make them highly promising in molecular imaging application.
There remain, however, challenges in designing peptide heterodimers, especially designing bivalent heterodimers. Better understanding of receptor expression pattern on tumor cells, such as the density of different receptors and the distance between two different binding epitopes is very important for the de novo design of bivalent heterodimer of high affinity and specificity. The length and rigidity of the linker connecting two ligands need to be optimized with the aid of molecular modeling to minimize tedious chemical synthesis and in vitro and in vivo screening.
Although peptide heterodimers are still in their early stage of applications in molecular imaging, they have already shown their power in tumor targeting. We expect to see more peptide heterodimers to be developed in the near future for molecular imaging and related applications.
References
- Aloj L, Morelli G. Design, synthesis and preclinical evaluation of radiolabeled peptides for diagnosis and therapy. Curr Pharm Des. 2004;10:3009–3031. doi: 10.2174/1381612043383511. [DOI] [PubMed] [Google Scholar]
- Benedetti E, Morelli G, Accardo A, Mansi R, Tesauro D, Aloj L. Criteria for the design and biological characterization of radiolabeled peptide-based pharmaceuticals. BioDrugs. 2004;18:279–295. doi: 10.2165/00063030-200418050-00001. [DOI] [PubMed] [Google Scholar]
- Handl HL, Vagner J, Han H, Mash E, Hruby VJ, Gillies RJ. Hitting multiple targets with multimeric ligands. Expert Opin Ther Targets. 2004;8:565–586. doi: 10.1517/14728222.8.6.565. [DOI] [PubMed] [Google Scholar]
- Josan JS, Vagner J, Handl HL, Sankaranarayanan R, Gillies RJ, Hruby VJ. Solid-phase synthesis of heterobivalent ligands targeted to melanocortin and cholecystokinin receptors. Int J Pept Res Ther. 2008;14:293–300. doi: 10.1007/s10989-008-9150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwekkeboom D, Krenning EP, de Jong M. Peptide receptor imaging and therapy. J Nucl Med. 2000;41:1704–1713. [PubMed] [Google Scholar]
- Li ZB, Cai W, Cao Q, et al. (64)Cu-labeled tetrameric and octameric RGD peptides for small-animal PET of tumor alpha(v)beta(3) integrin expression. J Nucl Med. 2007;48:1162–1171. doi: 10.2967/jnumed.107.039859. [DOI] [PubMed] [Google Scholar]
- Li ZB, Chen K, Chen X. (68)Ga-labeled multimeric RGD peptides for microPET imaging of integrin alpha(v)beta (3) expression. Eur J Nucl Med Mol Imaging. 2008a;35:1100–1108. doi: 10.1007/s00259-007-0692-y. [DOI] [PubMed] [Google Scholar]
- Li ZB, Wu Z, Chen K, Ryu EK, Chen X. 18F-labeled BBN-RGD heterodimer for prostate cancer imaging. J Nucl Med. 2008b;49:453–461. doi: 10.2967/jnumed.107.048009. [DOI] [PubMed] [Google Scholar]
- Liu S. Radiolabeled multimeric cyclic RGD peptides as integrin alphavbeta3 targeted radiotracers for tumor imaging. Mol Pharm. 2006;3:472–487. doi: 10.1021/mp060049x. [DOI] [PubMed] [Google Scholar]
- Liu S, Edwards DS, Ziegler MC, Harris AR, Hemingway SJ, Barrett JA. 99mTc-labeling of a hydrazinonicotinamide-conjugated vitronectin receptor antagonist useful for imaging tumors. Bioconjug Chem. 2001;12:624–629. doi: 10.1021/bc010012p. [DOI] [PubMed] [Google Scholar]
- Liu Z, Liu S, Wang F, Chen X. Noninvasive imaging of tumor integrin expression using (18)F-labeled RGD dimer peptide with PEG (4) linkers. Eur J Nucl Med Mol Imaging. 2009a;36:1296–1307. doi: 10.1007/s00259-009-1112-2. [DOI] [PubMed] [Google Scholar]
- Liu Z, Niu G, Shi J, Liu S, Wang F, Chen X. (68)Ga-labeled cyclic RGD dimers with Gly3 and PEG4 linkers: promising agents for tumor integrin alphavbeta3 PET imaging. Eur J Nucl Med Mol Imaging. 2009b;36:947–957. doi: 10.1007/s00259-008-1045-1. [DOI] [PubMed] [Google Scholar]
- Liu Z, Yan Y, Chin FT, Wang F, Chen X. Dual integrin and gastrin-releasing peptide receptor targeted tumor imaging using 18F-labeled PEGylated RGD-bombesin heterodimer 18F-FB-PEG3-Glu-RGD-BBN. J Med Chem. 2009c;52:425–432. doi: 10.1021/jm801285t. [DOI] [PubMed] [Google Scholar]
- Liu Z, Li ZB, Cao Q, Liu S, Wang F, Chen X. Small-animal PET of tumors with (64)Cu-labeled RGD-bombesin heterodimer. J Nucl Med. 2009d;50:1168–1177. doi: 10.2967/jnumed.108.061739. [DOI] [PubMed] [Google Scholar]
- Liu Z, Niu G, Wang F, Chen X. (68)Ga-labeled NOTA-RGD-BBN peptide for dual integrin and GRPR-targeted tumor imaging. Eur J Nucl Med Mol Imaging. 2009e;36:1483–1494. doi: 10.1007/s00259-009-1123-z. [DOI] [PubMed] [Google Scholar]
- Liu Z, Yan Y, Liu S, Wang F, Chen X. 18F, 64Cu, and 68 Ga labeled RGD-bombesin heterodimeric peptides for PET imaging of breast cancer. Bioconjug Chem. 2009f;20:1016–1025. doi: 10.1021/bc9000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen M, Choi S-K, Whitesides G. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Pillai R, Marinelli ER, Swenson RE. A flexible method for preparation of peptide homo- and heterodimers functionalized with affinity probes, chelating ligands, and latent conjugating groups. Biopolymers. 2006;84:576–585. doi: 10.1002/bip.20570. [DOI] [PubMed] [Google Scholar]
- Reubi JC, Waser B. Concomitant expression of several peptide receptors in neuroendocrine tumours: molecular basis for in vivo multireceptor tumour targeting. Eur J Nucl Med Mol Imaging. 2003;30:781–793. doi: 10.1007/s00259-003-1184-3. [DOI] [PubMed] [Google Scholar]
- Sharma SD, Jiang J, Hadley ME, Bentley DL, Hruby VJ. Melanotropic peptide-conjugated beads for microscopic visualization and characterization of melanoma melanotropin receptors. Proc Natl Acad Sci USA. 1996;93:13715–13720. doi: 10.1073/pnas.93.24.13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava A, von Wronski MA, Sato AK, et al. A distinct strategy to generate high-affinity peptide binders to receptor tyrosine kinases. Protein Eng Des Sel. 2005;18:417–424. doi: 10.1093/protein/gzi049. [DOI] [PubMed] [Google Scholar]
- Shrivastava A, Nunn AD, Tweedle MF. Designer peptides: learning from nature. Curr Pharm Des. 2009;15:675–681. doi: 10.2174/138161209787315620. [DOI] [PubMed] [Google Scholar]
- Tweedle MF. Adventures in multivalency, the Harry S. Fischer memorial lecture CMR 2005; Evian, France. Contrast Media Mol Imaging. 2006;1:2–9. doi: 10.1002/cmmi.91. [DOI] [PubMed] [Google Scholar]
- Tweedle MF. Peptide-targeted diagnostics and radiotherapeutics. Acc Chem Res. 2009;42:958–968. doi: 10.1021/ar800215p. [DOI] [PubMed] [Google Scholar]
- Vadas O, Rose K. Multivalency—a way to enhance binding avidities and bioactivity—preliminary applications to EPO. J Pept Sci. 2007;13:581–587. doi: 10.1002/psc.794. [DOI] [PubMed] [Google Scholar]
- Vagner J, Xu L, Handl HL, et al. Heterobivalent ligands crosslink multiple cell-surface receptors: the human melanocortin-4 and delta-opioid receptors. Angew Chem Int Ed Engl. 2008;47:1685–1688. doi: 10.1002/anie.200702770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zhang X, Xiong Z, et al. microPET imaging of glioma integrin {alpha}v{beta}3 expression using (64)Cu-labeled tetrameric RGD peptide. J Nucl Med. 2005;46:1707–1718. [PubMed] [Google Scholar]
- Wu Z, Li ZB, Chen K, et al. microPET of tumor integrin alphavbeta3 expression using 18F-labeled PEGylated tetrameric RGD peptide (18F-FPRGD4) J Nucl Med. 2007;48:1536–1544. doi: 10.2967/jnumed.107.040816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Vagner J, Josan J, et al. Enhanced targeting with heterobivalent ligands. Mol Cancer Ther. 2009;8:2356–2365. doi: 10.1158/1535-7163.MCT-08-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Bloch S, Xu B, Achilefu S. Design, synthesis, and evaluation of near infrared fluorescent multimeric RGD peptides for targeting tumors. J Med Chem. 2006;49:2268–2275. doi: 10.1021/jm050947h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccaro L, Del Gatto A, Pedone C, Saviano M. Peptides for tumour therapy and diagnosis: current status and future directions. Curr Med Chem. 2009;16:780–795. doi: 10.2174/092986709787549307. [DOI] [PubMed] [Google Scholar]
- Zhang X, Xiong Z, Wu Y, et al. Quantitative PET imaging of tumor integrin alphavbeta3 expression with 18F-FRGD2. J Nucl Med. 2006;47:113–121. [PMC free article] [PubMed] [Google Scholar]