

Abstract
Background:
This first-in-human, phase I clinical trial of p28 (NSC745104), a 28-amino-acid fragment of the cupredoxin azurin, investigated the safety, tolerability, pharmacokinetics and preliminary activity of p28 in patients with p53+ metastatic solid tumours.
Methods:
A total of 15 patients were administered p28 i.v. as a short infusion three times per week for 4 weeks followed by a 2-week rest under an accelerated titration 3+3 dose escalation design until either a grade 3-related adverse event occurred or the maximum tolerated dose (MTD) was reached. Single-dose and steady-state serum pharmacokinetics were characterised. Assessments included toxicity, best objective response by RECIST 1.1 Criteria, and overall survival.
Results:
No patients exhibited any dose-limiting toxicities (DLTs), significant adverse events or exhibited an immune response (IgG) to the peptide. The No Observed Adverse Effect Level (NOAEL) and MTD were not reached. Seven patients demonstrated stable disease for 7–61 weeks, three a partial response for 44–125 weeks, and one a complete response for 139 weeks. Three patients are still alive at 158, 140, and 110 weeks post therapy completion.
Conclusion:
p28 was tolerated with no significant adverse events. An MTD was not reached. Evidence of anti-tumour activity indicates a highly favourable therapeutic index and demonstrates proof of concept for this new class of non-HDM2-mediated peptide inhibitors of p53 ubiquitination.
Keywords: p28, p53, metastatic solid tumours
The tumour suppressor protein p53 (tp53) is expressed in most major types of solid tumours and haematological malignancies. Unfortunately, the p53 gene is mutated in∼50% of all p53+ human solid tumours. These tumours can express constitutively high levels of their mutant p53 due to a lack of feedback control of p53 protein levels (Midgley and Lane, 1997; Martin et al, 2002; Olivier et al, 2002). Mutations are concentrated within the p53 DNA-binding domain of ∼200 amino acids, a domain that is central to the function of p53 as a transcription factor (Bullock and Fersht, 2001; Olivier et al, 2002; Tan et al, 2009).
Strategies for restoration of p53 functions in tumours have focused on targeting wild-type p53 with the aim of protecting p53 from degradation by a major endogenous regulator, HDM2, a ubiquitin protein ligase that suppresses the transcriptional activity of p53 and accounts for a majority of p53 ubiquitination and subsequent proteosomal degradation (Vassilev et al, 2004; Tabernero et al, 2009; Andreeff et al, 2010).
A small molecule or peptide targeting overexpressed mutant or wild-type p53 in cancer cells should not affect wild-type p53 in normal cells because wild-type p53 is properly folded and expressed at low levels kept in check by HDM2. As mutant p53 is already activated in tumour cells, downstream pathways regulated by p53 are likely to remain intact. We therefore suggest that restoration of p53 function should have a therapeutic effect at any point in tumour development without an adverse effect on normal cells.
The cupredoxin family of copper containing, redox proteins represents a new and significant platform of potential anticancer agents (Yamada et al, 2002; Miao et al, 2005; Yang et al, 2005; Ye et al, 2005; Chaudhari et al, 2007). Amino acids 50–67 (p18) of azurin, a cupredoxin secreted by the opportunistic pathogen Pseudomonas aeruginosa, is the protein's transport domain. This discrete, amphipathic α-helical motif is responsible for the preferential penetration of azurin into human cancer cells (Hiraoka et al, 2005; Taylor et al, 2009). As such, it acts as a cell-penetrating peptide (Taylor et al, 2009). The preferential penetration of cancer cell lines exhibited by azurin and p18 is also shared by p28, amino acids 50–77 of azurin. After entry, p28 is processed to the nucleus (Taylor et al, 2009; Yamada et al, 2009), where it binds to a hydrophobic, non-mutable region within the DNA-binding domain of p53 and inhibits proteasomal degradation via an HDM2-independent pathway. This results in an increase in intracellular levels of wild-type and mutant p53 as well as its DNA-binding activity (Yamada et al, 2009; Bizzarri et al, 2011) and elevates the cyclin-dependent kinase inhibitors p21 and p27. These proteins, in turn, reduce the level of CDK2 and cyclin A1 and induce cell cycle arrest at G2/M (Figure 1) (Yamada et al, 2009).
Figure 1.
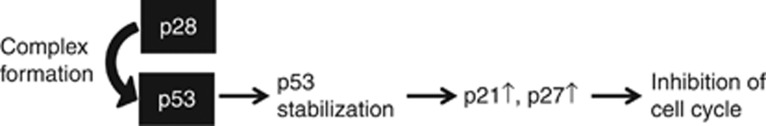
Model for stabilisation of p53 through a formation of p28 : p53 complex. p28 binds to a specific motif within the p53 DNA-binding domain that inhibits p53 proteasomal degradation and stabilizes p53, independently of HDM2. The increase in p53 transcriptionally regulates downstream genes, p21, p27, leading to inhibition of the cancer cell cycle at G2/M.
Preclinical pharmacological studies of p28 provided significant evidence for efficacy without attendent toxicity or immunogenicity (Jia et al, 2011) and prompted its entry into a clincial trial. The primary objective of this first-in-human, phase I, accelerated dose study was to determine the No Observed Adverse Effect Level (NOAEL) and maximum tolerated dose (MTD) of intravenous p28 given three times per week for 4 weeks in adult patients with p53-positive advanced solid tumours. Secondary objectives included establishing an appropriate dose for phase II studies, obtaining a pharmacokinetic profile, determining potential immunogenicity and if possible assessing preliminary antitumour activity.
Here we present safety, efficacy, and pharmacokinetic results from a phase I study of p28 in patients with p53+ advanced solid tumours, including one locoregionally advanced melanoma.
Patients and methods
Study design and endpoints
The primary objective was to determine the MTD and dose-limiting toxicity (DLT) of p28 (NSC 745104), a novel synthetic amino-acid peptide fragment of the cupredoxin azurin. The secondary objective was to assess pharmacokinetics and p28-induced immunogenicity in the study volunteers and additionally, if possible, to determine preliminary efficacy of p28 in these patients.
The trial used an enhanced accelerated titration design (Simon et al, 1997; Vickers et al, 2006; Vickers, 2006; Anderson et al, 2007), specifically an open-label, two-centre, single arm, phase I trial employing a three-plus-three dose-escalation design. The study design and number of patients entered at each dose level is illustrated in Figure 2. Patients were sequentially enrolled to receive p28 administered as an i.v. infusion over 15–30 min three times per week for 4 weeks followed by a 2-week rest period. Subjects were enrolled in groups of three, each starting at one of five progressively higher dosage groups (i.e., 0.83 mg kg−1, 1.66 mg kg−1, 2.5 mg kg−1, 3.33 mg kg−1 or 4.16 mg kg−1). The first group of three subjects received the starting dose of 0.83 mg kg−1 (10 mg kg−1 total dose), which was chosen to provide a 12-fold safety margin based on the highest dose tested in Cynomolgous sp. monkeys and exhibited significant preclinical efficacy against p53-positive xenograft tumours of similar type (Jia et al, 2011). Continuing patients then advanced to dose level 2 (1.66 mg kg−1) and three additional patients were added and began treatment at that dose; this same procedure was followed with each subsequent dose level (i.e., 2.5, 3.33, or 4.16 mg kg−1). All cumulative doses were within or above the levels for preclinical efficacy. Patients were treated and followed according to the protocol requirement or unacceptable toxicity or consent withdrawal.
Figure 2.

Escalating dose phase I clinical trial of p28 in patients (N=15) with advanced, refractory, p53-positive (>10% cells; IHC) solid tumours.
Approval for the trial was obtained from FDA (IND 77754) and the institutional review boards of two participating institutions. The study followed good clinical practice guidelines (http://www.clinicaltrials.gov identifier: NCT 00914914). All 15 patients provided written informed consent.
Patient selection
Adult patients aged⩾18 years with histologically or radiologically proven advanced solid tumours of diverse histogenesis and a possible life expectancy of ±6 months were deemed suitable for entry into this study.
Major inclusion criteria included: histologically proven solid tumour disease with documentation of measurable metastatic disease as defined by RECIST 1.1 (Eisenhauer et al, 2009); p53-positive tumours (primary or metastatic) proven by ICH (>10% of positive nuclear staining of the malignant cells); failure of prior standard treatments (chemotherapy and/or immunotherapy trials); distant metastatic disease or extensive regional disease deemed untreatable by either surgery, radiation therapy or chemotherapy or any of the combinations; a minimum of 4 weeks elapsed since completion of any form of prior therapy; adequate baseline organ function; adequate cardiac and pulmonary function. An exception of the prior therapy criteria was allowed for two metastatic melanoma patients who had previously refused to enter into any other extant clinical trials.
Adequate baseline organ function was assessed within 30 days before study entry. These included a granulocyte count >1500 per mm3, haematocrit >30%, and platelets >100 000 per mm3; calculated creatinine clearance >50 ml min−1, adequate liver function with SGOT, SGPT, LDH, and alkaline phosphatase <3 × the upper limit of normal; serum bilirubin <2.0 mg dl−1, PT and PTT not more than 1.5 times the upper limit of normal and adequate cardiac and pulmonary function. Patients with decreased LVEF or PFTs were evaluated by a cardiologist or pulmonologist before enrolment.
Major exclusion criteria included: patients undergoing chemo or immunotherapy, that is, cytokines and vaccines; p53-negative primary or metastatic tumour(s); any serious additional illness (HIV, hepatitis, untreated active infection or psychiatric illness that would prevent informed consent); any serious medical disorder (i.e., cardiovascular, insulin resistant diabetes, etc.); and concomitant brain metastases.
All questions regarding concomitant medications were referred to the study investigators. The following drugs and therapies were excluded during the study: hormonal therapy, chemotherapy, radiation therapy other than local for control of pain or for alleviation of neuropathy associated with metastases localised to spinal cord, any form of vaccine treatment for malignancy, mega dose vitamin therapy, and herbal medication.
The following immunocytochemical method was used to determine tumour p53 expression. Representative tissue sections of the patient's primary or metastatic tumour were stained for p53 status using a monoclonal antibody to p53 (Zhang, 1999) (DO-1, sk-126; Santa Cruz Biotech Inc., Santa Cruz, CA, USA) and visualised by biotinylated secondary antibody and ABC kit (Vector, Burlingame, CA, USA). Sections were counterstained with hematoxylin to identify tumour morphology. Ten separate areas from tumour slide were evaluated for p53-positive cells (minimum 1000 tumour cells counted for statistical analysis). All slides were evaluated by two independent pathologists without prior knowledge of patient status. Only cells with nuclear staining for p53 were considered positive; for a tumour to be classified as p53 positive, more than 10% of cells analysed (minimum 1000 cells counted) had to be considered p53 positive. For standard histology, 5 μm paraffin sections were stained with hematoxylin and eosin, and in some instances several other routine immunostains were also used.
Assessments
To determine the MTD, the trial design incorporated patients already in the study who previously received p28 and added three new patients at each level, until either the highest proposed dose was reached with no Grade 3 non-haematological or Grade 4 haematological toxicity or one-third of the patients experienced a Grade 3 non-haematological or Grade 4 haematological toxicity. In the former case, the preselected highest dose was to be considered as the MTD. In the latter case, that is, if one-third or more patients experienced Grade 3 non-haematological or Grade 4 haematological toxicity, the trial was to switch to standard design, that is, three new patients will be recruited at the next lower dose level.
Dose-limiting toxicity was defined as any of the following AEs attributable to the study drug that occurred during the first cycle of each treatment: grade 3 or more non-haematological toxicity, excluding alopecia and diarrhoea, nausea/vomiting in the absence of optimal prophylactic/supporting treatment; grade 4 thrombocytopenia or (any grade) requiring transfusion; anaemia lasting more than 7 days; febrile neutropenia (grade 3 or 4); or grade 4 neutropenia lasting more than 7 days.
Incidence and severity of AEs were collected at each study visit and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 3 (Trotti et al, 2003). Patients were evaluated with history, physical exam, vital signs, CBC, chemistries, urinalysis, and Eastern Cooperative Oncology Group (ECOG) performance status at baseline, weekly through 4 weeks of treatment and following a 2-week rest period before entering the next dose level.
Tumour status was assessed by CT CAP before and at week 5 of each treatment cycle. The Best Objective Response of p28 administration on the identified target lesions was measured according to RECIST 1.1 (Eisenhauer et al, 2009). When accessible, the target tumour was excised and standard histological studies were performed to assess the effect of p28. After study closure, measurement of target lesions was obtained from the scans performed by the treating physicians in the surviving patients. All tumour scans were retrospectively independently reviewed by a panel of independent radiologists who did not participate in the study.
Pharmacokinetic analysis
Bulk cGMP p28 was supplied by CS Bio Inc. (Menlo Park, CA, USA) and packaged as 50 mg sterile sealed vials with 5% sucrose by the NCI Division of Cancer Treatment under RAID grant NSC 745104. Blood samples were collected from the opposite arm. Serum was promptly harvested by centrifugation (1300 g, 4 °C, 10 min) and stored at −70 °C or less until assayed. Serum levels of p28 were determined from samples (collected pre-dose and at 5, 10, 20, 30, 60, and 120 min post dose on day one of each treatment cycle as well as pre-dose and 10 min post-dose once weekly thereafter) using a validated liquid chromatography tandem mass spectrometry assay with a lower limit of quantification of 20 ng ml−1 (Gorman et al, 2010; Jia et al, 2011). Individual patient plasma concentration–time data for each dose of p28 were analysed by standard non-compartmental methods. The maximum concentration of drug in plasma (Cmax) was based directly upon the actual assayed values of the study samples.
Statistical methods
All 15 patients received at least one full dose level (i.e., 12 doses) and are included in the safety analysis. These patients had one or more target lesions as defined by RECIST 1.1 and are included in the analysis of best objective response. All data in the p28 concentration–time plot in patients receiving increasing dose levels are presented as mean±s.e.m., and pharmacokinetics parameters of p28 in each patient were analysed by standard non-compartmental methods (Gorman et al, 2010; Jia et al, 2011). Survival of individual patients was measured from the date of first p28 treatment until death from any cause or last follow-up (Ramakrishna et al, 2010).
Results
Patient characteristics
Thirty-three patients (33) with advanced solid tumours with diverse diagnoses were screened, out of which 23 (70%) were positive for p53. Seventeen of the p53-positive patients were entered into the trial and two of these were withdrawn from the study due to inability to keep appointments, resulting in a total of fifteen patients who completed the trial. The other 16 screened patients were not eligible: 3 for concurrent brain metastases, 10 for testing p53-negative (i.e., <10% nuclear staining), and 3 for failing inclusion criteria.
The 15 patients with progressive, refractory, or recurrent solid tumours (7 melanoma, 4 colon cancer, and 1 of each of the following: soft tissue sarcoma; GIST, that is, gastrointestinal stromal cell tumour; prostate cancer; and pancreatic cancer) were enrolled between 27 April 2009 and 18 March 2011, at which time enrolment was closed.
All patients received at least one dose level of p28 i.v. as a short infusion 3 × per week for 4 weeks (followed by 2 weeks of rest) for a minimum total of 12 doses. Of the 15 patients, 8 (53%) received more than one dose level of therapy. Of those, three patients received two dose levels, three received three dose levels, and two received four dose levels, allowing assessment of long-term tolerability of p28.
All participants were followed for the duration of their participation in the study (minimum 6 weeks) until death (a range of 7–71 weeks). The data on the three still living patients is current as of 4 December 2012. Patient demographics and clinical characteristics are detailed in Table 1 and disease state and prior treatment in Table 2.
Table 1. Patient demographics and clinical characteristics.
| Demographic or clinical characteristic | No. of patients | Range | Median |
|---|---|---|---|
| Enrolled |
15 |
|
|
| Evaluable |
15 |
|
|
| Age, years |
|
47–80 |
62 |
| ECOG status |
|
0–2 |
1 |
|
Gender | |||
| Male | 11 | ||
| Female | 4 | ||
| Median number of prior anticancer therapy regimensa | 13 | 0–8 | 4 |
Abbreviation: ECOG=Eastern Cooperative Oncology Group.
Including immunotherapy.
Table 2. Patient disease and prior treatment.
| Survival in weeks | Prior treatment | p28 dose levels received | Disease at study entry |
|---|---|---|---|
| 158 (Alive) |
Refused |
2,3,4,5 |
Melanoma— left flank and lung nodules. Stage IV (M1b) |
| 140 (Alive) |
Allovectin-7; GP100 T-cell receptor clinical trial; IL-2; MART-I-F5 TCR clinical trial; RT to posterior neck and base of the occipital for loco-regional recurrence (4500 Gy, 250 Gy per session) |
3,4,5 |
Left post. Neck lesion; left post-nuchal lymphadenopathy. Stage III (unresectable) |
| 110 (Alive) |
Xeloda/Irinotecan; FOLFOX/Avastin; FOLFIRI/Avastin; Avastin/Erbitux; 5-FU/Leucovorin |
5 |
Multiple lung mets; retroperitoneal lymphadenopathy. Stage IV |
| 71 |
RT, external beam; Vectibix; Xeloda; Irinotecan; Irinotecan/Exurbia; FOLFIRI/Avastin; FOLFOX |
3,4,5 |
Bilateral lung mets; pelvic mass. Stage IV |
| 44 |
Refused |
4,5 |
Metastatic melanoma— lung. Stage IV |
| 32 |
DTIC |
2,3 |
Multiple dermal (subcutaneous) mets; left axillary LN's, liver lesion (right lobe). Stage IV |
| 32 |
High-dose IFN; DTIC, BCNU, Tamoxifen, IL-2; adjuvant biochemo; Biochemo |
2,3,4,5 |
Multiple liver mets, abdominal and sacral masses. Stage IV |
| 28 |
Nexavar; RT to sacrum; Lupron; Ketoconazole, OEstrogen patch, Leukine initiated; Docetaxel/Bevacizumab; Carboplatin/Etoposide |
4,5a |
Hepatic mets, multiple bone mets. Stage IV |
| 20 |
RT to right chest and axilla |
5 |
Multiple dermal/SC mets; liver and spleen. Stage IV |
| 17 |
Taxotere and Gemzar (2006); Taxotere/Gemzar (2008); RT pelvis |
1,2,3b |
Multiple lung mets, large abdominal mass and midline rectus muscle mass. Stage IV |
| 12 |
RT; 5-FU/Leucovorin; Erbitux; FOLFIRI/bevacizumab, CPT-11; S/P radiofrequency ablation; Rapamycin/grapefruit juice; FOLFOX/Bevacizumab |
1 |
Multiple lung mets. Stage IV |
| 12 |
Elesclomol clinical trial; DTIC; Taxol |
3 |
Multiple lung and liver mets; bone, abd/pelvis LN's. Stage IV |
| 10 |
Ifosfamide, Mesna, and Etoposide; S/P extensive resection; Gleevec; Vincristine/Adriamycin/Cytoxa; Sutent |
5 |
Leiomyosarcoma omentum and mesentery; GIST Stage IV unresectable |
| 7 |
Avastin; Vectibix; Rituxan; Oxaliplatin; Erbitux; CPT-11/5-FU/Leucovorin; CPT-11; Camptosar/5-FU/Leucovorin |
1 |
Multiple lung mets and large right hilar mass. Stage IV metastatic colon cancer |
| 7 | Gemcitabine and Tarceva; Xeloda/Oxaliplatin; Xeloda/Tarceva | 4 | Pulmonary, liver, and head of pancreas. Stage IV |
Abbreviations: 5-FU=fluorouracil; BCNU=Carmustine; CPT-11=Irinotecan; DTIC=Dacarbazine; FOLFIRI=FOL-folinic acid (leucovorin) and F-fluorouracil (5-FU) and IRI-irinotecan (Camptosar); FOLFOX=FOL-Folinic acid (leucovorin) and F-Fluorouracil (5-FU) and OX-Oxaliplatin (Eloxatin); IFN=interferon; RT=radiation therapy.
Completed 10 out of 12 doses.
Completed 9 out of 12 doses.
Dose-limiting toxicities, MTD and adverse events
The MTD was above the highest single (50 mg per kg total dose) and cumulative (140 mg per kg total dose; levels 2–5) doses studied. p28 did not elicit an antibody response in any patient during and following completion of the clinical trial (Figure 3).
Figure 3.

Lack of immunogenicity of p28 in patients. Serum samples taken after 12 injections of p28 at each treatment level were diluted 1 : 200 to 1 : 25 000 for each assay. O.D. at 450 nm was measured with substrate, PNPP. Values represent the mean±s.e.m. of each patient in three replicates. Pre-treatment control (black), Level 1 (white), Level 2 (red), Level 3 (blue), Level 4 (green), and Level 5 (orange). Standard curve for anti-p28 antibody at 1 : 70 000 dilution was generated by direct ELISA (inset). The 96-well plates were coated with p28 (0, 0.25, 2.5, 5.0, 10, 25, 50, 100, 250, 500, and 1000 ng per well).
There were no adverse laboratory events attributable to p28 by NCI Common Toxicity Criteria (Table 3). Consequently, the NOAEL was above the highest dose studied.
Table 3. Adverse events record during study.
| |
Grade |
Relationship to study drug |
|||||
|---|---|---|---|---|---|---|---|
| AE category | 1 | 2 | 3 | Probable | Possible | Unlikely | Unrelated |
| Pyrexia |
1 |
3 |
|
|
|
3 |
1 |
| Fatigue |
12 |
8 |
9 |
2 |
12 |
8 |
7 |
| Nausea and vomiting |
48 |
6 |
1 |
13 |
27 |
14 |
1 |
| Diarrhoea |
3 |
|
|
|
|
3 |
|
| Headache |
4 |
|
|
2 |
1 |
1 |
|
| Oedema |
3 |
3 |
|
|
|
5 |
1 |
| Cough; shortness of breath |
4 |
3 |
3 |
|
|
3 |
7 |
| Skin – injection site reaction |
5 |
|
|
|
3 |
2 |
|
| Anorexia |
3 |
6 |
2 |
|
3 |
6 |
2 |
| Myalgia |
2 |
|
|
|
1 |
1 |
|
| Constipation |
7 |
2 |
1 |
|
|
8 |
2 |
| Elevated alkaline phosphatase/bili |
|
1 |
1 |
|
|
|
2 |
| Alopecia |
1 |
|
|
|
|
1 |
|
| Ocular |
3 |
1 |
|
|
|
3 |
1 |
| Hyperkalemia |
1 |
|
|
|
|
1 |
|
| Hypertension – transient |
4 |
|
|
3 |
1 |
|
|
| Hypoalbuminemia |
1 |
5 |
|
|
2 |
2 |
2 |
| Hypoglycemia |
2 |
1 |
|
|
2 |
1 |
|
| Hypokalemia |
2 |
|
1 |
|
|
1 |
2 |
| Hyponatremia |
1 |
|
|
|
|
1 |
|
| Bacteremia |
|
|
1 |
|
|
1 |
|
| Coagulation – prolonged INR |
|
1 |
1 |
|
2 |
|
|
| Hypotension – transient |
|
1 |
2 |
|
|
1 |
2 |
| Other GI |
2 |
1 |
|
|
|
3 |
|
| Dizziness |
9 |
|
|
1 |
7 |
1 |
|
| Pain |
5 |
8 |
8 |
|
|
4 |
17 |
| Other metabolic |
2 |
|
1 |
|
|
2 |
1 |
| Urinary |
2 |
2 |
|
|
|
3 |
1 |
| Cardiac – transient |
|
1 |
|
|
|
|
1 |
| Leucopenia |
3 |
|
|
|
|
3 |
|
| Platelets |
6 |
|
|
|
|
6 |
|
| Vascular: deep vein thrombosis |
|
|
1 |
|
|
|
1 |
| Anaemia |
12 |
11 |
5 |
|
5 |
21 |
2 |
| Dehydration |
|
1 |
|
|
|
|
1 |
| Oral |
4 |
|
|
|
2 |
2 |
|
|
Totals |
152 |
65 |
37 |
21 |
68 |
111 |
54 |
| Percentage | 60% | 26% | 15% | 8% | 27% | 44% | 21% |
Abbreviations: AE=adverse event; GI=gastrointestinal; INR=international normalized ratio.
The AEs listed here are all those reported during all courses of treatment and for each visit. For this reason, the total number of AEs is much higher than the number of subjects (15) enrolled in the study. Very often, the higher frequency AEs represent recurring episodes in the same subject and reflect conditions already existing when the patient entered the study. The treating physician did not consider any of these adverse events directly related to p28 administration.
Antitumour response
All 15 patients were evaluable for survival and response to the target lesions during their participation in the trial and follow-up. Best objective responses as defined by RECIST 1.1 (Eisenhauer et al, 2009) of the target lesions during the course of the trial included one patient with a complete response, three patients with partial response, and seven patients (46%) with stable disease.
Figure 4A illustrates the best objective response of target lesions by patient during the entire trial period; waterfall plot analysis was used to illustrate response for each patient (Lara-Guerra et al, 2009; Faivre et al, 2011). Examples of target lesions in two living patients are shown in Figures 4B and C (CT scans of the head) and Figures 4D–F (complete regression of an intramuscular nodule before and after p28 treatment). Figure 4G shows a (pre-resection) post-study metastatic cancer recurrence.
Figure 4.

Tumour Response to p28. (A) Best response (%) of target lesions by patient. Before study entry, all patients underwent physical examination, laboratory measurements, and computed tomography scans as baseline. Tumour lesions were accurately measured in at least one dimension (longest diameter) with a minimum size of 10 mm by CT scan. Tumour response and progression were evaluated by Response Evaluation Criteria in Solid Tumours (RECIST 1.1). Complete Response (CR) was defined as disappearance of all target lesions. Partial response (PR) was defined as a 30% or above decrease in the sum of the longest diameters of target lesions, excluding complete disappearance of disease. Progressive disease (PD) was defined as a 20% or above increase in the sum of the longest diameters of target lesions. Stable disease (SD) was defined as small changes that did not meet the criteria for a PR or PD. M=melanoma, C=colon, P=prostate, S=sarcoma and Pa=pancreatic cancer. CR=green, PR=red, SD=blue, PD=black. *, Alive. All patients with tumour reduction evaluated as stable, PR and CR received either the highest or multiple levels of p28 (RECIST guideline, version 1.1; Eisenhauer et al, 2009). Values below each bar indicate the dose level (range) each patient received. #, progressive disease 28 December 2011, ‡, Cancer recurrence 23 July 2012. (B,C) Representative CT images of the head region (B) prior (baseline) and (C) post trial completion (dose level 3–5). (D–F) Another subject receiving dose levels 2–5 during the trial showed complete regression of an intramuscular nodule (D), red circle=target lesion; histological examinations of target lesion in left flank before p28 administration (E) and after receiving dose levels 2–5 during the trial (F). (G) An image of a polypoid metastatic lesion in the proximal ilium that recurred after completion of the study.
Survival
As of 4 December 2012, median overall survival was 28 weeks. With the exception of still living patients, patients exhibiting partial response during the trial survived for a range of 44–71 weeks, those exhibiting stable disease survived 7–32 weeks, and those exhibiting PD survived 12–32 weeks. Three patients (right three columns on Figure 5) showed best objective response of stable disease according to RECIST 1.1 guidelines during the trial period but survived only 7–10 weeks. Although a goal of the inclusion criteria was to select patients with anticipated 180 days survival, retrospective reviews of large phase I groups have demonstrated that up to 20% of patients die within the first 90 days of oncology phase I trials (Arkenau et al, 2008; Penel et al, 2010; Chau et al, 2011), as was the case with this trial.
Figure 5.

Patient overall survival as of 4 December 2012. Survival of each patient was measured from the date the first dose of p28 was administered until death from any cause or last follow-up. M=melanoma, C=colon, P=prostate, S=sarcoma and Pa=pancreatic cancer. *Alive (green bars). CR=complete response, PR=partial response, SD=stable and PD=progressive disease. Median overall survival: 28 weeks. Range alive: 110–158 weeks, median 140 weeks. Range of date of death (DOD) for deceased patients: 7–71 weeks, Median 19 weeks. Values within each bar represent the dose level (range) each patient received. #, progressive disease 28 December 2011. ‡, Cancer recurrence 23 July 2012.
Although overall survival was not one of the parameters identified as a goal of this trial, nevertheless it is of note that 3 of 15 patients (20%) of the patients are still living since the completion of the trial on 8 March 2011. One of the three (a melanoma patient) has been living for 158 weeks (3 years) after receiving dose levels 2–5 during the trial, without any additional melanoma related treatment, and remained in complete response for 139 weeks until 23 July 2012 when cancer in the proximal ilium (a polypoid metastatic lesion, the form most common in the GI tract)(Dasgupta and Brasfield, 1964) recurred, which was then resected. One other metastatic melanoma patient has survived and been living with relatively stable disease for 140 weeks (2.7 years). A patient with metastatic colon cancer has been living for 110 weeks (2.1 years) and had stable disease for 61 weeks until disease progressed on 28 December 2011. Survival for each patient with number of dose regimens they have received is shown in Figure 5.
Pharmacokinetics
Plasma samples were collected from all patients, but only 14 patients had adequate data for pharmacokinetic modelling at all dose levels (e.g., the level of p28 from one patient at dose level 1 was under the detection limit for the assay). At least 3 patients were sampled at each remaining dose level. Figure 6 represents the plasma concentration–time plot for p28 during day one of each dose level. In Table 4, the pharmacokinetic parameters in relation to p28 dosage are summarised. Maximum concentration, AUClast (h μg per ml) and t1/2γ (h) increased with the actual p28 dosage administered over the dosage range studied. The mean p28 half-life (t1/2), was 0.09 h (range 0.054–0.107 h). However, plasma clearance was maximal (1294 mg kg−1 per hour; range 1126–2294 mg kg−1 per hour) at dose level 3 as was the volume of distribution (529 ml kg−1; range 366–874 ml kg−1). No accumulation or alteration in distribution was observed in patients with repeat studies, which was expected given the short half-life and rapid plasma clearance of p28.
Figure 6.

Concentration profiles of p28 after i.v infusion. Plot of p28 concentration–time in patients receiving increasing dose levels. Serum samples were taken from patients at 0, 5, 10, 20, 30, 60, and 120 min after p28 i.v. infusion. p28 concentration in the serum was determined by LC/MS/MS assay.
Table 4. Pharmacokinetic parameters for p28 on phase I.
| Level 1 | Level 2 | Level 3 | Level 4 | Level 5 | |
|---|---|---|---|---|---|
| n=2 | n=4 | n=6 | n=6 | n=7 | |
| Dose (mg kg−1) |
0.83 |
1.66 |
2.50 |
3.33 |
4.16 |
| Dose escalation (%) |
— |
100 |
51 |
33 |
25 |
|
Cmax (μg ml−1) |
1.01 |
6.99 |
7.68 |
9.8 |
13.7 |
|
Tmax (h) |
0.17 |
0.08 |
0.08 |
0.08 |
0.08 |
|
t1/2 (h) |
0.144 |
0.054 |
0.102 |
0.107 |
0.105 |
|
t1/2α (h) |
0.144 |
0.014 |
0.019 |
0.014 |
0.010 |
|
t1/2β (h) |
NA |
0.089 |
0.053 |
0.048 |
0.035 |
|
t1/2γ (h) |
NA |
NA |
1.61 |
2.16 |
4.12 |
| AUClast (h μg per ml) |
0.24 |
1.32 |
1.93 |
2.83 |
3.70 |
| Cl (ml kg−1 per hour) |
3430 |
1259 |
1294 |
1176 |
1124 |
| Vdss (ml kg−1) | 547 | 270 | 529 | 468 | 431 |
Abbreviation: n=number of patients; NA, not available.
Pharmacokinetics parameters (Cmax=p28 maximum concentration in serum, Tmax=time to Cmax, t1/2=terminal half-life of p28, t1/2α=rapid distribution half-life, t1/2β=slow distribution half-life, t1/2γ=elimination half-life, AUClast=area-under curve, Cl=total clearance and Vdss=volume distribution at steady state) were calculated from the p28 concentrations in serum vs post-injection time. The concentration at 0 min is defined as 0 ng ml−1 Bold values indicate minimally effective dose.
Discussion
The accelerated titration study design described here is somewhat different than the traditional phase I trial design, wherein 3–6 patients are usually enrolled in each dose level. In our design, this feature is preserved—there are three new patients at each dose level. But to maximise each patient's chance to be treated at a potentially active dose, the accelerated titration design allows intra-patient dose escalation for a patient who remains on study and has no evidence of toxicity at the current dose (Simon et al, 1997; Vickers et al, 2006; Vickers, 2006; Anderson et al, 2007). At each dose level, in addition to the new patients, all continuing patients who survived the previous level are also treated.
Reports of regression of human tumours infected with microbial pathogens dates back more than 100 years (Coley, 1891, 1911). Since then, considerable effort has been spent in developing wild type or attenuated bacterial strains for the treatment of various types of cancers (Paglia and Guzman, 1998; Alexandroff et al, 1999; da Rocha et al, 2001). The results of these studies have been unsatisfactory given not only the significant toxicity and immune response but also a lack of sustained efficacy. In contrast, redox copper-containing protein azurin, a secretory product obtained from the opportunistic pathogen Pseudomonas aeruginosa, appears to be non-toxic and has shown sustainable anticancer effect (Yamada et al, 2002; Punj et al, 2004).
p28 (NSC745104) is a novel, synthetic 28-amino-acid peptide fragment of the cupredoxin azurin. p28 exhibits antiproliferative activity in a particular phase of the cell cycle, G2–M. This finding is important because a large number of chemotherapeutic drugs work only on cells that are actively proliferating, but not cells that are in the resting phase, G0. In mammalian cells, the tumour suppressor protein, p53 has long been implicated as significant in a cell's ability to either arrest cell cycle progression or to invoke apoptotic cell death. p28 binds to the DNA-binding domain of p53 and inhibits proteasomal degradation via an HDM2-independent pathway, inducing a post-translational increase in wild type and mutant p53 as well as its DNA-binding activity and elevating the cyclin-dependent kinase inhibitors p21 and p27. These proteins, in turn, reduce the level of CDK2, cyclin A1, and FOXM1 and induce cell cycle arrest at G2–M phase (Yamada et al, 2009). Thus, it stands to reason that continued prolonged use of p28 could be a model non-toxic therapeutic option for ultimate shrinkage of advanced/recurrent solid tumours resulting in long-term progression-free survival.
This first-in-human study of p28, a novel chemically synthesised 28-amino-acid fragment of the cupredoxin azurin, showed that this agent had evidence of antitumour activity and minimal toxicity. p28 is well tolerated in patients with a variety of advanced solid tumours over multiple cycles of treatment, with increasing doses. Neither an NOAEL or MTD was established, as no significant adverse effects nor toxicity was observed over the course of and following completion of the study. p28 did not elicit an immune response in any patient. There was no clear relationship between dose and any episode of transient nausea. Similar episodes of transient nausea that were unrelated to dose were occasionally seen in Cynomolgous sp. monkeys (Jia et al, 2011), suggesting a long-term tolerability for p28.
The pharmacokinetic behaviour of the tested formulation of p28 is characterised by rapid tissue uptake after i.v. administration, an increased terminal phase half-life, and low interpatient (dose level) variability. The distribution of p28 in the circulation following i.v. administration was rapid with a time to maximum concentration<10 min and did not increase with dosage above 10 mg kg−1 total dose. Systemic exposure increased with dosage, with an essentially linear relationship in Cmax, t1/2γ and AUC and actual dosage above 10 mg kg−1 total dose. However, we observed that clearance (ml kg−1 per hour) as well as Vdss (ml kg−1) between 20 and 50 mg kg−1 appeared maximal at 30 mg kg−1 total dose, suggesting that this was likely to be a minimally effective dose, particularly if administered over multiple courses (Figure 5, Table 4). This suggests the recommended dose for phase II clinical trials be set at 30 mg kg−1 (total dose), which is well within the range to elicit antitumour activity in vitro and in vivo (Jia et al, 2011; Mehta et al, 2011).
Early-phase clinical trials of noncytotoxic (e.g., antiangiogenic) agents in patients with advanced solid tumours can be complicated by several issues. Conventional end points in phase I trials are defined by predetermined toxicity criteria to define an MTD or, more recently, by biological end points to define a biologically effective dose. Single agents with little acute toxicity, administered sub-chronically, may be effective at doses well below the MTD or NOAEL and dose escalations to a MTD may be unnecessary. However, the definition of a biologically effective dose can be difficult if there is a lack of validated biological surrogate markers. This may be further complicated if there is a lack of understanding of its antitumour mechanisms, although this is not the case with p28 as its antitumour mechanisms are well documented (Olivier et al, 2002; Taylor et al, 2009; Yamada et al, 2009; Bizzarri et al, 2011).
There is preliminary evidence of activity of p28 in the patient population in this study, particularly in patients with melanoma or colon cancer, who had a 54-week overall survival that reflects preclinical efficacy data (Andreeff et al, 2010; Bizzarri et al, 2011; Jia et al, 2011). Although this could be attributed to the inconsistent growth rates of these tumours, all patients had recurrent, refractory, or progressive disease at study entry, and it suggests that p28 is comparable or improves upon results from additional early studies of agents designed to post-translationally increase the level of p53 in patients with advanced solid or haematological malignancies (Tabernero et al, 2009; Andreeff et al, 2010).
In addition, there is a clear suggestion that this antitumour activity is not only dose but duration related given that, in general, patients responded to single higher doses as well as cumulative doses of p28 across a wide series of tumour types (Figure 4). After entry, p28 binds to the p53 DNA-binding domain, leading to an increase in intracellular levels of wild-type and mutant p53 as well as its DNA-binding activity and elevating the cyclin-dependent kinase inhibitors, p21 and p27, inducing cell cycle arrest at G2/M (Yamada et al, 2009; Bizzarri et al, 2011). As p28 increases the DNA-binding activity as well as the level of mutated p53 (generally overexpressed) (Yamada et al, 2009; Bizzarri et al, 2011), there was no apparent relationship between the level of p53 expression and patient response or survival (Figure 7).
Figure 7.

Relationship between p53 status and patient response and survival. Complete response=green, partial response=red, stable disease=blue and progressive disease=black. *, Alive. Tissue sections representative of each patient's primary tumour were analysed for p53 status by IHC. #, Progressive disease 28 December 2011. ‡, Cancer recurrence 23 July 2012.
In summary, p28 is well tolerated in patients with recurrent, refractory, and progressive solid tumours at cumulative doses up to 140 mg kg−1 over a 48-week period with no immunogenicity or significant adverse effects that led to defining a NOAEL or MTD. Based on RECIST 1.1 criteria, p28 also appeared to have activity in refractory patients as well as two metastatic melanoma patients who had refused prior treatment. Additionally, pharmacokinetics data suggest that in adults a dose of 30 mg kg−1 can be administered for a prolonged period without toxicity and possibly with considerable clinical efficacy. An efficacy trial with long-term administration of p28 in advanced solid tumours is being planned.
Acknowledgments
This work was supported through a clinical trials agreement between CDG Therapeutics, Inc., and the University of Illinois at Chicago (UIC). This work was also supported by the NCI RAID program.
All terms of the research agreement between CDG Therapeutics, Inc., and UIC are managed by UIC in accordance with its conflict of interest management policies.
Footnotes
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Portions of the preliminary data in this article were presented in part at the 47th Annual American Society of Clinical Oncology Meeting, 3–7 June 2011, Chicago, IL, USA.
References
- Alexandroff AB, Jackson AM, O'Donnell MA, James K. BCG immunotherapy of bladder cancer: 20 years on. Lancet. 1999;353 (9165:1689–1694. doi: 10.1016/S0140-6736(98)07422-4. [DOI] [PubMed] [Google Scholar]
- Anderson KC, Hannah AL, Pazdur R, Farrell AT. A strategic framework for novel drug development in multiple myeloma. Br J Haematol. 2007;138 (2:153–159. doi: 10.1111/j.1365-2141.2007.06641.x. [DOI] [PubMed] [Google Scholar]
- Andreeff M, Kojima K, Padmanabhan S, Strair R, Kirschbaum M, Maslak P, Hillmen P, O'Brien S, Samaniego F, Borthakur G, Konopleva M, Vassilev L, Nichols G. A multi-center, open-label, phase i study of single agent RG7112, A first in class p53-MDM2 antagonist, in patients with relapsed/refractory acute myeloid and lymphoid leukemias (AML/ALL) and refractory chronic lymphocytic leukemia/small cell lymphocytic lymphomas (CLL/SCLL) Blood (ASH Annual Meeting Abstracts) 2010;116:657. [Google Scholar]
- Arkenau HT, Olmos D, Ang JE, Barriuso J, Karavasilis V, Ashley S, de Bono J, Judson I, Kaye S. 90-Days mortality rate in patients treated within the context of a phase-I trial: how should we identify patients who should not go on trial. Eur J Cancer. 2008;44 (11:1536–1540. doi: 10.1016/j.ejca.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Bizzarri AR, Santini S, Coppari E, Bucciantini M, Di Agostino S, Yamada T, Beattie CW, Cannistraro S. Interaction of an anticancer peptide fragment of azurin with p53 and its isolated domains studied by atomic force spectroscopy. Int J Nanomed. 2011;6:3011–3019. doi: 10.2147/IJN.S26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer. 2001;1 (1:68–76. doi: 10.1038/35094077. [DOI] [PubMed] [Google Scholar]
- Chau NG, Florescu A, Chan KK, Wang L, Chen EX, Bedard P, Oza AM, Siu LL. Early mortality and overall survival in oncology phase I trial participants: can we improve patient selection. BMC Cancer. 2011;11:426. doi: 10.1186/1471-2407-11-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari A, Mahfouz M, Fialho AM, Yamada T, Granja AT, Zhu Y, Hashimoto W, Schlarb-Ridley B, Cho W, Das Gupta TK, Chakrabarty AM. Cupredoxin-cancer interrelationship: azurin binding with EphB2, interference in EphB2 tyrosine phosphorylation, and inhibition of cancer growth. Biochemistry. 2007;46 (7:1799–1810. doi: 10.1021/bi061661x. [DOI] [PubMed] [Google Scholar]
- Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. Clin Orthop Relat Res. 1891;262:3–12. [PubMed] [Google Scholar]
- Coley WB. Surg Gynec Obstet. 1911. pp. 174–190.
- da Rocha AB, Lopes RM, Schwartsmann G. Natural products in anticancer therapy. Curr Opin Pharmacol. 2001;1 (4:364–369. doi: 10.1016/s1471-4892(01)00063-7. [DOI] [PubMed] [Google Scholar]
- Dasgupta TK, Brasfield RD. Metastatic melanoma of the gastrointestinal tract. Arch Surg. 1964;88:969–973. doi: 10.1001/archsurg.1964.01310240065013. [DOI] [PubMed] [Google Scholar]
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45 (2:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Faivre S, Zappa M, Vilgrain V, Boucher E, Douillard JY, Lim HY, Kim JS, Im SA, Kang YK, Bouattour M, Dokmak S, Dreyer C, Sablin MP, Serrate C, Cheng AL, Lanzalone S, Lin X, Lechuga MJ, Raymond E. Changes in tumor density in patients with advanced hepatocellular carcinoma treated with sunitinib. Clin Cancer Res. 2011;17 (13:4504–4512. doi: 10.1158/1078-0432.CCR-10-1708. [DOI] [PubMed] [Google Scholar]
- Gorman GS, Coward LU, Freeman L, Noker PE, Beattie CW, Jia L. A novel and rapid LC/MS/MS assay for bioanalysis of Azurin p28 in serum and its pharmacokinetics in mice. J Pharm Biomed Anal. 2010;53 (4:991–996. doi: 10.1016/j.jpba.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka Y, Granja AT, Fialho AM, Schlarb-Ridley BG, Das Gupta TK, Chakrabarty AM, Yamada T. Human cytochrome c enters murine J774 cells and causes G1 and G2/M cell cycle arrest and induction of apoptosis. Biochem Biophys Res Commun. 2005;338 (2:1284–1290. doi: 10.1016/j.bbrc.2005.10.078. [DOI] [PubMed] [Google Scholar]
- Jia L, Gorman GS, Coward LU, Noker PE, McCormick D, Horn TL, Harder JB, Muzzio M, Prabhakar B, Ganesh B, Das Gupta TK, Beattie CW. Preclinical pharmacokinetics, metabolism, and toxicity of azurin-p28 (NSC745104) a peptide inhibitor of p53 ubiquitination. Cancer Chemother Pharmacol. 2011;68 (2:513–524. doi: 10.1007/s00280-010-1518-3. [DOI] [PubMed] [Google Scholar]
- Lara-Guerra H, Waddell TK, Salvarrey MA, Joshua AM, Chung CT, Paul N, Boerner S, Sakurada A, Ludkovski O, Ma C, Squire J, Liu G, Shepherd FA, Tsao MS, Leighl NB. Phase II study of preoperative gefitinib in clinical stage I non-small-cell lung cancer. J Clin Oncol. 2009;27 (36:6229–6236. doi: 10.1200/JCO.2009.22.3370. [DOI] [PubMed] [Google Scholar]
- Martin AC, Facchiano AM, Cuff AL, Hernandez-Boussard T, Olivier M, Hainaut P, Thornton JM. Integrating mutation data and structural analysis of the TP53 tumor-suppressor protein. Hum Mutat. 2002;19 (2:149–164. doi: 10.1002/humu.10032. [DOI] [PubMed] [Google Scholar]
- Mehta RR, Yamada T, Taylor BN, Christov K, King ML, Majumdar D, Lekmine F, Tiruppathi C, Shilkaitis A, Bratescu L, Green A, Beattie CW, Das Gupta TK. A cell penetrating peptide derived from azurin inhibits angiogenesis and tumor growth by inhibiting phosphorylation of VEGFR-2, FAK and Akt. Angiogenesis. 2011;14 (3:355–369. doi: 10.1007/s10456-011-9220-6. [DOI] [PubMed] [Google Scholar]
- Miao XD, Ye ZM, Yang DS, Xu RZ, Li WX, Tao HM. [Cytotoxicity and apoptosis of human osteosarcoma U2OS cells induced by recombinant soluble AZURIN] Zhejiang Da Xue Xue Bao Yi Xue Ban. 2005;34 (5:384–389. doi: 10.3785/j.issn.1008-9292.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Midgley CA, Lane DP. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene. 1997;15 (10:1179–1189. doi: 10.1038/sj.onc.1201459. [DOI] [PubMed] [Google Scholar]
- Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19 (6:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- Paglia P, Guzman CA. Keeping the immune system alerted against cancer. Cancer Immunol Immunother. 1998;46 (2:88–92. doi: 10.1007/s002620050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penel N, Delord JP, Bonneterre ME, Bachelot T, Ray-Coquard I, Blay JY, Pascal LB, Borel C, Filleron T, Adenis A, Bonneterre J. Development and validation of a model that predicts early death among cancer patients participating in phase I clinical trials investigating cytotoxics. Invest New Drugs. 2010;28 (1:76–82. doi: 10.1007/s10637-009-9224-x. [DOI] [PubMed] [Google Scholar]
- Punj V, Bhattacharyya S, Saint-Dic D, Vasu C, Cunningham EA, Graves J, Yamada T, Constantinou AI, Christov K, White B, Li G, Majumdar D, Chakrabarty AM, Das Gupta TK. Bacterial cupredoxin azurin as an inducer of apoptosis and regression in human breast cancer. Oncogene. 2004;23 (13:2367–2378. doi: 10.1038/sj.onc.1207376. [DOI] [PubMed] [Google Scholar]
- Ramakrishna R, Barber J, Kennedy G, Rizvi A, Goodkin R, Winn RH, Ojemann GA, Berger MS, Spence AM, Rostomily RC. Imaging features of invasion and preoperative and postoperative tumor burden in previously untreated glioblastoma: Correlation with survival. Surgical neurology international. 2010;1 (pii:40. doi: 10.4103/2152-7806.68337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Freidlin B, Rubinstein L, Arbuck SG, Collins J, Christian MC. Accelerated titration designs for phase I clinical trials in oncology. J Natl Cancer Inst. 1997;89 (15:1138–1147. doi: 10.1093/jnci/89.15.1138. [DOI] [PubMed] [Google Scholar]
- Tabernero J, Dirix L, Schoffski P, Cervantes A, Capdevila J, Baselga J, van Beijsterveldt L, Winkler H, Kraljevic S, Zhuang SH. Phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of HDM-2 antagonist JNJ-26854165 in patients with advanced refractory solid tumors. J Clin Oncol. 2009;27:15. [Google Scholar]
- Tan YH, Chen YM, Ye X, Lu Q, Tretyachenko-Ladokhina V, Yang W, Senear DF, Luo R. Molecular mechanisms of functional rescue mediated by P53 tumor suppressor mutations. Biophys Chem. 2009;145 (1:37–44. doi: 10.1016/j.bpc.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BN, Mehta RR, Yamada T, Lekmine F, Christov K, Chakrabarty AM, Green A, Bratescu L, Shilkaitis A, Beattie CW, Das Gupta TK. Noncationic peptides obtained from azurin preferentially enter cancer cells. Cancer Res. 2009;69 (2:537–546. doi: 10.1158/0008-5472.CAN-08-2932. [DOI] [PubMed] [Google Scholar]
- Trotti A, Colevas AD, Setser A, Rusch V, Jaques D, Budach V, Langer C, Murphy B, Cumberlin R, Coleman CN, Rubin P. CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol. 2003;13 (3:176–181. doi: 10.1016/S1053-4296(03)00031-6. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303 (5659:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Vickers AJ. How to design a phase I trial of an anticancer botanical. J Soc Integr Oncol. 2006;4 (1:46–51. [PMC free article] [PubMed] [Google Scholar]
- Vickers AJ, Kuo J, Cassileth BR. Unconventional anticancer agents: a systematic review of clinical trials. J Clin Oncol. 2006;24 (1:136–140. doi: 10.1200/JCO.2005.03.8406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Goto M, Punj V, Zaborina O, Chen ML, Kimbara K, Majumdar D, Cunningham E, Das Gupta TK, Chakrabarty AM. Bacterial redox protein azurin, tumor suppressor protein p53, and regression of cancer. Proc Natl Acad Sci USA. 2002;99 (22:14098–14103. doi: 10.1073/pnas.222539699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Mehta RR, Lekmine F, Christov K, King ML, Majumdar D, Shilkaitis A, Green A, Bratescu L, Beattie CW, Das Gupta TK. A peptide fragment of azurin induces a p53-mediated cell cycle arrest in human breast cancer cells. Mol Cancer Ther. 2009;8 (10:2947–2958. doi: 10.1158/1535-7163.MCT-09-0444. [DOI] [PubMed] [Google Scholar]
- Yang DS, Miao XD, Ye ZM, Feng J, Xu RZ, Huang X, Ge FF. Bacterial redox protein azurin induce apoptosis in human osteosarcoma U2OS cells. Pharmacol Res. 2005;52 (5:413–421. doi: 10.1016/j.phrs.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Ye ZM, Miao XD, Yang DS, Xu RZ, Huang X, Ge FF. [Selective inducement effect of bacterial redox protein azurin on apoptosis of human osteosarcoma cell line U2OS] Ai zheng. 2005;24 (3:298–304. [PubMed] [Google Scholar]
- Zhang H. Evaluation of four antibodies in detecting p53 protein for predicting clinicopathological and prognostic significance in colorectal adenocarcinoma. Clin Cancer Res. 1999;5 (12:4126–4132. [PubMed] [Google Scholar]