

Abstract
Aims: Chronic hypoxia (CH) enhances depolarization-induced myofilament Ca2+ sensitization and resultant pulmonary arterial constriction through superoxide (O2−)-dependent stimulation of RhoA. Because NAD(P)H oxidase (NOX) has been implicated in the development of pulmonary hypertension, we hypothesized that vascular smooth muscle (VSM) depolarization increases NOX-derived O2− production leading to myofilament Ca2+ sensitization and augmented vasoconstrictor reactivity following CH. As epidermal growth factor receptor (EGFR) mediates Rac1-dependent NOX activation in renal mesangial cells, we further sought to examine the role EGFR plays in this response. Results: Vasoconstrictor responses to depolarizing concentrations of KCl were greater in lungs isolated from CH (4 wk, 0.5 atm) rats compared to normoxic controls, and this effect of CH was abolished by the general NOX inhibitor, apocynin. CH similarly augmented KCl-induced vasoconstriction and O2− generation (assessed using the fluorescent indicator, dihydroethidium) in Ca2+-permeabilized, pressurized small pulmonary arteries. These latter responses to CH were prevented by general inhibition of NOX isoforms (apocynin, diphenylene iodonium), and by selective inhibition of NOX 2 (gp91ds-tat), Rac1 (NSC 23766), and EGFR (AG 1478). Consistent with these observations, CH increased KCl-induced EGFR phosphorylation, and augmented depolarization-induced Rac1 activation in an EGFR-dependent manner. Innovation: This study establishes a novel signaling axis in VSM linking membrane depolarization to contraction that is independent of Ca2+ influx, and which mediates myofilament Ca2+ sensitization in the hypertensive pulmonary circulation. Conclusion: CH augments membrane depolarization-induced pulmonary VSM Ca2+ sensitization and vasoconstriction through EGFR-dependent stimulation of Rac1 and NOX 2. Antioxid. Redox Signal. 18, 1777–1788.
Introduction
Endogenous reactive oxygen species (ROS) are physiologically important intracellular second-messenger molecules that regulate vascular smooth muscle (VSM) phenotype (62) and contractility (4) in the normal pulmonary circulation. However, excessive ROS production from various enzymatic sources is considered to be a major contributing factor to both arterial remodeling (27, 36) and vasoconstrictor (21, 30, 36) components of chronic hypoxia (CH)-induced pulmonary hypertension (PH). Interestingly, recent evidence supports an important contribution of superoxide anion (O2-)-dependent RhoA activation to enhanced membrane depolarization-induced myofilament Ca2+ sensitization in hypertensive pulmonary arteries from CH rats (7). Although the signaling mechanism that links depolarization to RhoA-mediated VSM Ca2+ sensitization in this setting is unknown, evidence that depolarization stimulates NAD(P)H oxidase (NOX) to produce O2- in macula densa (38) and endothelial cells (10; 44; 60) suggests a potential role for NOX in this response.
Innovation.
This study establishes a novel signaling axis in VSM linking membrane depolarization to contraction that is independent of Ca2+ influx, and which mediates enhanced myofilament Ca2+ sensitization in the hypertensive pulmonary circulation. The concept of depolarization as a Ca2+-independent effector of EGFR-Rac1-NOX 2-RhoA signaling has potentially broad implications for understanding not only mechanisms of pulmonary vasoconstriction, but also for depolarization and oxidant regulation of cytoskeletal dynamics, motility, proliferation, apoptosis, and myogenicity in other cell systems.
NOX isoforms are multi-subunit enzymes found in the plasma membrane and on endosomes and have been implicated in the development of PH (27, 36, 46). NOX subtypes 1, 2, and 4, are the most abundant forms in VSM (41). The catalytic subunits of NOX 1 and 2 are activated by phosphorylation of the cytosolic subunits NOXO1 and NOXA1 in the case of NOX 1, and p47phox and p67phox in the case of NOX 2 (6, 12, 41). The small GTP-binding protein, Rac1, is also a critical signaling mediator of both NOX 1 and 2 activation (6, 12).
A potential upstream activator of NOX and Rac1 is the epidermal growth factor receptor (EGFR) (68), which transitions from an inactive monomeric form to an active homodimeric form upon phosphorylation of multiple tyrosine residues. EGFR has previously been implicated in the development of PH in rats (14, 45), and mediates PH in mice that overexpress the EGFR ligand, transforming growth factor alpha (33). Interestingly, depolarization can activate EGFR in both PC12 cells and cardiomyocytes (17, 63, 69). Furthermore, EGFR stimulation leads to Rac1 and NOX activation in glomerular mesangial cells (68), as well as RhoA activation in renal tubule epithelial cells (31). We therefore hypothesized that membrane depolarization increases NOX derived O2− following CH though activation of EGFR. We tested our hypothesis by assessing the roles of NOX, Rac-1, and EGFR in membrane depolarization-dependent vasoconstriction and O2- production in isolated small pulmonary arteries from CH and normoxic control rats. We also examined the contribution of NOX to depolarization-induced vasoconstriction in isolated lungs. Our findings reveal a unique role for VSM membrane depolarization to activate NOX 2 through EGFR-dependent stimulation of Rac1 in pulmonary arteries from CH rats, but not in those of control animals. Notably, this response represents a novel mechanism of depolarization-induced vasoconstriction occurring independently of changes in global intracellular Ca2+ concentration ([Ca2+]i).
Results
CH resulted in polycythemia, as indicated by a greater hematocrit (64±1%) in CH rats than in controls (47±1%). We have previously demonstrated that this 4 wk CH exposure protocol additionally results in PH, arterial remodeling, and right ventricular hypertrophy (3, 55, 56, 59).
CH augments depolarization-induced vasoconstriction but inhibits the Ca2+ response to KCl: Role of L-type Ca2+ channels
We have previously reported that CH augments vasoconstrictor reactivity to depolarizing concentrations of KCl in isolated lungs (7). To evaluate effects of CH on pulmonary arterial VSM reactivity and vessel wall [Ca2+]i responses to KCl, and to determine the contribution of L-type voltage-gated Ca2+ channels to this response, we performed concentration–response curves to KCl in endothelium-disrupted, pressurized pulmonary arteries [approximately 150–200 μm inner diameter (ID)] from CH and control rats. Arteries were loaded with the ratiometric Ca2+ indicator fura 2-AM, and superfused with physiological saline solution (PSS) (pH, Po2 and Pco2 of 7.39±0.02, 59.6±1.3, and 32.9±0.9, respectively). KCl-induced constriction was greater in CH compared to control arteries. Whereas KCl produced concentration-dependent increases in [Ca2+]i in control arteries, Ca2+ responses were largely attenuated following CH, with no significant changes occurring over the 30–60 mM range of KCl (Fig. 1A and B). Basal [Ca2+]i was not significantly different between vessels from control and CH rats. Background autofluorescence also was not significantly different between arteries from control [fluorescence emission at 340 nm excitation (F340)=233±16, fluorescence emission at 380 nm excitation (F380)=231±17; n=4) and CH (F340=268±14, F380=265±9; n=4) rats, and was approximately 7—8-fold less than resting emissions following fura 2 loading. Diltiazem, an L-type Ca2+ channel inhibitor, significantly lowered basal fura 2 emission ratios and diminished both vasoconstrictor and Ca2+ responses to KCl in each group. However, vasoconstrictor reactivity remained elevated in CH arteries compared to controls following L-type Ca2+ channel blockade, despite similar levels of [Ca2+]i between groups.
FIG. 1.

CH increases vasoconstriction to depolarizing concentrations of KCl in pressurized pulmonary arteries, but attenuates L-type Ca2+ channel-mediated increases in vessel wall [Ca2+]i. KCl-induced (30–120 mM) (A) vasoconstriction and (B) vessel wall [Ca2+]i [expressed as fura 2 340/380 nm emission ratios (F340/F380)] responses in isolated, pressurized (12 Torr), endothelium-disrupted CH and control pulmonary arteries in the presence or absence of the L-type Ca2+channel inhibitor diltiazem (50 μM). Values are means±SE of n=6–7 rats/group; *p<0.05 vs. respective control; #p<0.05 vs. respective vehicle.
O2−, but not H2O2, mediates CH-dependent increases in vasoconstrictor reactivity and VSM Ca2+ sensitization in response to KCl
Previous work from our laboratory has demonstrated that the O2− spin trap agent, tiron, prevents enhanced vasoconstriction to KCl following CH exposure (7). However, by scavenging O2−, tiron should also decrease H2O2 levels. Therefore, we sought to determine the relative contributions of O2− and H2O2 to enhanced depolarization-induced Ca2+ sensitization following CH. Endothelium-disrupted arteries were prepared as above and permeabilized to Ca2+ with ionomycin to clamp vessel wall and presumably VSM [Ca2+]i, allowing assessment of vasoconstriction independent of changes in [Ca2+]i as described previously (7, 53). Ca2+-clamp conditions were confirmed using fura-2. There were no differences in basal ID (control=162±4, CH=169±7 μm) or [Ca2+]i (control=1.23±0.05, CH=1.17±0.05; F340/F380) between groups. Experiments were conducted in the presence of the O2− scavenger, polyethylene glycol (PEG)-superoxide dismutase (SOD), or the H2O2 scavenger, PEG-catalase. Similar to tiron (7), PEG-SOD attenuated reactivity to KCl in pulmonary arteries from rats exposed to CH, while having no effect in control vessels (Fig. 2A). In contrast, PEG-catalase did not alter vasoconstriction to KCl in either group (Fig. 2B).
FIG. 2.

O2−, but not H2O2, mediates enhanced depolarization-induced myofilament Ca2+ sensitization and constriction in Ca2+-permeabilized arteries from CH rats. Vasoconstrictor responses to KCl in pressurized, endothelium-disrupted, Ca2+ permeabilized (3 μM ionomycin) pulmonary arteries from CH and control rats in the presence of (A) PEG-SOD (120 U/mL) or (B) PEG-catalase (250 U/mL). Values are means±SE of n=4–5 rats/group; *p<0.05 vs. control; #p<0.05 CH SOD vs. CH vehicle; τp<0.05 CH catalase vs. CH SOD.
NOX is required for enhanced depolarization-induced vasoconstriction and myofilament Ca2+ sensitization following CH
The role of NOX, as a source of O2−, in mediating enhanced membrane depolarization-dependent vasoconstriction following CH was tested in both nonpermeabilized pulmonary arteries and isolated, saline-perfused lungs using the NOX inhibitor apocynin. Apocynin decreased reactivity to KCl in CH arteries, while having no effect in control arteries (Fig. 3A).
FIG. 3.

Augmented KCl-induced vasoconstriction following CH is NOX-dependent in both nonpermeabilized arteries and isolated perfused rat lungs. (A) Vasoconstrictor responses to KCl in pressurized, endothelium-disrupted pulmonary arteries from CH and control rats in the presence or the NOX inhibitor apocynin (30 μM) or vehicle. (B) Vasoconstrictor responses to KCl (15–60 mM) in the presence of vehicle or apocynin (30 μM) in isolated, saline-perfused lungs from CH and control rats. Values are means±SE of n=5–7 rats/group; *p<0.05 vs. respective control; #p<0.05 CH apocynin vs. CH vehicle.
Consistent with responses in isolated arteries, vasoconstrictor responses to depolarizing concentrations of KCl were greater in isolated lungs from CH rats than from control lungs (Fig. 3B), as previously reported (7). Apocynin decreased vasoreactivity to KCl in CH lungs, while having no effect in those of control rats. Perfusate Po2, Pco2, and pH were not different between groups and treatments (data not shown).
To address more directly the role of NOX in enhanced depolarization-dependent VSM Ca2+ sensitization following CH, we further examined effects of NOX inhibitors on KCl-dependent vasoconstriction in Ca2+-permeabilized arteries. Similar to effects of PEG-SOD (Fig. 2A), the nonselective NOX inhibitors apocynin (Fig. 4A) and diphenyleneiodonium (DPI) (Fig. 4B) attenuated KCl-dependent vasoconstriction in arteries from CH, but not control rats, and normalized responses between groups. Similar inhibitory effects were observed using the specific NOX 2 inhibitory peptide, gp91ds-tat [also known as Nox2ds-tat (13)], compared to the scrambled peptide (Fig. 4C).
FIG. 4.

NOX2 is required for greater KCl-mediated Ca2+ sensitization and constriction in Ca2+-permeabilized arteries from CH rats compared to controls. Vasoconstrictor responses to KCl in pressurized, Ca2+ permeabilized pulmonary arteries from CH and control rats in the presence of the NOX inhibitors (A) apocynin (30 μM), (B) DPI (10 μM), (C) gp91ds-tat (50 μM), their respective vehicles, or the scrambled control peptide for gp91ds-tat. All arteries were endothelium-disrupted. Values are means±SE of n=4–5 rats/group. *p<0.05 vs. control; #p<0.05 vs. CH vehicle.
CH-induced increases in basal and KCl-stimulated O2− levels are NOX dependent
O2− production was measured by fluorescence detection of dihydroethidium (DHE) oxidation in endothelium-disrupted, pressurized, Ca2+-permeabilized arteries from control and CH rats using previously described methods (7, 30, 53). CH exposure resulted in elevated basal and KCl-induced O2− levels in pressurized pulmonary arteries (Fig. 5) as previously described (7). In contrast, KCl did not alter O2− production in control arteries. In arteries from CH animals, apocynin lowered basal DHE fluorescence to the level of controls, while both apocynin and DPI prevented KCl-dependent increases in O2− production (Fig. 5A and 5B, respectively). gp91ds-tat also decreased basal DHE fluorescence compared to the scrambled peptide in arteries from CH rats, and prevented the effect of KCl to elevate DHE fluorescence in these vessels (Fig. 5C), suggesting that NOX 2 plays a primary role in depolarization-induced O2− generation and pulmonary vasoconstriction.
FIG. 5.

CH-induced increases in basal and depolarization-stimulated O2− levels in pressurized arteries are NOX 2-dependent. DHE fluorescence under basal conditions and following administration of KCl (60 mM) in pressurized, endothelium-disrupted, Ca2+-permeabilized pulmonary arteries from CH and control rats in the presence (A) apocynin, (B) DPI, (C) gp91ds-tat, their respective vehicles, or the scrambled control peptide for gp91ds-tat. KCl was administered 1 min into recording. Values are means±SE of n=4–5 rats/group; *p<0.05 vs. control at all time points. #p<0.05 vs. CH vehicle; τp<0.05 vs. CH vehicle at time 0.
VSM membrane potential is not altered by NOX inhibition
To confirm that inhibitory influences of gp91ds-tat on KCl-induced vasoconstriction and O2− production in CH arteries do not result from potential effects of the peptide to cause VSM membrane hyperpolarization, we next measured membrane potential with sharp electrodes in pressurized arteries from each group in the presence of gp91ds-tat or its scrambled control peptide. VSM membrane potential was depolarized in CH arteries compared to controls under both basal and KCl-stimulated conditions (Fig. 6) as previously reported (7). However, NOX 2 inhibition was without effect on membrane potential in either group.
FIG. 6.

NOX 2 inhibition does not alter vascular smooth muscle (VSM) membrane potential in pressurized arteries from either control or CH rats. VSM membrane potential responses to KCl (60 mM) in the presence of the NOX 2 inhibitory peptide, gp91ds-tat (50 μM), or its scrambled control in pressurized, endothelium-disrupted, Ca2+-permeabilized pulmonary arteries. Values are means±SE of n=4 rats/group; *p<0.05 vs. respective control; #p<0.05 vs. corresponding basal measurement.
Pulmonary arterial NOX 2 expression following CH
To address whether greater KCl-dependent pulmonary vasoreactivity and O2− production following CH are associated with an increase in NOX 2 expression, we measured levels of the catalytic subunit NOX 2 (gp91phox) in intrapulmonary arteries from CH and control rats. However, we observed no difference in NOX 2 levels between CH and control arteries (Fig. 7).
FIG. 7.

Pulmonary arterial NOX 2 expression is unaltered following CH. Representative Western blots and mean densitometric data for NOX 2 and β actin in intrapulmonary arteries from CH and control rats. n=4 rats/group. There are no significant differences.
EGFR-induced Rac1 activation contributes to augmented depolarization-induced vasoconstriction and O2− generation following CH
The contribution of EGFR and Rac1 to augmented depolarization-induced Ca2+ sensitization and O2− generation following CH was assessed by measuring KCl-dependent vasoconstriction and DHE fluorescence in arteries from each group in the presence or absence of the Rac1 inhibitor, NSC 23766 (39), or the EGFR inhibitor, AG1478 (68). Consistent with our hypothesis, both Rac1 and EGFR inhibition prevented KCl-mediated increases vasoreactivity (Fig. 8A and C) and DHE fluorescence (Fig. 8B and D) in arteries from CH rats, while having no effect in control arteries. Similar to effects of O2− scavenging (7, 30) and NOX inhibition (Fig. 5), Rac1 inhibition reduced basal O2− levels in CH arteries (Fig. 8B). In contrast, EGFR inhibition was without effect on basal DHE fluorescence in either group (Fig. 8D).
FIG. 8.

Rac1 and EGFR contribute to increased KCl-induced Ca2+ sensitization, vasoconstriction and O2− generation following CH. Vasoconstrictor responses to KCl in pulmonary arteries from control and CH rats in the presence of (A) the Rac1 inhibitor, NSC 23766 (50 μM), (C) the EGFR inhibitor, AG 1478 (1 μM), or their respective vehicles. DHE fluorescence responses to KCl (60 mM) in the presence of (B) NSC 23766 or (D) AG 1478 in pressurized CH and control arteries. KCl was administered 1 min into recording. Values are means±SE of n=4 rats/group; *p<0.05 vs. control; #p<0.05 vs. CH vehicle; τP<0.05 vs. CH vehicle at time 0.
We next sought to define the signaling relationship between EGFR and KCl-dependent Rac1 activation in ionomycin-treated intrapulmonary arteries from control and CH rats using a Rac1-GTP pull-down assay. Consistent with effects of Rac1 inhibition on vasoreactivity and O2− production (Fig. 8A and B), CH tended to increase basal Rac1 activity (GTP-bound Rac1/total Rac1), although this effect did not achieve statistical significance (Fig. 9). Furthermore, KCl produced an increase in Rac1-GTP levels in arteries from CH but not control rats, and this response to KCl was abolished by EGFR inhibition. EGFR activity, assessed as the ratio of phosphorylated to total EGFR levels, was similarly increased by KCl only in CH arteries (Fig. 10). Basal levels of phosphorylated EGFR, however, were not different between control and CH arteries. No differences in either total Rac1 or total EGFR (each normalized to β actin) were observed between groups (data not shown).
FIG. 9.

Depolarization mediates EGFR-dependent Rac1 activation in arteries from CH but not control rats. Representative Western blots and mean densitometric data for GTP bound (active) Rac1 normalized to total Rac1 in homogenates from control and CH intrapulmonary arteries. Active Rac1 was measured under baseline conditions and following 10 min stimulation with KCl (60 mM) or KCl plus the EGFR inhibitor, AG1478 (1 μM). Values are means±SE of n=4 rats/group; *p<0.05 vs. control KCl; #p<0.05 vs.CH vehicle; τp<0.05 vs. CH KCl.
FIG. 10.

EGFR phosphorylation is induced by KCl in arteries from CH but not control rats. Representative Western blots and mean densitometric data for phosphorylated (active) EGFR normalized to total EGFR in homogenates from control and CH intrapulmonary arteries. Phosphorylated and total EGFR were measured under baseline conditions and following 10 min stimulation with KCl (60 mM). Values are means±SE of n=7 rats/group; *p<0.05 vs. control KCl; #p<0.05 vs.CH vehicle.
Discussion
The goal of the present study was to identify signaling mechanisms linking VSM membrane depolarization to O2− production and enhanced myofilament Ca2+ sensitization in the hypertensive pulmonary circulation of CH rats. The major findings from this study are that: 1) enhanced membrane depolarization-induced O2− production, myofilament Ca2+ sensitization, and pulmonary vasoconstriction following CH require NOX 2; 2) CH augments KCl-dependent O2− generation and vasoconstriction through EGFR-mediated activation of Rac1; and 3) Rac1, but not EGFR, contributes to elevated basal O2− levels following CH. Together, these findings demonstrate a novel mechanism of CH to facilitate membrane depolarization-induced activation of NOX 2 through EGFR-dependent Rac1 signaling in pulmonary VSM, which may contribute to augmented vasoconstrictor sensitivity and PH in this setting.
Exposure to CH increases basal pulmonary arterial tone (8, 49) and augments vasoconstrictor sensitivity to both receptor-mediated agonists (1, 30, 52) and depolarizing stimuli (7, 49). Although mechanisms involving enhanced Ca2+ influx may contribute to greater pulmonary vasoreactivity following CH (28, 34, 64), it is clear that myofilament Ca2+ sensitization mediated by RhoA provides a major contribution to these vasoconstrictor responses (7, 8, 30, 49, 65). Interestingly, the effect of depolarization to activate RhoA in hypertensive pulmonary arteries is thought to occur through a mechanism that is independent of L-type Ca2+ channel stimulation or global changes in VSM [Ca2+]i (7). Consistent with this possibility are our current findings that enhanced vasoconstrictor reactivity to KCl following CH is associated with a markedly blunted vessel wall Ca2+ response compared to control arteries, as previously reported (7). Furthermore, although Ca2+ influx through L-type Ca2+ channels contributes to KCl-mediated vasoconstriction and vessel wall [Ca2+]i in arteries from both control and CH rats, KCl-mediated vasoconstriction persisted following L-type Ca2+ channel blockade in hypertensive arteries, despite similar [Ca2+]i levels between groups. Diltiazem also significantly lowered basal [Ca2+]i in arteries from both control and CH rats, suggesting that L-type Ca2+ channels contribute to resting [Ca2+]i in pressurized small pulmonary arteries. It is noteworthy that, whereas the magnitude of vasoconstrictor responses to KCl in Ca2+-clamped arteries was much greater than the residual vasoconstrictor responses to KCl following L-type Ca2+ channel blockade in nonpermeabilized vessels, this difference is likely the result of the higher [Ca2+]i levels achieved in Ca2+-permeabilized compared to diltiazem-treated nonpermeabilized arteries. Therefore, under physiological conditions, where resting [Ca2+]i levels are not clamped at a higher value, and where depolarization is only comparable to that achieved with 30 mM KCl, the contribution of enhanced Ca2+ sensitivity to depolarization-induced vasoconstriction may be less.
Endogenous ROS derived from multiple enzymatic sources and cell types are widely considered to play an important role in the pathogenesis of various forms of PH (11, 25, 52), including CH-induced PH (21, 27, 36). While the mechanisms by which ROS contribute to PH are likely multifaceted, it is clear that ROS signaling is central to the vasoconstrictor component of CH-induced PH. Such ROS-dependent vasoconstrictor mechanisms may involve endothelial dysfunction (22, 67), scavenging of NO by O2− (29), and direct effects of O2− and H2O2 to promote VSM contraction through intracellular second messenger pathways (7, 30, 32, 35). Indeed, recent studies from our laboratory have demonstrated a link between O2− (or reactive products of O2−) and myofilament Ca2+ sensitization in mediating CH-induced increases in pulmonary vasoconstrictor reactivity to both receptor-mediated agonists and depolarizing stimuli (7, 30). However, neither the specific ROS involved, the enzymatic source of O2−, nor the signaling mechanisms linking depolarization to O2− generation have been previously addressed.
O2− and H2O2 are the primary ROS known to be involved in regulation of pulmonary vascular tone. In pulmonary VSM, O2− mediates a contractile effect through activation of Rho kinase and subsequent myofilament Ca2+ sensitization (32), whereas H2O2 is reported to have both contractile (35) and relaxant (9) properties. We have previously demonstrated that tiron, a spin-trap agent that selectively scavenges O2− and thus would be expected to lower both O2− and H2O2 levels, prevents enhanced depolarization-induced vasoconstriction, O2− production, and RhoA activation following CH (7). Therefore, we sought to determine the relative contributions of these ROS species to enhanced myofilament Ca2+ sensitivity following CH. Our observation that greater vasoreactivity to KCl in Ca2+-permeabilized arteries from CH rats is prevented by PEG-SOD, but not PEG-catalase, supports a role for O2− in this response.
NAD(P)H oxidases have been implicated in the development of both systemic (23, 43) and pulmonary (27, 36, 46) hypertension, and thus represent a potential source of O2− mediating enhanced depolarization-induced pulmonary VSM Ca2+ sensitization following CH. In agreement with this possibility is evidence that CH-mediated increases in pulmonary vasoreactivity, ROS production and PH are attenuated in mice deficient in the catalytic subunit of NOX 2 (36). Additionally, NOX 2 contributes to impaired endothelium-dependent pulmonary vasodilation in mice (22) and neonatal piglets (21) with CH-induced PH. Furthermore, CH increases NOX-derived ROS production, NOX 1 expression, and p67phox levels in the membrane fraction of small pulmonary arteries from piglets, suggesting elevated NOX 1 activity in this neonatal model of PH (16, 21). NOX 4 gene and protein expression are also elevated following CH exposure in murine pulmonary arteries and lung tissue (46, 51) and are similarly upregulated by CH in cultured human pulmonary arterial endothelial cells (40). However, the contribution of NOX 4 to the development of PH remains poorly defined. A role for NOX in augmented depolarization-induced Ca2+ sensitization following CH is demonstrated by our present findings that apocynin decreased the vasoconstrictor response to KCl in nonpermeabilized arteries and isolated lungs from CH rats, but not controls. This argument is further supported by evidence from Ca2+permeabilized arteries that nonspecific NOX inhibition with apocynin or DPI attenuated both vasoreactivity and O2− generation in response to KCl selectively in arteries from CH rats. Similar results were observed using the highly selective NOX 2 inhibitor, gp91ds-tat, which competitively inhibits p47phox binding to the catalytic subunit of NOX 2, and thus prevents assembly of the active enzyme complex (58). These data therefore support a distinct contribution of NOX 2-derived O2− to enhanced depolarization-induced vasoconstriction independent of changes in global vessel wall [Ca2+]i following CH. Although we cannot exclude the possibility that localized, subsarcolemmal Ca2+ events contribute to the observed responses, we are confident that, within the sensitivity limits of the system, global [Ca2+]i is effectively clamped by ionomycin in this preparation. An additional limitation of this system is the inability to selectively measure VSM [Ca2+]i, as other cell types within the vascular wall (e.g., fibroblasts) likely contribute to the fura 2 signal. Furthermore, adventitial remodeling resulting from CH exposure may increase the contribution of this layer to the overall signal.
It is unlikely that greater basal and depolarization-stimulated O2− production following CH are a function of increased arterial NOX 2 expression, since levels of the NOX 2 catalytic subunit pg91phox were similar between arteries from control and CH rats. Indeed, the lack of effect of NOX inhibition on either vasoreactivity or O2− levels in control arteries suggests CH facilitates a coupling of depolarization to NOX 2 activation that is not present in the normal pulmonary circulation. Nevertheless, we cannot exclude a possible contribution of altered expression of other components of the NOX 2 complex to this response.
Rac1 is an important mediator of NOX 1 and 2 activation (6, 12), and thus represents a potential signaling factor that links membrane depolarization to stimulation of NOX 2 following CH. Consistent with this possibility is evidence that depolarization activates Rac1 and NOX in endothelial and macula densa cells (10, 38, 39, 44, 60). Here we demonstrate that KCl-mediated membrane depolarization activates Rac1 selectively in arteries from pulmonary hypertensive rats. Our findings that basal and KCl-mediated O2− generation and associated vasoconstriction are diminished by NSC 23766 only in CH arteries further suggest that Rac1 activation contributes to both elevated resting O2− levels and depolarization-stimulated O2− production and vasoconstriction following CH. However, it is additionally possible that the phosphorylation state of the NOX subunits p47phox or p67phox is altered by membrane depolarization following CH, as phosphorylation of these subunits can also contribute to NOX activation (6, 12, 41).
Based on evidence that depolarization leads to EGFR activation in PC12 cells, and that membrane stretch stimulates EGFR-dependent NOX activation in mesangial cells (68), we next examined EGFR as a proximal mediator of depolarization-induced, Rac1-dependent NOX 2 activation in CH arteries. A role for EGFR in this response is supported by our present findings that EGFR phosphorylation was induced by a depolarizing stimulus only in CH arteries, and that selective EGFR inhibition, like Rac1 and NOX 2 inhibition, prevented effects of CH to augment both depolarization-induced vasoconstriction and vascular O2− production. We additionally found that depolarization-mediated Rac1 activation in intrapulmonary arteries from CH rats was prevented with AG 1478, suggesting EGFR signals upstream of Rac1 to promote O2− generation and vasoconstriction following CH. However, unlike responses to Rac1 and NOX 2 inhibition, EGFR inhibition had no effect to reduce basal production in vessels from CH rats, suggesting that whereas EGFR is required for KCl-mediated O2− production, CH elevates basal O2− levels through an EGFR-independent mechanism. Further supporting this possibility is our present finding that basal levels of phosphorylated EGFR were unaltered following exposure to CH. The mechanism by which CH increases basal O2− production in pulmonary arteries is unknown, but may involve coupling of transmural pressure-induced VSM stretch to NOX activation.
Although the mechanism by which depolarization mediates EGFR activation in hypertensive pulmonary arteries remains to be established, it is possible that CH alters the composition of membrane signaling platforms, including caveolae or other lipid rafts, to permit regulation of EGFR by voltage-sensitive proteins. Metabotropic signaling by ion channels (5, 20), G protein-coupled receptors (37, 42), and voltage-sensitive phosphatases (19, 26, 47, 48) represent possible mediators of depolarization-induced VSM Ca2+ sensitization. It is alternatively possible that EGFR expresses a membrane potential-sensitive domain that imparts direct voltage-sensitive enzymatic activity.
As previously reported following inhibition of Rho kinase, PKC, or scavenging of O2− (7), a persistent vasoconstrictor response to KCl was observed even after inhibition of NOX, Rac1, or EGFR in arteries from both groups. Although the mechanism of this residual vasoconstriction is unknown, it is possible that a Ca2+-independent myosin light chain kinase, such as integrin-linked kinase (15) or ZIP kinase (50), contributes to this response. Future studies are also needed to determine the contribution of membrane depolarization-induced NOX 2 activation and subsequent Ca2+ sensitization to enhanced agonist-induced pulmonary vasoconstrictor sensitivity (1, 30) and myogenic vasoconstriction (8) following CH, and the potential role of dysregulation of endogenous antioxidant systems (54) in these responses.
In conclusion, we have identified a unique signaling pathway in pulmonary vascular smooth muscle involving EGFR-induced Rac1 and NOX 2 activation that couples membrane depolarization to myofilament Ca2+ sensitization and vasoconstriction. Furthermore, whereas this pathway is not functional in the normotensive pulmonary circulation, it is induced by long-term exposure to hypoxia, and may provide a basis for understanding mechanisms of vasoconstriction that contribute to CH-induced PH.
Methods
All protocols and surgical procedures in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of New Mexico Health Sciences Center (Albuquerque, NM).
Experimental groups
Male Sprague-Dawley rats (body wt. 250–350 g, age 3–4 mo; Harlan Industries, Indianapolis, IN) were exposed to 4 wk CH by placement in a hypobaric chamber maintained at ∼380 Torr as previously described (8, 30, 57).
Isolated lung protocol: Contribution of NOX to KCl-dependent vasoconstriction
Lungs were isolated from rats, perfused with PSS containing 4% bovine serum albumin as a colloid, and ventilated with a 21% O2, 6% CO2 gas mixture as previously described (57). Following stabilization of baseline pressure, a cumulative concentration–response relationship to depolarizing concentrations of KCl (15–60 mM) was assessed (7). This PSS had the same osmolality as the control solution achieved by exchanging NaCl for equimolar concentrations of KCl. Experiments were conducted in the presence or absence of the general NOX inhibitor apocynin (30 μM, Sigma, St. Louis, MO) (61) or vehicle (PSS).
Isolated pulmonary artery protocols: Assessment of vasoreactivity, vessel wall [Ca2+]i, O2− levels, and membrane potential
Small pulmonary arteries were isolated and cannulated for simultaneous assessment of vasoreactivity and VSM [Ca2+]i, O2− or membrane potential, as previously described (7). All arteries were studied at a transmural pressure of 12 Torr, as we have previously demonstrated that reactivity to KCl following CH is similarly augmented at 12 or 35 Torr (7). All isolated arteries were studied under normoxic conditions for the pulmonary arterial circulation (equilibrated with a 10% O2, 6% CO2, and balance N2 gas mixture) and endothelium disrupted to directly evaluate effects of CH on VSM reactivity to KCl independent of endothelial influences. Background-subtracted fura-2 F340/F380 emission (510 nm) ratios were calculated with IonOptix Ion Wizard software and recorded continuously throughout the experiment to measure vessel wall [Ca2+]i, with simultaneous measurement of ID from red wavelength bright-field images. Disruption of the endothelium was confirmed by lack of response to ACh following a UTP constriction (8, 30). To directly assess mechanisms of myofilament Ca2+ sensitization independent of changes in vessel wall [Ca2+]i, we clamped vessel wall [Ca2+]i in some arteries by permeabilizing with the Ca2+ ionophore, ionomycin (3 μM, Sigma), as previously described (30). All Ca2+ permeabilized vessels were equilibrated with PSS containing a calculated free Ca2+ concentration of 300 nM. This concentration of Ca2+ was chosen to provide optimal vasoreactivity to KCl while having minimal effects on resting tone based on preliminary studies.
Contribution of L-type Ca2+ channels and NOX to KCl-dependent vasoconstriction
KCl-induced (30, 60, and 120 mM) vasoconstrictor and Ca2+ responses were measured in nonpermeabilized arteries from CH and control rats in the presence of the L-type Ca2+ channel inhibitor diltiazem (50 μM) (7), the NOX inhibitor apocynin (30 μM) or their vehicles. All inhibitors used for isolated vessel experiments were added to the superfusate 20 min prior to the beginning of experimentation and were present throughout the protocol.
Role of O2−, H2O2, NOX, Rac1, and EGFR in KCl-dependent VSM Ca2+ sensitization
Vasoconstrictor responses to increasing concentrations of KCl (30, 60, and 120 mM) were assessed in Ca2+ permeabilized arteries from CH and control rats in the presence of PEG-SOD (120 U/mL, Sigma) (22), PEG-catalase (250 U/mL, Sigma) (16), the NOX inhibitors apocynin (30 μM) (61), DPI (10 μM, Sigma) (66), or their vehicle controls. Parallel protocols were conducted in the presence of the specific NOX 2 inhibitor gp91ds-tat (50 μM, Tufts, Boston, MA) (58), its corresponding scrambled control peptide, the Rac1 inhibitor NSC 23766 (50 μM, Cayman, Ann Arbor, MI; a guanine nucleotide exchange factor inhibitor chosen for its selectivity for Rac1 over the similar G-protein RhoA) (39), the selective EGFR inhibitor AG1478 (1 μM, Cayman) (68), or appropriate vehicles. Fura-2 ratios were monitored throughout all experiments to confirm calcium clamp.
Role of NOX, Rac1, and EGFR in KCl-dependent O2− production
DHE (10 μM, Molecular Probes, Eugene, OR) fluorescence was used to assess depolarization-induced O2− generation in endothelium-disrupted, pressurized, Ca2+-permeabilized arteries from each group, as previously described (7). After establishing a baseline value, 60 mM KCl was administered and DHE fluorescence was measured over 12 minutes. Arteries were exposed to DPI, apocynin, gp91ds-tat, NSC23766, AG1478, or the respective vehicle to determine the role of NOX, Rac1, and EGFR in KCl-stimulated O2− production.
Effects of NOX 2 inhibition on VSM membrane potential
VSM cell membrane potential was measured in arteries prepared as above in the presence of gp91ds-tat or scramb-tat using sharp electrodes, as previously described (7).
Western blot analysis
NOX 2 expression
NOX 2 expression was compared between intrapulmonary arteries of CH and control rats by Western blotting, using methods similar to those previously described (7). Pulmonary arterial NOX 2 catalytic subunit expression was measured using a mouse monoclonal NOX 2 (gp91phox) antibody (1:2000, BD Biosciences, Lexington, KY) (18). NOX 2 expression was normalized to β actin expression.
Rac1 and EGFR activation
Intrapulmonary arteries from CH and control rats were isolated and incubated with the Ca2+ ionophore ionomycin (37°C; 30 min). Some arteries were additionally treated with KCl (60 mM) or KCl plus the EGFR inhibitor AG 1478 (1 μM). Arteries were homogenized as previously described (7). Rac1 activity was assessed using a Rac1 activation assay kit (Cytoskeleton, Denver, CO) that detects levels of GTP-bound Rac1 (68). Levels of GTP-bound Rac1 were normalized to total Rac1 protein levels determined from separate Western blots. Homogenates were also probed for levels of phosphorylated EGFR. A polyclonal anti-phospho EGFR antibody (1:500, Cell Signaling, Beverly, MA) (68) was used to detect EGFR phosphorylated on the tyrosine 1068 residue (2, 24). Phosphorylated EGFR levels were normalized to total EGFR levels (1:500, Cell Signaling) for comparison.
Calculations and statistics
For isolated lung experiments, total pulmonary vascular resistance was calculated as the difference between pulmonary arterial and pulmonary venous pressures (Pa and Pv respectively) divided by flow (30 ml·min−1·kg body wt−1). Vasoconstrictor responses were calculated as a percentage of baseline ID. All data are presented as means±SE, and n refers to the number of animals in each group. A t-test, one-way ANOVA, two-way ANOVA, or one-way repeated measures ANOVA was used to make comparisons when appropriate. If differences were detected by ANOVA, individual groups were compared using the Student-Newman-Keuls or Bonferroni test. A probability of p<0.05 was considered significant for all comparisons.
Abbreviations Used
- [Ca2+]i
free intracellular Ca2+ concentration
- CH
chronic hypoxia
- DHE
dihydroethidium
- EGFR
epidermal growth factor receptor
- ID
inner diameter
- NOX
NAD(P)H oxidase
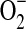
superoxide anion
- PH
pulmonary hypertension
- PSS
physiological saline solution
- ROS
reactive oxygen species
- VSM
vascular smooth muscle
Acknowledgments
The authors thank Minerva Murphy and Dr. Jessica Snow for technical assistance. This work was supported by National Institutes of Health Grants HL-92598 (N. L. Jernigan), HL-88192 (T.C. Resta), HL-07736 and HL-95640 (B. R. Walker), and American Heart Association grants 0755775Z (T. C. Resta) and 0625647Z (B.R.S. Broughton).
Author Disclosure Statement
The authors have no competing financial interests.
References
- 1.Barman SA. Vasoconstrictor effect of endothelin-1 on hypertensive pulmonary arterial smooth muxcle involves Rho-kinase and protein kinase C. Am J Physiol Lung Cell Mol Physiol. 2007;293:L472–L479. doi: 10.1152/ajplung.00101.2006. [DOI] [PubMed] [Google Scholar]
- 2.Batzer A. Rotin D. Urena J. Skolnik E. Schlessinger J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol Cell Biol. 1994;14:5192–5201. doi: 10.1128/mcb.14.8.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bierer R. Nitta C. Friedman J. Codianni S. de Frutos S. Dominguez-Bautista J. Howard T. Resta TC. Bosc L. NFATc3 is required for chronic hypoxia-induced pulmonary hypertension in adult and neonatal mice. Am J Physiol Lung Cell Mol Physiol. 2011;301:L872–L880. doi: 10.1152/ajplung.00405.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Billaud M. Marthan R. Savineau J. Guibert C. Vascular smooth muscle modulates endothelial control of vasoreactivity via reactive oxygen species production through myoendothelial communications. PLoS ONE. 2009;4:e6432. doi: 10.1371/journal.pone.0006432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Billups D. Billups B. Challiss R. Nahorski S. Modulation of Gq-protein-coupled inositol triphosphate and Ca2+ signaling by the membrane potential. J Neurosci. 2006;26:9983–9995. doi: 10.1523/JNEUROSCI.2773-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandes RP. Kreuzer J. Vascular NADPH oxidases: Molecular mechanisms of activation. Cardiovasc Res. 2004;65:16–27. doi: 10.1016/j.cardiores.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Broughton BR. Jernigan NL. Norton CE. Walker BR. Resta TC. Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am J Physiol Lung Cell Mol Physiol. 2010;298:L232–L242. doi: 10.1152/ajplung.00276.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broughton BR. Walker BR. Resta TC. Chronic hypoxia induces Rho kinase-dependent myogenic tone in small pulmonary arteries. Am J Physiol Lung Cell Mol Physiol. 2008;294:L797–L806. doi: 10.1152/ajplung.00253.2007. [DOI] [PubMed] [Google Scholar]
- 9.Burke TM. Wolin MS. Hydrogen peroxide elicits pulmonary arterial relaxation and guanylate cyclase activation. Am J Physiol. 1987;252:H721–H732. doi: 10.1152/ajpheart.1987.252.4.H721. [DOI] [PubMed] [Google Scholar]
- 10.Chatterjee S. Browning EA. Hong N. Debolt K. Sorokina EM. Liu W. Birnbaum MJ. Fisher AB. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am J Physiol Heart Circ Physiol. 2012;302:H105–H114. doi: 10.1152/ajpheart.00298.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen MJ. Chiang LY. Lai YL. Reactive oxygen species and substance P in monocrotaline-induced pulmonary hypertension. Toxicol Appl Pharmacol. 2001;171:165–173. doi: 10.1006/taap.2000.9117. [DOI] [PubMed] [Google Scholar]
- 12.Clempus RE. Griendling KK. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc Res. 2006;71:216–225. doi: 10.1016/j.cardiores.2006.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Csanyi G. Cifuentes-Pagano E. Ghouleh IA. Ranayhossaini DJ. Egena L. Lopes LR. Jackson HM. Kelly EE. Pagano PJ. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase NOX 2. Free Rad Biol Med. 2011;51:1116–1125. doi: 10.1016/j.freeradbiomed.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dahal B. Cornitescu T. Tretyn A. Pullamsetti S. Kosanovic D. Dumitrascu R. Ghofrani HA. Weissmann N. Voswinckel R. Banat GSW. Grimminger F. Schermuly RT. Role of epidermal growth factor inhibition in experimental pulmonary hypertension. Am J Respir Crit Care Med. 2010;181:158–159. doi: 10.1164/rccm.200811-1682OC. [DOI] [PubMed] [Google Scholar]
- 15.Deng J. Van Lierop J. Sutherland C. Walsh M. Ca2+-independent smooth muscle contraction. A novel function for integrin-linked kinase. J Biol Chem. 2001;276:16365–16373. doi: 10.1074/jbc.M011634200. [DOI] [PubMed] [Google Scholar]
- 16.Dennis KE. Aschner JL. Milatovic D. Shmidt JW. Aschner M. Kaplowitz MR. Zhang Y. Fike CD. NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol. 2009;297:L596–L607. doi: 10.1152/ajplung.90568.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Egea J. Espinet C. Comella JX. Calmodulin modulates mitogen-activated protein kinase activation in response to membrane depolarization in PC12 cells. J Neurochem. 1998;70:2554–2564. doi: 10.1046/j.1471-4159.1998.70062554.x. [DOI] [PubMed] [Google Scholar]
- 18.El-Adway MS. Ansari HR. Fil D. Tilley SL. Mustafa SJ. NADPH oxidase pathway is involved in aortic contraction induced by A3 adenosine receptor in mice. J Pharmacol Exper Therapeut. 2011;338:711–717. doi: 10.1124/jpet.111.180828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Falkenburger B. Jensen J. Hille B. Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J Gen Physiol. 2010;135:99–114. doi: 10.1085/jgp.200910345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez-Tenorio M. Porras-Gonzalez C. Castellana A. Del Valle-Rodriguez A. Lopez-Barneo J. Urena J. Metabotropic regulation of RhoA/Rho-associated kinase by L-type Ca2+ channels: New mechanism for depolarization-evoked mammalian arterial contraction. Circ Res. 2011;108:1348–1357. doi: 10.1161/CIRCRESAHA.111.240127. [DOI] [PubMed] [Google Scholar]
- 21.Fike CD. Slaughter J. Kaplowitz MR. Zhang Y. Aschner JL. Reactive oxygen species from NADPH oxidase contribute to altered pulmonary vascular responses in piglets with chronic hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2008;295:L881–L888. doi: 10.1152/ajplung.00047.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fresquet F. Pourageaud F. Leblais V. Brandes RP. Savineau J. Marthan R. Muller B. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br J Pharmacol. 2006;148:714–723. doi: 10.1038/sj.bjp.0706779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gavazzi G. Banfi B. Deffert C. Fiette L. Schappi M. Herrmann F. Krause KH. Decreased blood pressure in NOX-1-deficient mice. FEBS Lett. 2006;580:497–504. doi: 10.1016/j.febslet.2005.12.049. [DOI] [PubMed] [Google Scholar]
- 24.Gay B. Suarez S. Weber C. Rahuel J. Fabbro D. Furet P. Caravatti G. Schoepter J. Effect of potent and selective inhibitors of the Grb2 SH2 domain on cell motility. J Biol Chem. 1999;274:23311–23315. doi: 10.1074/jbc.274.33.23311. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez Bosc LV. Resta TC. Walker BR. Kanagy NL. Mechanisms of intermittent hypoxia induced hypertension. J Cell Mol Med. 2010;14:3–17. doi: 10.1111/j.1582-4934.2009.00929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halaszovich C. Schreiber D. Oliver D. Ci-VSP is a depolarization-activated phosphatidylinositol-4,5-bisphoste and phosphatidylinositol-3,4,5 triposphate 5'-phosphatase. J Biol Chem. 2009;284:2106–2113. doi: 10.1074/jbc.M803543200. [DOI] [PubMed] [Google Scholar]
- 27.Ismail S. Sturrock A. Wu B. Cahill B. Norman K. Huecksteadt T. Sanders K. Kennedy T. Hoidal J. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: The role of autocrine production of transforming growth factor-{beta}1 and insulin-like growth factor binding protien-3. Am J Physiol Lung Cell Mol Physiol. 2009;296:L489–L499. doi: 10.1152/ajplung.90488.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jernigan NL. Herbert L. Walker BR. Resta TC. Chronic hypoxia upregulates pulmonary arterial ASIC1: A novel mechanism of enhanced store-operated Ca2+ entry and receptor-dependent vasoconstriction. Am J Physiol Cell Physiol. 2012;302:C931–C940. doi: 10.1152/ajpcell.00332.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jernigan NL. Walker BR. Resta TC. Endothelium-derived reactive oxygen species and endothelin-1 attenuate NO-dependent pulmonary vasodilation following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2004;287:L801–L808. doi: 10.1152/ajplung.00443.2003. [DOI] [PubMed] [Google Scholar]
- 30.Jernigan NL. Walker BR. Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2008;295:L515–L529. doi: 10.1152/ajplung.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kakiashvili E. Dan Q. Vandermeer M. Zhang Y. Waheed F. Pharm M. Szaszi K. The epidermal growth factor receptor mediates tumor necrosis factor-alpha-induced activation of ERK/GEF-H1/RhoA pathway in tubular epithelium. J Biol Chem. 2011;286:9268–9279. doi: 10.1074/jbc.M110.179903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knock G. Snetkov V. Shaifta Y. Connolly M. Drndarski S. Noah A. Pourmahram G. Becker S. Aaronson P. Ward J. Superoxide constricts rat pulmonary arteries via Rho-kinase-mediated Ca2+ sensitization. Free Radic Biol Med. 2009;46:633–642. doi: 10.1016/j.freeradbiomed.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Cras TD. Hardie WD. Fagan KA. Whisett JA. Korfhagen TR. Disrupted pulmonary vascular development and pulmonary hypertension in transgenic mice overexpressing tranforming growth factor-alpha. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1046–L1054. doi: 10.1152/ajplung.00045.2003. [DOI] [PubMed] [Google Scholar]
- 34.Lin MJ. Leung GP. Zhang WM. Yang XR. Yip KP. Tse CM. Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: A novel mechanism of hypoxic pulmonary hypertension. Circ Res. 2004;95:496–505. doi: 10.1161/01.RES.0000138952.16382.ad. [DOI] [PubMed] [Google Scholar]
- 35.Lin MJ. Yang XR. Cao Y. Sham JS. Hydrogen peroxide-induced Ca2+ mobilization in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1598–L1601. doi: 10.1152/ajplung.00323.2006. [DOI] [PubMed] [Google Scholar]
- 36.Liu JQ. Zelko IN. Erbynn EM. Sham JS. Folz RJ. Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox) Am J Physiol Lung Cell Mol Physiol. 2006;290:L2–L10. doi: 10.1152/ajplung.00135.2005. [DOI] [PubMed] [Google Scholar]
- 37.Liu Q. Zheng Y. Korde A. Yadav V. Rathore R. Wess J. Wang Y. Membrane depolarization causes a direct activation of G protein-coupled receptors leading to local Ca2+ release in smooth muscle. Proc Natl Acad Sci USA. 2009;106:11418–11423. doi: 10.1073/pnas.0813307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu R. Garvin J. Ren Y. Pagano P. Carretero O. Depolarization of the macula densa induces superoxide production via NAD(P)H oxidase. Am J Physiol Renal Physiol. 2007;292:F1867–F1872. doi: 10.1152/ajprenal.00515.2006. [DOI] [PubMed] [Google Scholar]
- 39.Liu R. Juncos LA. GTPase-Rac enhances depolarization induced superoxide production by the macula densa during tubuloglomerular feedback. Am J Physiol Regul Integr Comp Physiol. 2010;298:R453–R458. doi: 10.1152/ajpregu.00622.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu X. Murphy T. Nanes M. Art CM. PPAR {gamma} regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-{kappa}B. Am J Physiol Lung Cell Mol Physiol. 2010;299:L559–L566. doi: 10.1152/ajplung.00090.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manea A. NADPH oxidase-derived reactive oxygen species: Involvement in vascular physiology and pathology. Cell Tissue Res. 2010;342:325–339. doi: 10.1007/s00441-010-1060-y. [DOI] [PubMed] [Google Scholar]
- 42.Martinez-Pinna J. Gurung I. Mahaut-Smith M. Morales A. Direct voltage control of endogenous lysophosphatidic acid G-protein-coupled receptors in Xenopus oocytes. J Physiol. 2010;588:1683–1693. doi: 10.1113/jphysiol.2009.183418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsuno K. Yamanda H. Iwata K. Jin D. Katsuyama M. Matsuki M. Takai S. Yaminishi K. Miyazaki M. Matsubara H. Yabe-Nishimura C. Nox 1 is involved in angiotensin II-mediated hypertension: A study in Nox 1-deficient mice. Circulation. 2005;112:2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- 44.Matsuzaki I. Chatterjee S. Debolt K. Manevich Y. Zhang Q. Fisher AB. Membrane Depolarization and NADPH oxidase activation in aortic endothelium during ischemia reflect altered mechanotransduction. Am J Physiol Heart Circ Physiol. 2005;288:H336–H343. doi: 10.1152/ajpheart.00025.2004. [DOI] [PubMed] [Google Scholar]
- 45.Merklinger SL. Jones PL. Martinez EC. Rabinovitch M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circ Res. 2005;112:423–431. doi: 10.1161/CIRCULATIONAHA.105.540542. [DOI] [PubMed] [Google Scholar]
- 46.Mittal M. Roth M. Konig P. Hofmann E. Dony E. Goyal P. Selbitz AC. Schermuly RT. Ghofrani HA. Kwapiszewska G. Kummer W. Klepetko W. Hoda MA. Fink L. Hanze J. Seeger W. Grimminger F. Schmidt H. Weissmann N. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res. 2007;101:258–267. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 47.Murata Y. Iwasaki H. Sasaki M. Inaba K. Okamura Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature. 2005;435:1239–1243. doi: 10.1038/nature03650. [DOI] [PubMed] [Google Scholar]
- 48.Murata Y. Okamura Y. Depolarization activates the phosphoinositide phosphatase Ci-VSP, as detected in Xenopus oocutes coexpressing sensors of PIP2. J Physiol. 2007;583:875–889. doi: 10.1113/jphysiol.2007.134775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagaoka T. Morio Y. Casanova N. Bauer N. Gebb SA. McMurtry IF. Oka M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 2004;287:L665–L672. doi: 10.1152/ajplung.00050.2003. [DOI] [PubMed] [Google Scholar]
- 50.Niiro N. Ikebe M. Zipper-interacting protein kinase induces Ca2+ free smooth muscle contraction via myosin light chain phosphorylation. J Biol Chem. 2001;276:29567–29574. doi: 10.1074/jbc.M102753200. [DOI] [PubMed] [Google Scholar]
- 51.Nisbet RE. Bland J. Kleinhenz DJ. Mitchell PO. Walp E. Sutliff RL. Hart CM. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am J Respir Cell Mol Biol. 2010;42:482–490. doi: 10.1165/rcmb.2008-0132OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nisbet RE. Graves AS. Kleinhenz DJ. Rupnow HL. Reed AL. Fan TH. Mitchell PO. Sutliff RL. Hart CM. The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am J Respir Cell Mol Biol. 2009;40:601–609. doi: 10.1165/rcmb.2008-0145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Norton CE. Jernigan NL. Kanagy NL. Walker BR. Resta TC. Intermittent hypoxia augments pulmonary vascular smooth muscle reactivity to NO: Regulation by reactive oxygen species. J Appl Physiol. 2011;111:980–988. doi: 10.1152/japplphysiol.01286.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nozik-Grayck E. Suliman HB. Majka S. Albietz J. Van Rheen Z. Roush K. Stenmark KR. Lung EC-SOD overexpression attenuates hypoxic induction of Erg-1 and chronic hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol. 2008;295:L422–L430. doi: 10.1152/ajplung.90293.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Resta TC. Chicoine LG. Omdahl JL. Walker BR. Maintained upregulation of pulmonary eNOS gene and protein expression during recovery from chronic hypoxia. Am J Physiol. 1999;276:H699–H708. doi: 10.1152/ajpheart.1999.276.2.H699. [DOI] [PubMed] [Google Scholar]
- 56.Resta TC. Kanagy NL. Walker BR. Estradiol-induced attenuation of pulmonary hypertension is not associated with altered eNOS expression. Am J Physiol Lung Cell Mol Physiol. 2001;280:L88–L97. doi: 10.1152/ajplung.2001.280.1.L88. [DOI] [PubMed] [Google Scholar]
- 57.Resta TC. Walker BR. Chronic hypoxia selectively augments endothelium-dependent pulmonary arterial vasodilation. Am J Physiol. 1996;270:H888–H896. doi: 10.1152/ajpheart.1996.270.3.H888. [DOI] [PubMed] [Google Scholar]
- 58.Rey FE. Cifuentes ME. Kiarash MT. Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2- and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 59.Snow JB. Kitzis V. Norton CE. Torres SN. Johnson KD. Kanagy NL. Walker BR. Resta TC. Differential effects of chronic hypoxia and intermittent hypocapnic and eucapnic hypoxia on pulmonary vasoreactivity. J Appl Physiol. 2008;104:110–118. doi: 10.1152/japplphysiol.00698.2005. [DOI] [PubMed] [Google Scholar]
- 60.Sohn HY. Keller M. Gloe T. Morawietz H. Rueckschloss U. Ohl U. The small G-protein Rac mediates depolarization-induced superoxide formation in human endothelial cells. J Biol Chem. 2000;275:18745–18750. doi: 10.1074/jbc.M000026200. [DOI] [PubMed] [Google Scholar]
- 61.Stolk J. Hiltermann TJ. Dijkman JH. Verhoeven AJ. Characteristics of inhition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- 62.Su B. Miltra S. Gregg H. Flavahan S. Chotani M. Clark K. Goldschmidt-Clermont P. Flavahan N. Redox regulation of vascular smooth muscle cell differentiation. Circ Res. 2001;89:39–46. doi: 10.1161/hh1301.093615. [DOI] [PubMed] [Google Scholar]
- 63.Tahara S. Fukuda K. Kodama H. Kato T. Miyoshi S. Ogawa S. Potassium channel blocker activates extracellular signal-regulated kinases through Pyk2 and epidermal growth factor receptor in rats. J Am Coll Cardiol. 2001;38:1554–1563. doi: 10.1016/s0735-1097(01)01558-3. [DOI] [PubMed] [Google Scholar]
- 64.Wang J. Weigand L. Lu W. Sylvester JT. Semenza G. Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle. Circ Res. 2006;98:1528–1537. doi: 10.1161/01.RES.0000227551.68124.98. [DOI] [PubMed] [Google Scholar]
- 65.Weigand L. Sylvester JT. Shimoda LA. Mechanisms of endothelin-1-induced contraction in pulmonary arteries from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 2006;290:L284–L290. doi: 10.1152/ajplung.00449.2004. [DOI] [PubMed] [Google Scholar]
- 66.Wind S. Beuerlein K. Armitage ME. Taye A. Kumar A. Janowitz D. Neff C. Shaw AM. Wingler K. Schmidt H. Oxidative stress and endothelial dysfunction in aorta of aged spontaneously hypertensive rats by NOX1/2 is reversed by NADPH oxidase inhibition. Hypertension. 2010;56:490–497. doi: 10.1161/HYPERTENSIONAHA.109.149187. [DOI] [PubMed] [Google Scholar]
- 67.Zhang M. Song P. Xu J. Zou MH. Activation of NAD(P)H oxidase by thromboxane A2 receptor uncouples endothelial nitric oxide synthase. Arterioscler Throm Vasc Biol. 2011;31:125–132. doi: 10.1161/ATVBAHA.110.207712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y. Peng F. Gao B. Ingram AJ. Krepinsky JC. Mechanical strain-induced RhoA activation requires NADPH oxidase-mediated ROS generation in caveolae. Antioxid Redox Signal. 2010;13:959–973. doi: 10.1089/ars.2009.2908. [DOI] [PubMed] [Google Scholar]
- 69.Zwick E. Daub H. Aoki N. Yamaguchi-Aoki Y. Tinofer I. Ullrich A. Critical role of calcium-dependent epidermal growth factor receptor transactivation in PC12 cell membrane depolarization and bradykinin signaling. J Biol Chem. 1997;272:24767–24770. doi: 10.1074/jbc.272.40.24767. [DOI] [PubMed] [Google Scholar]