

Abstract
Vaccinia DNA topoisomerase IB (TopIB) relaxes supercoils by forming and resealing a covalent DNA-(3′-phosphotyrosyl274)-enzyme intermediate. Conserved active site side chains promote the attack of Tyr274 on the scissile phosphodiester via transition state stabilization and general acid catalysis. Two essential side chains, Lys167 and Arg130, act in concert to protonate and expel the 5′-O leaving group. Here we gained new insights to catalysis through chemical mutagenesis of Lys167. Changing Lys167 to cysteine crippled the DNA cleavage and religation transesterification steps (kcl = 4.3 × 10−4 s−1; krel = 9 × 10−4 s−1). The transesterification activities of the K167C enzyme were revived by in vitro alkylation with 2-bromoethylamine (kcl = 0.031 s−1; krel ≥ 0.4 s−1) and 3-bromopropylamine (kcl = 0.013 s−1; krel = 0.22 s−1), which convert the cysteine to γ-thialysine and γ-thiahomolysine, respectively. These chemically installed lysine analogs were more effective than a genetically programmed arginine-167 substitution characterized previously. The modest differences in the transesterification rates of the 2-bromoethylamine and 3-bromopropylamine-treated enzymes highlights that TopIB is tolerant of a longer homolysine side chain for assembly of the active site and formation of the transition state.
Type IB DNA topoisomerases (TopIB) relax DNA supercoils by repeatedly breaking and rejoining one strand of the DNA duplex through a covalent DNA-(3′-phosphotyrosyl)-enzyme intermediate. Vaccinia virus TopIB, which displays stringent specificity for cleavage at the sequence 5′ (C/T)CCTT↓ in the scissile strand1–5, has been an instructive model system for mechanistic studies of the TopIB family. The cleavage and religation transesterification reactions are driven by four amino acid side chains (Arg130, Lys167, Arg223, His265) that comprise the active site and catalyze the attack of Tyr274 on the scissile phosphodiester.6–10 The two arginines and the histidine interact directly with the scissile phosphodiester in the crystal structure of the covalent TopIB-DNA intermediate (Fig. 1) and are proposed to stabilize the developing negative charge on a pentacoordinate phosphorane transition state.11–15 Lys167 serves together with Arg220 as a general acid to expel the 5′-OH leaving group during the cleavage reaction.16,17
Figure 1. Structural basis of TopIB transesterification chemistry.
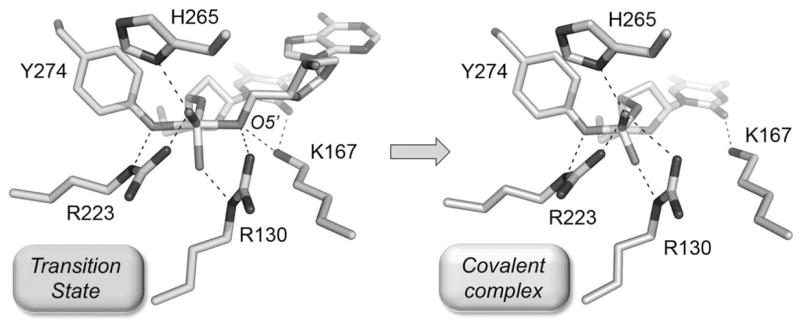
The active sites of the pentavalent vanadate complex of poxvirus TopIB (a transition state mimetic; PDB 3IGC; shown at left) and the covalent tyrosyl-DNA intermediate of poxvirus TopIB (PDB 2H7F; shown at right) were superimposed and then offset laterally. The catalytic amino acid side chains are indicated. Atomic contacts are denoted by dashed lines.
Van Duyne and colleagues18 exploited vanadate as a mimetic to capture a crystal structure of the pentavalent transition state of poxvirus TopIB (Fig. 1), which adopts a trigonal bipyramidal geometry with the attacking tyrosine and the O5′ atom of the leaving strand at the apices, as expected for the proposed associative mechanism.11 This structure confirmed the atomic contacts to the nonbridging and 5′-bridging phosphate oxygens that were surmised from functional studies employing DNA substrates containing stereospecific methylphosphonate or bridging 5′ phosphorothioate substitutions.12,13,16,17 The O5′ leaving atom in the transition state is coordinated simultaneously by the Arg130 and Lys167 side chains implicated as general acid catalysts, suggesting a concerted mechanism of leaving group expulsion.
Comparison of the transition state18 to the covalent intermediate15 and the apoenzyme19 of poxvirus TopIB highlights the mobility of the Lys167 side chain, which is located at the tip of a β-hairpin loop. Lys167 in the apoenzyme is located far away from its position near the scissile phosphodiester of the transition state and is therefore out of position to perform transesterification chemistry. The act of DNA binding elicits a major conformational switch that brings Lys167 (and also Arg130 and Tyr274) into the active site. After transesterification, Lys167 moves away from the leaving O5′ atom and becomes “parked” in the DNA minor groove via its contact with the O2 carbonyl atom of the +1T base (Fig. 1).
Traditional mutagenesis methods have illuminated structure-activity relations for the components of the vaccinia TopIB active site.6–10,13,16,17,20–22 In the case of Lys167, an alanine substitution causes a 10−4 decrement in the rate of transesterification, which is substantially revived when Lys167 is replaced by arginine9,16 or when the 5′-bridging oxygen of the scissile phosphodiester is replaced by sulfur.16,17 The residual ~102 difference in transesterification rates between the wild-type TopIB and the K167R mutant could reflect the predicted 2 log unit difference in the pKa values for the Lys and Arg side chains.23 Alternatively, there might be steric constraints on the larger arginine side chain that preclude full restoration of catalytic power. The genetically programmable protein “tool kit” offers no additional options to instructively modify this functional group at the TopIB active site.
A recent innovation in the analysis of TopIB mechanism is the use of “chemical mutagenesis” to introduce unnatural analogs are in lieu of a catalytic amino acid. As implemented by Hecht and colleagues24–27 for vaccinia TopIB and human TopIB, this entails in vitro translation of the TopIB protein, from an mRNA with a stop codon in lieu of the codon for the catalytic tyrosine nucleophile, in the presence of a chemically misacylated suppressor tRNA that incorporates a tyrosine analog. Studies of the effects of meta-substituted analogs of Tyr274 on vaccinia TopIB revealed that a meta-OH reduced the rate of single-turnover DNA cleavage 130-fold without affecting the rate of single-turnover religation. By contrast, meta-OCH3 and NO2 groups elicited only a 6-fold decrement in cleavage rate. It was proposed that the meta-OH uniquely antagonizes deprotonation of the para-OH nucleophile during the cleavage transesterification step.26
In the present study, we take a different approach to chemical mutagenesis, by producing recombinant vaccinia TopIB with cysteine in lieu of Lys167 and then reacting the K167C protein in vitro with haloalkyl compounds to install lysine-like analogs. The successful chemical reactivation of the K167C mutant provides insights to TopIB general acid catalysis.
EXPERIMENTAL PROCEDURES
Recombinant TopIB
Vaccinia virus TopIB was produced in E. coli with a C-terminal His6 tag. The expression plasmid pET21-TopoHis was constructed as follows. The TopIB open reading frame was amplified by PCR with an antisense primer designed to introduce a XhoI restriction site at the translation stop codon while converting it to serine codon. The PCR product was digested with NdeI (at the translation initiation codon) and XhoI and then inserted into a T7-based expression vector pET21b that had been digested with NdeI and XhoI. This maneuver fused the 314-aa TopIB polypeptide in-frame to 9-aa His-tag, SLEHHHHHH. Single and compound missense mutations K167A, K167C, C100A-C211A, C100A-C211A-K167A and C100A-C211A-K167C were introduced into the TopIB gene by two-stage PCR overlap extension. The inserts of the wild-type and mutant pET21-TopHis plasmids were sequenced to exclude the acquisition of unwanted changes during amplification and cloning. The pET21-TopoHis plasmids were transformed into Escherichia coli BL21(DE3). IPTG-induction of TopIB expression and purification of the TopIB proteins from soluble bacterial extracts by sequential Ni-agarose and phosphocellulose chromatography were performed as described previously.26 Protein concentrations were determined by using the BioRad dye reagent with BSA as the standard.
Chemical modification of vaccinia TopIB at Cys167
Reaction mixtures containing (per 20 μl) 100 mM Tris-HCl (pH 8.0), 3 μg of vaccinia TopIB, and 100 mM of freshly prepared alkylating agent as specified were incubated at room temperature in the dark for 2 h. The cysteine alkylation reactions were terminated by adding 2-mercaptoethanol to 200 mM final concentration. Mock-treated control reactions lacking the alkylating agent were performed in parallel. The treated and mock-treated TopIB proteins were prepared freshly for each experiment shown. The alkylating agents 2-bromoethylamine, 3-bromopropylamine, 2-bromoethanol, and 2-bromoacetate were purchased from Sigma-Aldrich.
Single-turnover DNA cleavage
An 18-mer CCCTT-containing DNA oligonucleotide was 5′-end-labeled by enzymatic phosphorylation in the presence of [γ32P]ATP and T4 polynucleotide kinase, then gel-purified and hybridized to a complementary 30-mer strand. Cleavage reaction mixtures containing (per 20 μl) 50 mM Tris-HCl (pH 7.5), 0.3 pmol 32P-labeled 18mer/30mer DNA, and 150 ng (4 pmol) of TopIB (mock-treated or treated with alkylating agent) were incubated at 37°C. The cleavage reactions were initiated by adding TopIB to pre-warmed reaction mixtures. The reactions were quenched at the times specified by adding SDS to 1% final concentration. The samples were analyzed by electrophoresis through a 10% polyacrylamide gel containing 0.1% SDS. Covalent complex formation was revealed by transfer of radiolabeled DNA to the TopIB protein. The extent of covalent complex formation was quantified by scanning the dried gel with a Fujifilm BAS-2500 imager.
RESULTS
Chemical mutagenesis of Lys167 by post-synthetic installation of a lysine analog
The chemical approach to introducing a lysine-like functional group at a specific site in a recombinant protein exploits the spontaneous reactivity of a cysteine side chain with 2-bromoethylamine to form the lysine analog γ-thialysine28–42 (Fig. 2B). The analog is structurally and chemically similar to lysine; the replacement of a methylene group with a sulfur increases side chain length by about 0.3 Å and lowers the pKa of Nζ by ~1.1 compared to lysine.32,39 The chemical modification is quite specific for cysteine at mildly alkaline pH.39 Optimal use of this method to illuminate structure-activity relations at an essential lysine in an enzyme active site entails the following steps (Fig. 2): (i) elimination of other cysteine residues in the protein by mutation to alanine; (ii) mutation of the lysine of interest to cysteine, which is now the only cysteine in the polypeptide; (iii) verification that the K-to-C change ablates enzyme activity; (iv) chemical aminoethylation of the cysteine to form γ-thialysine; and (v) interrogation of whether enzymatic activity is restored as a consequence of the cysteine aminoethylation maneuver. An advantage of this approach is that it can be applied to purified recombinant cysteine-containing enzyme and thereby yield greater quantities of chemically modified protein for functional studies than what can be obtained by in vitro translation.
Figure 2. Chemical mutagenesis of vaccinia virus TopIB Lys167.

The mutagenesis strategy is outlined. (A) Aliquots (5 μg) of the indicated phosphocellulose preparations of vaccinia TopIB were analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (kDa) of marker polypeptides are indicated on the left. (B) Chemical reaction of 2-bromoethylamine with cysteine to form γ-thialysine. (C) Chemical structures of other bromoalkyl compounds tested.
To execute this agenda, we first generated a cysteine-less C100A-C211A mutant of vaccinia TopIB. Then we replaced Lys167 with alanine or cysteine in the context of the wild-type and C100A-C211A proteins. The wild-type and mutated topoisomerases were produced in E. coli with His6 tags and purified from soluble bacterial extracts by Ni-agarose and phosphocellulose chromatography steps. SDS-PAGE analysis verified that the recombinant proteins were of similar high purity with respect to the TopIB polypeptide (Fig. 2A).
The purified proteins were reacted for 2 h at 22°C with 100 mM 2-bromoethylamine (2-BEA) in 100 mM Tris-HCl (pH 8.0). Control “mock-treated” protein samples were incubated in the same buffer without 2-BEA. The 2-BEA and mock reactions were quenched by adjustment to 200 mM 2-mercaptoethanol. The mock-treated and 2-BEA-modified proteins were then tested for DNA transesterification under single turnover conditions using a “suicide” substrate containing a single CCCTT site. The suicide substrate (Fig. 3) consisted of a 5′ 32P-labeled 18-mer scissile strand 5′-pCGTGTCGCCCTTATTCCC annealed to an unlabeled 30-mer strand. The cleavage transesterification reaction of mock-treated wild-type vaccinia TopIB and the cysteine-less C100A-C211A mutant resulted in covalent attachment of the 32P-labeled 12-mer 5′-pCGTGTCGCCCTTp to the enzyme and spontaneous dissociation of the unlabeled 6-mer 5′-OH leaving strand from the TopIB-DNA complex. The reaction yielded a discrete SDS-stable protein-DNA adduct that was resolved during SDS-PAGE from the free DNA that migrated near the electrophoretic front (Fig. 3, left panel, lanes 1 and 4). Quantification of the protein-DNA adduct and free DNA showed that 90% of the labeled DNA strand was transferred to the mock-treated wild-type and C100A-C211A topoisomerases during a 10 min reaction (Fig. 4). The extents of transesterification by the 2-BEA-treated wild-type and C100A-C211A proteins were the same as the mock-treated controls (Fig. 3, middle panel, lanes 1 and 4; Fig. 4).
Figure 3. Aminoalkylation of Cys167 restores DNA cleavage activity.
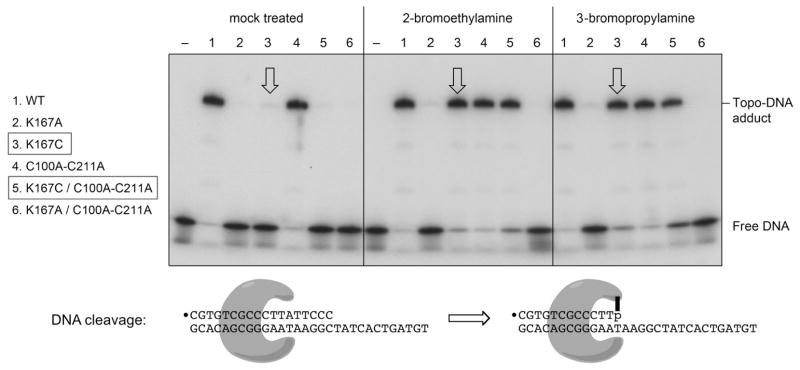
The single-turnover cleavage reaction is depicted at the bottom of the figure. The 18-mer/30-mer suicide cleavage substrate is shown with the 5′ 32P-label denoted by ●. The poxvirus TopIB forms a C-shaped protein clamp around the CCCTTp↓A recognition site. Transesterification results in the formation of a covalent 32P-labeled pCGTGTCGCCCTTp-(TopIB) adduct and dissociation of the unlabeled HOATTCCC leaving strand. Cleavage reaction mixtures (20 μl) containing 50 mM Tris-HCl (pH 7.5), 0.3 pmol 5′ 32P-labeled DNA substrate and 150 ng (4 pmol) of TopIB as specified (by numbers at left and above the gel lanes) were incubated at 37°C for 10 min. The reactions were quenched with 1% SDS. The products were analyzed by SDS-PAGE. An autoradiogram of the dried gel is shown. Free DNA migrated near the dye front. Covalent complex formation was revealed by transfer of radiolabeled DNA to the TopIB protein. The arrows overlying lanes 3 highlight the successful chemical rescue of K167C DNA cleavage activity by 2-bromoethylamine and 3-bromopropylamine.
Figure 4. Tests of chemical rescue of K167C by haloalkyl compounds.

Single-turnover cleavage by mock-treated and haloalkyl-treated TopIB proteins was performed as described in the legend to Fig. 3. The extents of covalent complex formation were quantified by scanning the dried SDS gels with a Fujifilm BAS-2500 imager. Each datum in the bar graph is the average of three independent experiments ± SEM. The alkylating agents were: 2-bromoethylamine (2-BEA), 3-bromoprolylamine (3-BPA), 2-bromoethanol (2-BE), and 2-bromoacetate (2-BA).
Changing Lys167 to either alanine or cysteine suppressed DNA cleavage, whether in the context of the otherwise wild-type TopIB (Fig. 3 left panel, lanes 2 and 3) or the C100A-C211A mutant (Fig. 3 left panel, lanes 2 and 3) (Fig. 4, mock). To gauge and compare the impact of the K167A and K167C mutations, we employed a longer duplex DNA substrate containing a single CCCTT site to assay transesterification under equilibrium conditions4 (Fig. 5). We refer to this DNA as an equilibrium substrate because the 22-mer leaving strand generated upon cleavage at CCCTT remains associated with the topoisomerase DNA complex via base pairing to the nonscissile strand. The K167A mutant displayed a slow approach to equilibrium over 4 h (Fig. 5) and achieved a much higher extent of covalent adduct formation at equilibrium (84%) than what one sees for wild-type TopIB.4 The increase in the cleavage equilibrium constant (Keq) attests that the K167A mutation has relatively less impact on the rate of cleavage (kcl) than on the rate of religation (krel). A nonlinear regression fit of the data to a pseudo-first order one-phase association in Prism (Fig. 5) yielded a rate constant (kobs) of 1.9 × 10−4 s−1 for the approach to equilibrium. Solving the equations kobs = kcl + krel and Kcl = kcl/krel yielded the component rate constants kcl =1.6 × 10−4 s−1 and krel = 3.2 × 10−5 s−1 for K167A, which are in the same range as those derived previously for K167A with a 60-bp equilibrium cleavage substrate.9 The K167C mutant displayed a faster approach to equilibrium (kobs = 1.3 × 10−3 s−1) and a lower endpoint cleavage value compared to K167A (Fig. 5). From these data, we derived component rate constants kcl = 4.3 × 10−4 s−1 and krel = 9 × 10−4 s−1 for K167C. Whereas cysteine in lieu of alanine had minor restorative effect (~2.5-fold) on forward cleavage, it elicited an ~30-fold enhancement of religation (though K167C remained 1000-fold slower at religation than wild-type vaccinia TopIB).
Figure 5. Kinetics of equilibrium DNA cleavage by K167A and K167C.
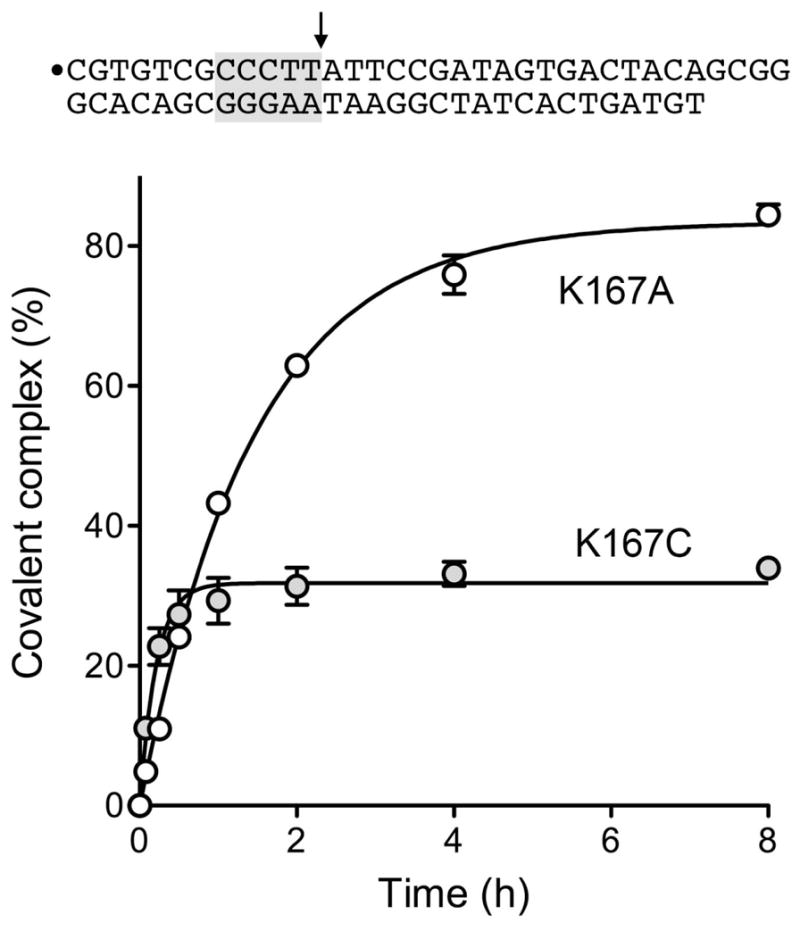
The 34-mer/30-mer equilibrium cleavage substrate is depicted at the top with the 5′ 32P-label denoted by ●, the CCCTT sequence shaded gray, and the cleavage site indicated by the arrow. Reaction mixtures containing (per 20 μl) 50 mM Tris-HCl (pH 7.5), 0.3 pmol of 32P-labeled DNA, and 150 ng (4 pmol) of purified K167A or K167C TopIB were incubated at 37°C. The reactions were initiated by adding TopIB to pre-warmed reaction mixtures. Aliquots (20 μl) were withdrawn at the times specified and the reactions were quenched immediately with SDS. The quenched reactions were treated with 10 μg of proteinase K for 60 min at 37°C, then mixed with an equal volume of 95% formamide/20 mM EDTA, and heat-denatured prior to electrophoresis through a 17% polyacrylamide gel containing 7 M urea in TBE (90 mM Tris-borate, 2.5 mM EDTA). The radiolabeled DNAs and the DNA-peptide adducts derived by proteolysis of the covalent TopIB-DNA complex were quantified by scanning the gel. Covalent adduct formation (expressed as the percent of the total labeled DNA) is plotted as a function of reaction time. Each datum is the average of three separate experiments ±SEM. Non-linear regression curve fits to a pseudo-first order exponential function (executed in Prism) are shown.
The instructive finding was that treatment with 2-BEA restored single-turnover DNA cleavage activity to the K167C and C100A-C211A-K167C proteins (Fig. 3, middle panel, lanes 3 and 5; Fig. 4). By contrast, 2-BEA treatment had no salutary effect on the K167A and C100A-C211A-K167A proteins (Fig. 3, middle panel, lanes 2 and 6; Fig. 4). Thus, the chemical rescue maneuver works for TopIB. This experiment told us also that: (i) we did not need to convert the two native cysteines of vaccinia TopIB (C100 and C211) to alanine in order to conduct a successful chemical mutagenesis study of Lys167; and (ii) we did not need to denature the protein with urea30–32,37 in order to chemically modify Cys167, a finding consistent with the location of amino acid 167 on a surface-exposed β-hairpin loop of the TopIB apoenzyme.19
Test of other cysteine alkylating agents for chemical rescue of DNA cleavage activity
Having achieved a successful chemical rescue with 2-BEA, we sought to modify Cys167 with other haloalkyl compounds (Fig. 2B) to create a series of non-native side chains and then evaluate which (if any) restore activity and to what extent they do so. We applied this strategy with 3-bromopropylamine (3-BPA), and found that: (i) 3-BPA treatment had no effect on the DNA cleavage activity of wild-type TopIB or C100A-C211A (Fig. 3, right panel, lanes 1 and 4); (ii) 3-BPA treatment rescued the DNA cleavage activity of the K167C and C100A-C211A-K167C topoisomerases (Fig. 3, right panel, lanes 3 and 5); and (iii) 3-BPA did not rescue the transesterification defects of the K167A and C100A-C211A-K167A proteins (Fig. 3, right panel, lanes 2 and 6) (Fig. 4). We conclude that TopIB catalysis can accommodate an unnatural γ-thiahomolysine side chain with a longer alkyl spacer between the main-chain and the amino functional group that serves as the general acid.
We also treated the Cys167-containing TopIB proteins with 2-bromoethanol (2-BE) and 2-bromoacetate (2-BA) (Fig. 2C), to install hydroxyl and carboxylate functional groups with lysine-like spacers. Whereas treatment with 2-BE or 2-BA did not adversely affect the extent of DNA cleavage by wild-type TopIB and C100A-C211A, it also failed to revive the activity of the K167A and C100A-C211A-K167A proteins (Fig. 4). These results attest to the critical role of the 167 alkylamine moiety in TopIB catalysis.
Effects of γ-thialysine and γ-thiahomolysine on the rate of DNA cleavage
We conducted a kinetic analysis of the suicide cleavage reactions of the 2-BEA- and 3-BPA-modified K167C proteins in order to gauge the impact of the lysine analogs on TopIB catalysis. Mock-treated K167C and wild-type TopIB served as controls. The TopIB proteins were added in 13-fold molar excess over the 32P-labeled DNA substrate. The pertinent feature of the suicide cleavage reaction is that the unlabeled 6-mer 5′-OH leaving strand ATTCCC dissociates spontaneously from the covalent TopIB-DNA complex. Loss of the leaving strand drives the reaction toward the covalent state, so that the reaction can be treated kinetically as a first-order unidirectional process. As expected, the K167C protein was virtually unreactive within the 5 min time frame of the experiment (Fig. 6A; <1% DNA cleaved). The wild-type TopIB cleavage reaction attained 93% of the endpoint value at 5 s, the earliest time tested, consistent with a kcl of ~0.4–0.5 s−1 as reported previously.4 The 2-BEA and 3-BPA modified K167C proteins displayed a steady increase in covalent adduct formation and attained extents of DNA cleavage at 5 min (87% and 84%, respectively) similar to that of wild-type TopIB (90%) (Fig. 6A). Fitting the data in Fig. 6A to a single exponential yielded kcl values of 0.031 s−1 and 0.013 s−1 for the 2-BEA and 3-BPA modified K167C proteins.
Figure 6. Kinetics of single-turnover cleavage by 2-BEA- and 3-BPA-modified TopIB.
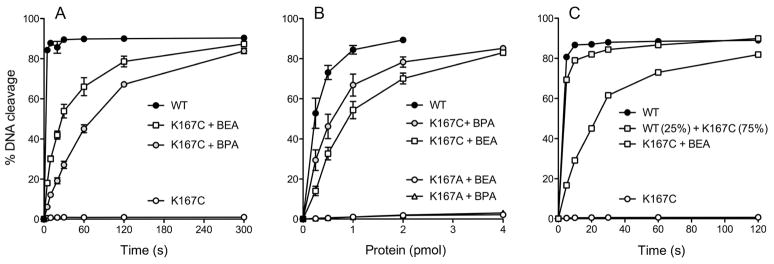
(A) Kinetic analysis of single turnover cleavage of the 18-mer/30-mer substrate was performed as described under Experimental Procedures. The percentage of input DNA cleaved was plotted as a function of reaction time for the indicated TopIB preparations. Each datum in the graph is the average of three independent time course experiments ± SEM. The cleavage rate constants were derived by nonlinear regression fitting of the data to an exponential one-phase association in Prism. (B) Cleavage reactions mixtures containing 0.3 pmol 5′ 32P-labeled 18-mer/30-mer DNA substrate and increasing amounts of TopIB as specified were incubated for 15 min at 37°C. The percentage of input DNA cleaved was plotted as a function of input TopIB. Each datum in the graph is the average of three independent protein titration experiments ± SEM. (C) The kinetics of single-turnover cleavage of 0.3 pmol 5′ 32P-labeled 18-mer/30-mer DNA substrate by 150 ng (4 pmol) of either pure wild-type TopIB (WT), pure K167C, 2-BEA-treated K167C, or a 25%/75% mixture of WT and K167C.
Having shown that the yield of covalent adduct by the chemically modified K167C proteins under single-turnover conditions was comparable to wild-type TopIB, we could gauge the extent of Cys167 chemical modification by performing an enzyme titration in the suicide cleavage assay. Fig. 6B shows a plot of the yields of covalent TopIB-DNA adduct in a 15 min DNA cleavage reaction with 0.3 pmol of 32P-labeled DNA substrate versus the amounts of input wild-type, and 2-BEA- and 3-BPA-treated K167C and K167A proteins. As expected, the 2-BEA- and 3-BPA-treated K167A proteins were inactive. By contrast, the wild-type TopIB generated near-stoichiometric yields of covalent adduct, i.e., 0.25 pmol of input TopIB, the lowest level tested, formed 0.15 pmol of covalent protein-DNA complex (Fig. 6B). The titration profiles of the 2-BEA- and 3-BPA-treated K167C proteins were shifted to right compared to wild-type enzyme. By comparing the slopes of titration curves, we estimated that half of the 3-BPA treated enzyme was catalytically active (i.e., contained γ-thiahomolysine in lieu of Cys167) and one-fourth of the 2-BEA treated enzyme was catalytically active (i.e., contained γ-thialysine at position 167).
With this information in hand, we queried whether the presence of significant residual K167C might have affected the kinetics of single-turnover cleavage by the chemically re-activated 2-BEA-modified TopIB containing γ-thialysine. To model this scenario, we pre-mixed wild-type TopIB and the K167C mutant (to achieve 25% wild-type and 75% K167C, thus mimicking the composition of the 2-BEA treated TopIB). The kinetics of single-turnover DNA cleavage by the WT/K167C mixture are shown in Fig. 6C. The presence of the K167C protein had no effect on the cleavage reaction endpoint, but resulted in a slight reduction of the extent of cleavage at the 5 s time point vis à vis pure wild-type TopIB. The apparent kcl values of the wild-type TopIB and WT/K167C mixtures were 0.44 s−1 and 0.32 s−1, respectively (Fig. 6C). [The apparent kcl of a 2-BEA treated K167C preparation assayed in parallel was 0.043 s−1 (Fig. 6C), which is close the to the value of 0.031 s−1 derived from the experiments in Fig. 6A.] We regard the apparent kcl of the WT/K167C mixture as the upper bound for full restoration of activity, and the kcl of the pure K167C mutant (derived from the experiment in Fig. 5) as the baseline activity against which the activities of the treated K167C preparations (comprising mixtures of chemically modified K167C and residual unmodified K167C) can be compared. Accordingly (using the kcl data from Fig. 6A), the conversion of Cys167 to γ-thialysine elicited a ~70-fold enhancement in the rate of DNA cleavage and thereby restored the cleavage rate to within a factor of 10 of the Lys167-containing enzyme. Modification of the Cys167 to γ-thiahomolysine increased kcl by ~30-fold. The finding that the rate of DNA cleavage by the 2-BEA-modified K167C protein was only 2.4-fold faster than that of the 3-BPA-modified K167C signifies that TopIB is relatively tolerant of the extra methylene substituent of the homolysine side chain during the cleavage transesterification step. A salient point is that γ-thialysine (kcl = 0.031 s−1) and γ-thiahomolysine (kcl = 0.013 s−1) at position 167 promote faster catalysis of DNA cleavage than does arginine: the reported9 K167R kcl value being 0.0067 s−1.
Effects of γ-thialysine and γ-thiahomolysine on the rate of DNA religation
The effects of the lysine-167 analogs on the religation transesterification reaction were studied under single-turnover conditions by assaying the ability of preformed suicide intermediates to transfer the covalently held 5′ 32P-labeled 12-mer strand to a 5′ OH-terminated 18-mer strand to generate a 30-mer product (Fig. 7). After forming the suicide intermediate on the 18-mer/30-mer DNA substrate, the religation reaction was initiated by adding a 50-fold molar excess of the 18-mer DNA acceptor strand. The sequence of the added 18-mer is fully complementary to the 5′ single-stranded tail of the suicide intermediate. The ionic strength was adjusted simultaneously to 0.5 M NaCl to promote dissociation of the topoisomerase after strand ligation and prevent recleavage of the 32P-labeled 30-mer strand transfer product. Aliquots were withdrawn immediately prior to the addition of 18-mer and NaCl (defined as time 0) and at various times afterward. The extent of religation at each time point was quantified as the fraction of the 32P-labeled DNA present as covalent adduct at time zero that was converted to 30-mer strand transfer product (Fig. 7). The religation reaction of wild-type TopIB was effectively complete by 5 s, the earliest time analyzed, in keeping with prior measurements of krel of 1.2 to 1.4 s−1 for vaccinia TopIB.9,43 The religation profile of the 2-BEA-modified TopIB(K167C)-DNA intermediate was indistinguishable from wild-type TopIB, within the limits of the assay (i.e., krel was ≥0.4 s−1), signifying that the installation of γ-thialysine in lieu of Lys167 had less than a 3- to 4-fold effect on the rate of religation. Taking the krel value of untreated K167C (from the experiment in Fig, 5) as a baseline, the conversion of Cys167 to γ-thialysine increased the rate of religation by at least 400-fold. The religation profile of the 2-BPA-modified TopIB(K167C)-DNA intermediate was detectably slowed (krel 0.22 s−1), indicating that the longer γ-thiahomolysine side chain exerted only on modest (at least 2-fold, but likely less than 6-fold) effect on the rate of the religation transesterification reaction. Note that γ-thialysine and γ-thiahomolysine at position 167 are at least an order of magnitude faster in catalysis of religation than is arginine: the K167R krel value9 being 0.012 to 0.022 s−1.
Figure 7. Effects lysine analogs on DNA religation.

Cleavage reaction mixtures containing (per 20 μl) 50 mM Tris-HCl (pH 7.5), 0.3 pmol 5′ 32P-labeled 18-mer/30-mer DNA substrate, and 150 ng (4 pmol) of TopIB as specified were incubated at 37°C for 5 min to form the suicide intermediate depicted at the top of the figure. Religation was initiated by the simultaneous addition of NaCl to 0.5 M and a 5′-OH 18-mer acceptor strand d(ATTCCGATAGTGACTACA) to a concentration of 15 pmol/22 μl (a 50-fold molar excess over the input DNA substrate). Aliquots (20 μl) were withdrawn at the times specified and quenched immediately with 1% SDS. A time 0 sample was withdrawn before the addition of the acceptor strand. The samples were digested for 1 h at 37°C with 10 μg of proteinase K, then mixed with an equal volume of 95% formamide/20 mM EDTA, heat-denatured, and analyzed by electrophoresis through a 17% polyacrylamide gel containing 7 M urea in TBE. The radiolabeled DNAs and DNA-peptide adducts (derived by proteolysis of the covalent TopIB-DNA complex) were quantified by scanning the gel. Religation of the covalently bound 12-mer strand to the 18-mer acceptor DNA yielded a 5′ 32P-labeled 30-mer strand transfer product. The extents of religation (expressed as the percent of the covalent intermediate converted into 30-mer) are plotted as a function of reaction time for each TopIB protein. Each datum for the alkylated K167C proteins is the average of three separate experiments ±SEM. Each datum for wild-type TopIB is the average of five experiments ±SEM.
Effects of γ-thialysine and γ-thiahomolysine on supercoil relaxation
The wild-type, K167C and aminoalkylated K167C proteins were also assayed for their rates of supercoil relaxation (Fig. 8). The K167C protein failed to relax the supercoiled DNA within the 10 min time frame of the experiment, as expected from its feeble DNA cleavage and religation kinetics. The wild-type TopIB relaxed all of the input DNA within 1 to 2 min. The 2-BEA treated K167C relaxed the supercoils at about one-tenth the rate of wild-type TopIB and the 2-BPA-treated K167C relaxed at about one-twentieth the wild-type rate. After correcting for the fractions of alkylated enzymes in the 2-BEA and 3-BPA-treated K167C preparations, we estimate that γ-thialysine and γ-thiahomolysine promote supercoil release one-third and one-tenth as well as the native lysine. The hierarchy of alkylation effects on relaxation was concordant with the effects on single-turnover DNA cleavage.
Figure 8. Aminoalkylation of Cys167 restores supercoil relaxation activity.
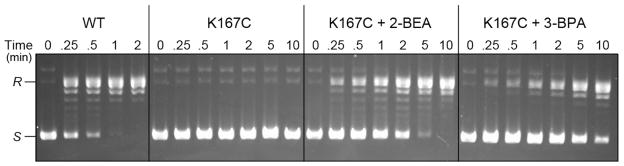
Relaxation reaction mixtures containing (per 20 μl) 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 2.5 mM EDTA, 300 ng of supercoiled pUC19 DNA, and 2.25 ng of WT, K167C, 2-BEA-treated K167C or 3-BPA-treated K167C TopIB as specified were incubated at 37°C. The reactions were initiated by the addition of enzyme. Aliquots (20 μl) were withdrawn at the times specified and quenched immediately with SDS. The time 0 samples were taken prior to addition of enzyme. The reaction products were analyzed by 1% agarose horizontal gel electrophoresis. The gels were stained with ethidium bromide and DNA was visualized under UV illumination.
DISCUSSION
Here we successfully applied post-synthetic chemical modification of a defective K167C mutant TopIB to show that lysine analogs γ-thialysine and γ-thiahomolysine rescue the DNA cleavage, DNA religation, and DNA supercoil relaxation activities of vaccinia TopIB. The chemically installed lysine analogs are more effective than a genetically programmed conservative arginine substitution9 in promoting the DNA transesterification steps. The modest rate differential between γ-thialysine and γ-thiahomolysine attests to the ability of TopIB to accommodate a longer alkylamino side chain during assembly of the active site and adoption of a transition-state in which the lysine or homolysine side chain coordinates the O5′ leaving group. The functional evidence for Lys167 catalysis of leaving group expulsion is well established.16,17 The incomplete restoration of function by arginine,9 and the failure of 2-bromoethanol to revive cleavage activity of K167C, are consistent with the model of general acid catalysis, whereby the arginine guanidinium (pKa 12.5) is less amenable to proton donation and an ε-OH analog of lysine (the product of cysteine alkylation by 2-bromoethanol, with a pKa ~15) is much worse in this respect.
It is not immediately obvious why γ-thialysine, with a pKa 1.1 unit lower than lysine,39 would slow the single-turnover cleavage rate by an order of magnitude. Assuming that Lys167 has a “consensus” pKa of 10.5, then γ-thialysine-167 would have a pKa of 9.4 and Nζ of thialysine would still be fully protonated in the ground state at pH 7.5. However, it is possible that the pKa of Lys167 is perturbed downward, either in the apoenzyme or the non-covalent TopIB·DNA complex, so that a further reduction by 1.1 units for γ-thialysine-167 could result in a significant fraction of the amino moiety being unprotonated and hence unavailable for general acid catalysis of DNA cleavage. Alternatively, the reduced cleavage activity of the γ-thialysine-167 TopIB could be caused by unfavorable contacts or conformational effects of the larger Sγ atom, independent of a pKa effect.
Lys167 in its unprotonated state is presumed, by microscopic reversibility, to function (along with Arg130) in abstracting a proton from the attacking 5′-OH DNA strand during the religation transesterification reaction. This scenario is consistent with the apparently benign effect of γ-thialysine on the rate of religation. The K167A mutation slows the rate religation by more than four orders of magnitude; the present observation that the K167C enzyme is 3-fold faster at religation than K167A suggests that the cysteine sulfur can function, albeit quite poorly, in lieu of the lysine or γ-thialysine nitrogen as a proton acceptor during religation. The pKa of cysteine in proteins can vary widely around an average pKa value of 6.8.23 If the average value pertains to Cys167 in TopIB, then it would be largely unprotonated under our assay conditions and potentially suitable for function as a general base. It is conceivable that the shorter distance of the cysteine sulfur atom from the main-chain (compared to the nitrogen of lysine or homolysine) limits its access to the O5′ atom in the TopIB active site (Fig. 1) and thereby accounts for the feeble activity of Cys167 in DNA religation compared to Lys167.
Acknowledgments
This work was supported by NIH grant GM46330.
References
- 1.Shuman S, Prescott J. Specific DNA cleavage and binding by vaccinia virus DNA topoisomerase I. J Biol Chem. 1990;265:17826–17836. [PubMed] [Google Scholar]
- 2.Shuman S. Site-specific interaction of vaccinia virus DNA topoisomerase I with duplex DNA: minimal DNA substrate for strand cleavage in vitro. J Biol Chem. 1991;266:11372–11379. [PubMed] [Google Scholar]
- 3.Shuman S, Turner J. Site-specific interaction of vaccinia virus topoisomerase I with base and sugar moieties in duplex DNA. J Biol Chem. 1993;268:18943–18950. [PubMed] [Google Scholar]
- 4.Tian L, Sayer JM, Jerina DM, Shuman S. Individual nucleotide bases, not base pairs, are critical for triggering site-specific DNA cleavage by vaccinia topoisomerase. J Biol Chem. 2004;279:39718–39726. doi: 10.1074/jbc.M407376200. [DOI] [PubMed] [Google Scholar]
- 5.Minkah N, Hwang Y, Perry K, Van Duyne GD, Hendrickson R, Lefkowitz EJ, Hannenhalli S, Bushman FD. Variola virus topoisomerase: DNA cleavage specificity and distribution of sites in poxvirus genomes. Virology. 2007;365:60–69. doi: 10.1016/j.virol.2007.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shuman S, Kane EM, Morham SG. Mapping the active site tyrosine of vaccinia virus DNA topoisomerase I. Proc Natl Acad Sci USA. 1989;86:9793–9797. doi: 10.1073/pnas.86.24.9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morham SG, Shuman S. Phenotypic selection and characterization of mutant alleles of a eukaryotic DNA topoisomerase I. Genes Dev. 1990;4:515–524. doi: 10.1101/gad.4.4.515. [DOI] [PubMed] [Google Scholar]
- 8.Cheng C, Wang LK, Sekiguchi J, Shuman S. Mutational analysis of 39 residues of vaccinia DNA topoisomerase identifies Lys-220, Arg-223, and Asn-228 as important for covalent catalysis. J Biol Chem. 1997;272:8263–8269. doi: 10.1074/jbc.272.13.8263. [DOI] [PubMed] [Google Scholar]
- 9.Wittschieben J, Shuman S. Mechanism of DNA transesterification by vaccinia topoisomerase: catalytic contributions of essential residues Arg130, Gly132, Tyr136, and Lys167. Nucleic Acids Res. 1997;25:3001–3008. doi: 10.1093/nar/25.15.3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petersen BØ, Shuman S. Histidine-265 is important for covalent catalysis by vaccinia topoisomerase and is conserved in all eukaryotic type I enzymes. J Biol Chem. 1997;272:3891–3896. doi: 10.1074/jbc.272.7.3891. [DOI] [PubMed] [Google Scholar]
- 11.Stivers JT, Jagadeesh GJ, Nawrot B, Stec WJ, Shuman S. Stereochemical outcome and kinetic effects of Rp and Sp phosphorothioate substitutions at the cleavage site of vaccinia type I DNA topoisomerase. Biochemistry. 2000;39:5561–5572. doi: 10.1021/bi992429c. [DOI] [PubMed] [Google Scholar]
- 12.Tian L, Claeboe CD, Hecht SM, Shuman S. Guarding the genome: electrostatic repulsion of water by DNA suppresses a potent nuclease activity of topoisomerase IB. Mol Cell. 2003;12:199–208. doi: 10.1016/s1097-2765(03)00263-6. [DOI] [PubMed] [Google Scholar]
- 13.Tian L, Claeboe CD, Hecht SM, Shuman S. Mechanistic plasticity of DNA topoisomerase IB: phosphate electrostatics dictate the need for a catalytic arginine. Structure. 2005;13:513–520. doi: 10.1016/j.str.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 14.Nagarajan R, Kwon K, Nawrot B, Stec WJ, Stivers JT. Catalytic interaction of topoisomerase IB. Biochemistry. 2005;44:11476–11485. doi: 10.1021/bi050796k. [DOI] [PubMed] [Google Scholar]
- 15.Perry K, Hwang Y, Bushman FD, Van Duyne GD. Structural basis for specificity in the poxvirus topoisomerase. Mol Cell. 2006;23:343–354. doi: 10.1016/j.molcel.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 16.Krogh BO, Shuman S. Catalytic mechanism of DNA topoisomerase IB. Molecular Cell. 2000;5:1035–1041. doi: 10.1016/s1097-2765(00)80268-3. [DOI] [PubMed] [Google Scholar]
- 17.Krogh BO, Shuman S. Proton relay mechanism of general acid catalysis by DNA topoisomerase IB. J Biol Chem. 2002;277:5711–5714. doi: 10.1074/jbc.C100681200. [DOI] [PubMed] [Google Scholar]
- 18.Perry K, Hwang Y, Bushman FD, Van Duyne GD. Insights from the structure of a smallpox virus topoisomerase-DNA transition state mimetic. Structure. 2010;18:127–137. doi: 10.1016/j.str.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng C, Kussie P, Pavletich N, Shuman S. Conservation of structure and mechanism between eukaryotic topoisomerase I and site-specific recombinases. Cell. 1998;92:841–850. doi: 10.1016/s0092-8674(00)81411-7. [DOI] [PubMed] [Google Scholar]
- 20.Shuman S. Polynucleotide ligase activity of eukaryotic topoisomerase I. Mol Cell. 1998;1:741–748. doi: 10.1016/s1097-2765(00)80073-8. [DOI] [PubMed] [Google Scholar]
- 21.Wittschieben J, Petersen BØ, Shuman S. Replacement of the active site tyrosine of vaccinia DNA topoisomerase by glutamate, cysteine, or histidine converts the enzyme into a site-specific endonuclease. Nucleic Acids Res. 1998;26:490–496. doi: 10.1093/nar/26.2.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang Y, Minkah N, Perry K, Van Duyne GD, Bushman FD. Regulation of catalysis by the smallpox virus topoisomerase. J Biol Chem. 2006;281:38052–38060. doi: 10.1074/jbc.M608858200. [DOI] [PubMed] [Google Scholar]
- 23.Pace CN, Grimsley GR, Scholtz JM. Protein ionizable groups: pK values and their contribution to protein stability and solubility. J Biol Chem. 2009;284:13285–13289. doi: 10.1074/jbc.R800080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao R, Zhang Y, Choudhury AK, Dedkova LM, Hecht SM. Analogues of vaccinia virus DNA topoisomerase I modified at the active site tyrosine. J Am Chem Soc. 2005;127:3321–3331. doi: 10.1021/ja044182z. [DOI] [PubMed] [Google Scholar]
- 25.Gao R, Zhang Y, Dedkova LM, Choudhury AK, Rahier NJ, Hecht SM. Effects of modification of the active site tyrosine of human DNA topoisomerase I. Biochemistry. 2006;45:8402–8410. doi: 10.1021/bi0605179. [DOI] [PubMed] [Google Scholar]
- 26.Yakovleva L, Chen S, Hecht SM, Shuman S. Chemical and traditional mutagenesis of vaccinia DNA topoisomerase provide insights to cleavage site recognition and transesterification chemistry. J Biol Chem. 2008;283:16093–16103. doi: 10.1074/jbc.M801595200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen S, Zhang Y, Hecht SM. p-Thiophenylalanine-induced DNA cleavage and religation activity of a modified vaccinia topoisomerase IB. Biochemistry. 2011;50:9340–9351. doi: 10.1021/bi201291p. [DOI] [PubMed] [Google Scholar]
- 28.Smith HB, Hartman FC. Restoration of activity to catalytically deficient mutants of ribulosebisphosphate carboxylase/oxygenase by aminoethylation. J Biol Chem. 1988;263:4921–4925. [PubMed] [Google Scholar]
- 29.Planas A, Kirsch JF. Reengineering the catalytic lysine of aspartate aminotransferase by chemical elaboration of a genetically introduced cysteine. Biochemistry. 1991;30:8268–8276. doi: 10.1021/bi00247a023. [DOI] [PubMed] [Google Scholar]
- 30.Smiley JA, Jones ME. A unique catalytic and inhibitor-binding role for Lys93 or yeast orotidylate decarboxylase. Biochemistry. 1992;31:12162–12168. doi: 10.1021/bi00163a027. [DOI] [PubMed] [Google Scholar]
- 31.Kim DW, Yoshimura T, Esaki N, Satoh E, Soda K. Studies of the active-site lysyl residue of thermostable aspartate aminotransferase: combination of site-directed mutagenesis and chemical modification. J Biochem. 1994;115:93–97. doi: 10.1093/oxfordjournals.jbchem.a124311. [DOI] [PubMed] [Google Scholar]
- 32.Gloss LM, Kirsch JF. Decreasing the basicity of the active site base, Lys-258, of Escherichia coli aspartate aminotransferase by replacement with γ-thialysine. Biochemistry. 1995;34:3990–3998. doi: 10.1021/bi00012a017. [DOI] [PubMed] [Google Scholar]
- 33.Gloss LM, Kirsch JF. Examining the structural and chemical flexibility of the active site base, Lys-258, of Escherichia coli aspartate aminotransferase by replacement with unnatural amino acids. Biochemistry. 1995;34:12323–12332. doi: 10.1021/bi00038a028. [DOI] [PubMed] [Google Scholar]
- 34.Highbarger LA, Gertl JA, Kenyon GL. Mechanism of the reaction catalyzed by acetoacetate decarboxylase: importance of lysine 116 is determining the pKa of active site lysine 115. Biochemistry. 1996;35:41–46. doi: 10.1021/bi9518306. [DOI] [PubMed] [Google Scholar]
- 35.Paetzel M, Strynadka NCJ, Tschantz WR, Casareno R, Bullinger PR, Dalbey RE. Use of site-directed chemical modification to study an essential lysine in Escherichia coli leader peptidase. J Biol Chem. 1997;272:9994–10003. doi: 10.1074/jbc.272.15.9994. [DOI] [PubMed] [Google Scholar]
- 36.Nash HM, Lu R, Lane WS, Verdine GL. The critical active-site amino of the human 8-oxoguanine DNA glycosylase, hOgg1: direct identification, ablation and chemical reconstitution. Chem Biol. 1997;4:693–702. doi: 10.1016/s1074-5521(97)90225-8. [DOI] [PubMed] [Google Scholar]
- 37.Bochar DA, Tabernero L, Stauffacher CV, Rodwell VW. Aminoethylcysteine can replace the function of the essential active site lysine of Pseudomonas mevalonii 3′-hydroxy-3-methylglutaryl coenzyme A reductase. Biochemistry. 1999;38:8879–8883. doi: 10.1021/bi9902687. [DOI] [PubMed] [Google Scholar]
- 38.Hopkins CE, O’Connor PB, Allen KN, Costello CE, Tolan DR. Chemical-modification rescue assessed by mass spectrometry demonstrates that γ-thialysine yields the same activity as lysine in aldolase. Prot Sci. 2002;11:1591–1599. doi: 10.1110/ps.3900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hopkins CE, Hernandez G, Lee JP, Tolan DR. Aminoethylation in model peptides reveals conditions for maximizing thiol specificity. Arch Biochem Biophys. 2005;443:1–10. doi: 10.1016/j.abb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 40.Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng R, Dam TK, Brewer CF, Blanchard JS. Active site residues in Mycobacterium tuberculosis pantothenate synthetase required in the formation and stabilization of the adenylate intermediate. Biochemistry. 2004;43:7171–7178. doi: 10.1021/bi049676n. [DOI] [PubMed] [Google Scholar]
- 42.Woodyer R, Wheatley JL, Relyea HA, Rimkus S, van der Donk WA. Site-directed mutagenesis of active site residues of phosphite dehydrogenase. Biochemistry. 2005;44:4765–4774. doi: 10.1021/bi047868c. [DOI] [PubMed] [Google Scholar]
- 43.Nagarajan R, Stivers JT. Major groove interactions of vaccinia Topo I provide specificity by optimally positioning the covalent phosphotyrosine linkage. Biochemistry. 2006;45:5775–5782. doi: 10.1021/bi060133i. [DOI] [PMC free article] [PubMed] [Google Scholar]