

Abstract
MicroRNAs (miRNAs) are evolutionary conserved noncoding molecules that regulate gene expression. They influence a number of diverse biological functions, such as development and differentiation. However, their dysregulation has been shown to be associated with disease states, such as cancer. Genes and pathways regulating their biogenesis remain unknown and are highly sought after. For this purpose, we have validated a multiplexed high-content assay strategy to screen for such modulators. Here, we describe its implementation that makes use of a cell-based gain-of-function reporter assay monitoring enhanced green fluorescent protein expression under the control of miRNA 21 (miR-21); combined with measures of both cell metabolic activities through the use of Alamar Blue and cell death through imaged Hoechst-stained nuclei. The strategy was validated using a panel of known genes and enabled us to successfully progress to and complete an arrayed genome-wide short interfering RNA (siRNA) screen against the Ambion Silencer Select v4.0 library containing 64,755 siRNA duplexes covering 21,565 genes. We applied a high-stringency hit analysis method, referred to as the Bhinder–Djaballah analysis method, leading to the nomination of 1,273 genes as candidate inhibitors of the miR-21 biogenesis pathway; after several iterations eliminating those genes with only one active duplex and those enriched in seed sequence mediated off-target effects. Biological classifications revealed four major control junctions among them vesicular transport via clathrin-mediated endocytosis. Altogether, our screen has uncovered a number of novel candidate regulators that are potentially good druggable targets allowing for the discovery and development of small molecules for regulating miRNA function.
Introduction
MicroRNAs (miRNAs) are small, noncoding RNA molecules composed of 20–23 nucleotide (nt) length that regulate gene expression at the mRNA post-transcriptional level resulting in translational repression or degradation. Discovered in 1993 from nematode Caenorhabditis elegans, Lee and coworkers found that lin-4, an important gene for postembryonic development of C. elegans does not code for a protein; instead, it is transcribed into a 22-nt RNA molecule.1 Seven years later, a second miRNA, let-7 also emerged as a heterochronic switch gene from C. elegans genetic screening2 and the discovery of many more miRNAs in their biological roles has continued thereafter. A major milestone in 2002 established association of miRNAs with cancer when Calin and coworkers demonstrated deletion and downregulation of miR-15 and miR-16 in lymphocytic leukemia.3 miRNAs are now implicated in a number of diverse biological functions, including cell proliferation, development, differentiation, and metabolism.
According to the current biogenesis model,4,5 miRNAs are transcribed by RNA polymerase II or RNA polymerase III inside the nucleus into primary miRNA (pri-miRNA). The pri-miRNA undergoes endonucleolytic cleavage by the microprocessor complex6 composed of DROSHA7,8 and DGCR8.9 The resulting 60–110-nt long precursor-miRNA (pre-miRNA) is exported into the cytoplasm by Exportin-5 (XPO5) in complex with Ran-GTP, where it gets loaded onto the RNA-induced silencing complex (RISC).10 RISC is a multiprotein complex composed of DICER, tar-RNA binding protein (TRBP), and PIWI family of argonaute proteins. TRBP initiates conformational changes in DICER;11 activating it for cleavage of pre-miRNA into 20–22 nt long mature miRNA composed of a guide strand and its complementary passenger strand. The mature miRNA are loaded into RISC along with EIF2C2 to target mRNA.12 The complementary seed region of the miRNA binds to either the upstream 3′ untranslated region (UTR) or poly A tail of the mRNA sequence resulting in translational repression or degradation.
Gene regulation by miRNA is essential for normal development and strong evidence supports its role in underlying disease states, including cancer.13,14 Although the mode of action has been extensively studied in the past decade, we have only begun to understand how genes and pathways regulate miRNA biogenesis and expression. Recent research has introduced several new proteins that interact with the miRNA biogenesis, for example, transforming growth factor beta (TGF-β) and bone morphogenetic protein (BMP) regulate DROSHA-mediated pri-miRNA processing of miR-21 through binding of SMAD proteins.15 In another study, Fukuda and coworkers identified DEAD-box RNA helicase subunits, p68 and p72, in the pri-miRNA processing.16 Briefly, miRNAs are now recognized as important regulators of gene expression of which dysregulation leads to dramatic consequences and emerging studies are now driven toward understanding their biogenesis.
RNA interference (RNAi) is emerging as a powerful tool for functional genomics analysis to identify genes involved in diverse cellular processes. In particular, synthetic 20–25-nt double-stranded RNA molecules or short interfering RNA duplexes (siRNA) are commonly employed in focused and genome-wide screening applications to uncover how genes are involved in biological processes. Recently, a genome-wide siRNA screen identified RBMX to be essential in the repair of DNA double-stranded breaks.17 Additionally, a kinome-focused siRNA screen identified PDGFR as a sensitizer of pancreatic cancer cells.18 As such, we made use of a recently described reporter cell line of miRNA activity for chemical screening; adapting the multiplexing potential of the assay to search for modulator genes using siRNA technology to interrogate and dissect the miRNA biogenesis pathway by loss-of-function analysis.19 Our strategy makes use of a multiplexed-approach to score for genes that modulate miR-21 biogenesis by monitoring enhanced green fluorescent protein (EGFP) expression, while simultaneously assessing cell death through imaged Hoechst-stained nuclei (NUCL), and the cellular metabolic activity through the use of the Alamar Blue (AB).
In this report, we first describe the implementation and validation of our multiplexed high-content screening strategy against a panel of known and reported genes of the miRNA biogenesis pathway. Next, we report on the execution and performance of our arrayed genome-wide siRNA screen against the Ambion Silencer Select v4.0 library containing 64,755 siRNA duplexes covering 21,565 genes. We applied a high-stringency hit nomination method encompassing criteria of at least two active duplexes per gene and filtered for potential off-target effects (OTEs), referred to as the Bhinder–Djaballah analysis (BDA) method,20 and leading to the identification of 1,273 inhibitor gene candidates of the miR-21 biogenesis pathway; the knockdown of which resulted in enhancement of the EGFP fluorescence signal output. Here, we present the results of our screen together with the application of the BDA method to identify active siRNA duplexes and nominate their gene as potential modulators of the miRNA biogenesis pathway in general, and discuss how these newly identified genes and pathways would provide a better understanding of miRNA production and regulation. We also provide a list of 121 high-confidence gene candidates.
Material and Methods
Cell Culture and Materials
The reporter cell line harboring EGFP under miRNA regulation was generated as previously reported.19 In brief, HeLaS3 cells were seeded into a 24-well plate at 200,000 cells per well in 500 μL of growth media containing Dulbecco's modified Eagle's medium (DMEM), high glucose with L-glutamine, D-glucose, and sodium pyruvate supplemented with 10% heat-inactivated fetal bovine serum (FBS). After 24 h, pcDNA/TO/EGFPmiR21 (Addgene, Cambridge, MA) was diluted with Opti-MEM media to 0.2 μg/well; in which Lipofectamine 2000 transfection solution was added at a final concentration of 0.4 μL/well, and incubated at room temperature for 15 min to promote transfection reagent complex formation. Next, 100 μL of transfection reagent complex was added into the wells and incubated for an additional 24 h. Cells were passaged, and then selected with Zeocin at a concentration of 200 μg/mL for 6 days. Zeocin-resistant cells were harvested and cell stocks were stored at −170°C.19 Cells were grown at 37°C and 5% CO2 in complete growth media containing DMEM, high glucose with L-glutamine, D-glucose, and sodium pyruvate supplemented with 10% heat-inactivated FBS, and 200 μg/mL of Zeocin. All cell culture supplies were from Life Technologies (Carlsbad, CA) and Sigma-Aldrich (St Louis, MO).
Liquid Dispensing and Automation System
Several liquid dispensing devices were used throughout this study. siRNA duplexes were plated and transferred using a 384 stainless steel head with disposable low-volume polypropylene tips on a PP-384-M Personal Pipettor (Apricot Designs, Monrovia, CA). The addition of cell suspensions and growth media was performed using the Multidrop 384 (Thermo Fisher Scientific, Waltham, MA) and AB addition was performed using the FlexDrop IV (Perkin Elmer, Waltham, MA). Cell fixation and staining was performed using the ELx405 automated washer (Biotek, Winooski, VT). Assay plates were incubated in the Steri-Cult automated incubator (Thermo Fisher Scientific) under controlled humidity at 37°C and 5% CO2. The assay was performed on a fully automated linear track robotic platform (Thermo Fisher Scientific) using several integrated peripherals for plate handling, cell incubators, liquid dispensing, and detection systems.
Assay Validation with Focused Panel of Genes
Assay optimization and adaptation into 384-well plate format was performed as previously described.19 To validate the reporter assay for RNAi screening, our optimized conditions were tested with a focused panel targeting 17 genes: (1) known and active components of the miRNA pathway: DGCR8, DICER1, DROSHA, EIF2C1, EIF2C2, EIF2C3, EIF2C4, DHX9, MOV10, TARBP2, TNRC6A, TNRC6B, TNRC6C, XPO5, (2) cell cycle regulators as functional controls for transfection efficiency: KIF11, PLK1, and (3) an unrelated gene as a negative functional control: SCPEP1. Three siRNA duplexes per gene were obtained from the Ambion Silencer Select v4.0 library, including the Silencer Select Negative Control siRNA #1 (4390843) as the assay negative control. The siRNA duplexes were diluted from stock solutions in nuclease-free water and 5 μL was transferred into assay plates to achieve the desired final concentration of 50 nM. Next, 10 μL/well Opti-MEM media was added followed by 15 μL/well Lipofectamine RNAi Max transfection solution at a final concentration of 0.1 μL/well and incubated at room temperature for 20 min to promote siRNA-transfection reagent complex formation. Next, cell suspensions, 500 cells per well, were dispensed into the assay plates in 50 μL growth media. At day 5 post-transfection, 8 μL AB reagent was added to the cells and further incubated for another day. The resulting fluorescence intensity was read on the LEADseeker Multimodality Imaging System (GE Healthcare, Piscataway, NJ). Cells were then fixed in 4% paraformaldehyde (w/v) for 20 min followed by nuclei staining in a solution containing 1 μM Hoechst and 0.05% Triton X-100 (v/v) for 15 min. Cells were washed twice in PBS and assay plates stored at 4°C until imaging. Images of cells in assay plates were acquired on the IN Cell Analyzer 3000 (INCA3000) for EGFP fluorescence intensity and Hoechst-stained nuclei.
Gene Expression Analysis Using Quantitative Polymerase Chain Reaction
Cells were treated with siRNA duplexes targeting specified genes at 5 nM using the Lipofectamine RNAi Max transfection solution for 2 days. Total RNA was extracted from these siRNA-treated cells and TaqMan gene expression assays were run according to the manufacturer's specifications (Life Technologies). As a reference control, 18S rRNA was used for normalization of quantitative real-time polymerase chain reaction (PCR) data.21
miRNA Expression Analysis Using Quantitative PCR
Cells were treated as described above with siRNA duplexes targeting EIF2C2, DGCR8, DROSHA, DICER1, and PLK1. Total RNA was extracted from these siRNA-treated cells with the miRCURY RNA Isolation Kit (Exiqon, Woburn, MA) and 10 ng of RNA was used for first-strand cDNA synthesis with the Universal cDNA Synthesis Kit (Exiqon). For detection of miRNA expression, PCR amplification was carried out in a final volume of 10 μL containing 4 μL of cDNA diluted 80× from the cDNA synthesis, 5 μL of SYBR Green master mix (Exiqon), and 1 μL of hsa-miR-21 PCR primer mix (Exiqon). The following conditions were used in the PCR amplification assay: denaturation at 95°C for 10 min, followed by an amplification step of 40 cycles at 95°C for 10 s, and finally a 60°C step for 1 min on the ABI 7900HT (Life Technologies). As a reference control, 5S rRNA was used for normalization of quantitative real-time PCR data.22
Genome-Wide siRNA Library
The Ambion Silencer Select v4.0 library was purchased from Life Technologies. The arrayed genome-wide siRNA library contains 64,755 siRNA duplexes covering 21,565 genes with an approximate redundancy of three duplexes per gene and plated across 186 384-well polypropylene source plates with columns 13 and 14 empty for controls. The siRNA duplexes have been chemically modified to enhance their specificity and based on antisense strand analysis; 99.9% of the sequences are unique with a 0.1% degree of redundancy. In addition, the library has undergone a 5-step bioinformatics filtering process to predict and eliminate siRNA duplexes with OTEs.23 For quality control, the siRNA duplexes are purified, analyzed by MALDI-TOF mass spectrometry, re-annealed, and then checked by gel electrophoresis to ensure proper annealing.
Genome-Wide siRNA Library Screening for Modulators of miRNA Biogenesis
For the genome-wide screen, the siRNA library was diluted from 20 μM stock solutions in nuclease-free water to 1 μM concentration and 5 μL was transferred into assay plates to achieve a final concentration of 50 nM. For internal reference, each assay plate contained Silencer Select Negative Control #1 siRNA (4390843) as negative control in column 13 and EIF2C2 siRNA (s25931) as the positive control in column 14 at a final concentration of 50 nM. Next, 10 μL/well Opti-MEM medium was added followed by 15 μL/well Lipofectamine RNAi Max transfection solution at a final concentration of 0.1 μL/well and incubated at room temperature for 20 min to promote siRNA-transfection reagent complex formation. Next, cell suspensions, 500 cells per well, were dispensed into the assay plates in 50 μL growth media. At day 5 post-transfection, 8 μL AB was added to the cells and read on the LEADseeker following a 1-day incubation as described above. Cells were then fixed and stained followed by imaging on the INCA3000 for EGFP fluorescence intensity and Hoechst-stained nuclei. The genome-wide screen was performed in duplicate to assess reproducibility in a total of 372 assay plates.
Image Acquisition, Analysis, and Screening Data Management
Images were acquired with an exposure time of 1.5 ms on the automated laser confocal INCA3000 microscope at the following wavelengths: 364 nm excitation/450 nm emission in the blue channel for Hoechst-stained nuclei, and 488 nm excitation/535 nm emission in the green channel for the EGFP signal. For assay development, nine images per well were collected using a 40× magnifying objective covering 90% of the well and required 45 s per well, with a total imaging time of 180 min for a complete 384-well microtiter plate. For screening, four images per well were collected using a 40× magnifying objective covering 40% of the well and required 10 s per well, with a total imaging time of 60 min for a complete 384-well microtiter plate. Images were analyzed using the Raven 1.0 software's built-in object intensity analysis module to assess EGFP fluorescence intensity per well and count the number of Hoechst-stained nuclei. Analysis of the EGFP signal and the NUCL count required approximately 20 min for a complete 384-well microtiter plate. Screening data files were loaded into Oncology Research Informatics System, a custom-built suite of modules for RNAi registration, plating, and data management.
Data Analysis by the BDA Method
Assay validation and genome-wide screening data were scored by applying the BDA method on the multiplexed readouts for EGFP signal enhancement, NUCL count, and AB signal output. The BDA method comprises of 5 steps to analyze and score for the active siRNA duplexes and genes; namely, (1) active duplex identification, (2) active gene identification, (3) OTE filtering, (4) re-scoring, and (5) biological classifications.20 To identify modulators of miRNA biogenesis, siRNA duplexes were individually scored as active using an EGFP signal threshold based on 2 standard deviations (2σ) from the mean of the negative control with the Silencer Select Negative Control #1 siRNA. The active genes were nominated from the active siRNA duplexes using a hit rate per gene (H score) of ≥60. H score is defined as follows:
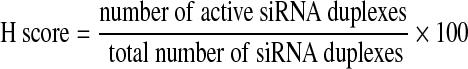 |
(1) |
To identify cytotoxic genes, siRNA duplexes were individually scored for NUCL count and AB signal using a threshold of 80% inhibition relative to the data median. Genes were nominated as cytotoxic using an H score of ≥60. Active genes were further classified: noncytotoxic active genes were categorized as nonessential genes and cytotoxic active genes were categorized as essential genes. The essential and nonessential genes were subjected to OTE filtering, followed by re-scoring of the active genes after removing siRNA duplexes, which qualified as high confidence-OTE. The genes deemed as active after re-scoring were further analyzed for their biological classification. Enriched gene ontology (GO) functional categories and InterPro (IPR) protein domains were found using the DAVID functional annotation tool. A functional activity map was created after removing redundant GO and IPR terms for the biological functions, molecular functions, and protein domains in nonessential genes and essential genes. The statistical data analysis was done using Perl. The human 3′UTR sequences were obtained from the University of California at Santa Cruz genome browser: human genome assembly GRCh37/hg19 (http://genome.ucsc.edu).24 The sequences for human miRNAs were obtained from miRBase release 18 (www.mirbase.org).25 The drug and druggable target information was downloaded from DrugBank (www.drugbank.ca).26
Results
Multiplexed High-Content Assay to Identify Modulators of miRNA Biogenesis
To screen for genes and pathways that modulate miRNA biogenesis, we adapted a previously described cell-based high-content assay.19 The reporter cell line expresses a plasmid encoding EGFP fused to a sequence with perfect complementarity to miR-21 in its 3′ UTR region, whereby miR-21 endogenously generated by the HeLa S3 cells specifically destabilizes the EGFP mRNA resulting in low-level protein expression of EGFP; hence, low fluorescence output. By knocking down genes potentially involved in the miR-21 biogenesis, the miR-21 levels will be reduced; thus, allowing for the expression and stabilization of the EGFP mRNA resulting in an increase production of the EGFP protein and its fluorescence output (Fig. 1A). This gain-of-function assay strategy was successfully implemented for chemical screening19 and we hypothesize that RNAi-mediated knockdown of genes involved in the biogenesis pathway would result in rescue of EGFP expression, and resulting in a gain of a measurable EGFP fluorescence signal output; hence, a perfect surrogate reporter to study the cellular miRNA biogenesis activity (Fig. 1B).
Fig. 1.

Multiplexed-assay strategy to identify gene and pathway modulators of microRNA (miRNA) biogenesis. (A) Principle of the cell-based reporter assay. Cells expressing a plasmid encoding an enhanced green fluorescent protein (EGFP) fused to a sequence with perfect complementarity to miR-21 in its 3′ untranslated region (UTR) results in low protein expression levels; hence, low fluorescence intensity output; the EGFP mRNA destabilization is caused by endogenous levels of miR-21. Depletion of miR-21 levels by knocking down genes potentially involved in its biogenesis, would allow for restoring the EGFP mRNA leading to the production of the EGFP protein, and resulting in higher fluorescence signal output. (B) Adaptation of the cell-based assay to RNAi screening. Cells were reverse transfected with siRNA duplexes for 6 days. siRNA knockdown of gene targets that inhibit miRNA biogenesis results in an increased EGFP fluorescence signal. siRNA knockdown of gene targets that do not effect miRNA biogenesis have no change in EGFP fluorescence signal. (C) Workflow of the multiplexed-assay and imaging analysis. Cells (500 cells per well) are reverse-transfected with 50 nM of siRNA duplexes from the Ambion Silencer Select v4.0 library for 6 days. Cells are also incubated with Alamar Blue (AB) reagent to measure metabolic activity. Automated image analysis scored EGFP fluorescence intensity as readout for modulators of miRNA biogenesis and Hoechst-stained nuclei for cytotoxicity.
As a proof of concept, we adapted the strategy for large-scale siRNA screening. To achieve the set goals of identifying genes and pathways that modulate the miRNA biogenesis pathway, we have devised a simple monitoring of cell metabolic activity using AB together with dual image acquisition for both EGFP signal and Hoechst-stained nuclei. By combining this multiplexed approach with an arrayed siRNA library, we could score on-the-fly for enhancement of EGFP fluorescence intensity as the primary readout of inhibition of miR-21 production, processing, and function; and cell death by two measures of metabolic activities with AB and cell death through remaining nuclei per assayed well (Fig. 1C). To our knowledge, this is the first RNAi screening strategy for modulators of miRNA biogenesis with multiplexed readout enabling us to score for both nonessential and essential genes in host cells.
Validation of a Multiplexed High-Content Assay
As previously described, we have optimized cell seeding and transfection conditions in 384-well plate format to identify modulators of the miRNA biogenesis pathway.19 To evaluate the performance of the reporter cell line, we validated the assay following an optimized workflow (Table 1), against a focused panel of siRNA duplexes targeting 17 genes; with some reported to be active components of the miRNA biogenesis pathway. The library consisted of 14 genes involved in the miRNA pathway (DGCR8, DHX9,27 DICER1, DROSHA, EIF2C1,28 EIF2C2, EIF2C3,28 EIF2C4,28 MOV10,29 TARBP2,30 TNRC6A,31 TNRC6B,31 TNRC6C,31 and XPO5), an unrelated gene (SCPEP132) as a negative functional control, and two genes were selected as phenotypic controls targeting cell cycle regulation (KIF1133 and PLK134). KIF11 is a motor protein involved in chromosome positioning during cell mitosis and PLK1 is a key regulator of cell division whose knockdown results in cell death. In addition, we included the Silencer Select Negative Control #1 siRNA containing a scrambled ineffective siRNA sequence to measure background EGFP fluorescence intensity.
Table 1.
Workflow of Multiplexed-Assay
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | siRNA plating–arrayed format | 5 μL | siRNA duplexes diluted in water at 1 μM concentration |
| 2 | siRNA dilution | 10 μL | Opti-MEM media |
| 3 | Transfection reagent | 15 μL | Lipofectamine RNAi Max at 0.1 μL/well in Opti-MEM media |
| 4 | Complex formation | 20 min | Complex formation for siRNA delivery into cells |
| 5 | Cell plating | 50 μL | 500 cells in growth media |
| 6 | Incubation time | 5 day | 37°C, 5% CO2 |
| 7 | Alamar Blue | 8 μL | Alamar Blue addition |
| 8 | Incubation time | 1 day | 37°C, 5% CO2 |
| 9 | Alamar Blue readout | Epi mirror: FLINT Emission mirror: Cy3/Cy3B | LEADseeker multimodality imaging system |
| 10 | Fix | 50 μL | 4% PFA (w/v) for 20 min |
| 11 | Nuclear staining | 50 μL | 1 μM Hoechst solution with 0.05% Triton X-100 (v/v) |
| 12 | Assay readout | 364 nm/450 nm & 488 nm/535 nm (ex/em) | INCA3000 automated microscope |
| 13 | Image analysis | — | Multiparametric analysis using Raven 1.0 software |
Step Notes
1. Dispensing on the PP-384-M Personal Pipettor using a custom 384 head; 30 sec per 384-well microplate.
2. Dispensing into assay plate with Multidrop 384.
3. Dispensing into assay plate with Multidrop 384.
4. Assay plates at room temperature.
5. Cells prepared in media and dispensing into 384-well assay plate with Multidrop 384.
6. Assay plates stored in the Steri-Cult; an automated incubator.
7. Dispensing into assay plate with FlexDrop IV.
8. Assay plates stored in the Steri-Cult.
9. Alamar Blue fluorescence signal quantified with FLINT epi mirror and Cy3/Cy3B emission mirror.
10–11. Aspirating on the ELx405 automated washer and dispensing with Multidrop 384; 1 min per 384-well microtiter plate.
12. For assay development, nine images per well for 45 sec per well with total imaging time of 180 min per 384-well microtiter plate. For screening, four images per well for 10 sec per well with total imaging time of 60 min per 384-well microtiter plate.
13. Analysis of EGFP signal output and Hoechst-stained nuclei, 20 min per 384-well microtiter plate.
INCA3000, IN CELL Analyzer 3000.
Three siRNA duplexes were tested for each gene at 50 nM concentration and individually scored as a modulator of miRNA biogenesis if the EGFP fluorescence intensity was above a threshold based on 2σ from the mean of the Silencer Select Negative Control #1 siRNA (Table 2). siRNA duplexes targeting EIF2C2, DGCR8, DROSHA, TNRC6B, DICER1, TARBP2, EIF2C3, and MOV10 lead to an upregulation above the threshold of 20% EGFP signal gain. Of the reported active components of miRNA biogenesis, our multiplexed-assay identified EIF2C2, DGCR8, DROSHA, DICER1, and EIF2C3 with at least 2/3 active siRNA duplexes, while TNRC6B, TARBP2, and MOV10 had only 1/3 active siRNA duplex. As expected, siRNA duplexes for SCPEP1 had no effect on EGFP signal; and more importantly, KIF11 and PLK1 resulted in cell death as measured by AB and Hoechst-stained nuclei. Representative images obtained for each duplex tested for the following genes: EIF2C2, DGCR8, DROSHA, DICER1, XPO5, and PLK1 are shown in Figure 2A. The overall acquired images for the validation are found in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/adt).
Table 2.
Validation of Multiplexed-Assay
| Gene | EGFP gaina | Cell viabilityb | Gene knockdownc | Reference |
|---|---|---|---|---|
| EIF2C2 (Ago2) | 2/3 siRNA | No effect | 2/3 siRNA | 12 |
| DGCR8 | 3/3 siRNA | 1/3 siRNA toxic | 1/3 siRNA | 9 |
| DROSHA (RNASEN) | 2/3 siRNA | 1/3 siRNA toxic | 3/3 siRNA | 7,8 |
| TNRC6B | 1/3 siRNA | No effect | 3/3 siRNA | 31 |
| DICER1 | 2/3 siRNA | 2/3 siRNA toxic | 3/3 siRNA | 11 |
| TARBP2 | 1/3 siRNA | 1/3 siRNA toxic | 3/3 siRNA | 30 |
| EIF2C1 (Ago1) | No gain | 1/3 siRNA toxic | 3/3 siRNA | 28 |
| EIF2C3 (Ago3) | 2/3 siRNA | 1/3 siRNA toxic | 0/3 siRNA | 28 |
| EIF2C4 (Ago4) | No gain | No effect | 2/3 siRNA | 28 |
| TNRC6A | No gain | No effect | 2/3 siRNA | 31 |
| TNRC6C | No gain | No effect | 2/3 siRNA | 31 |
| DHX9 | No gain | 1/3 siRNA toxic | 3/3 siRNA | 27 |
| XPO5 | No gain | No effect | 3/3 siRNA | 10 |
| MOV10 | 1/3 siRNA | 1/3 siRNA toxic | 3/3 siRNA | 29 |
| SCPEP1 | No gain | 1/3 siRNA toxic | 3/3 siRNA | 32 |
| KIF11 | No gain | 3/3 siRNA toxic | 3/3 siRNA | 33 |
| PLK1 | 1/3 siRNA | 3/3 siRNA toxic | 3/3 siRNA | 34 |
Number of siRNA for given gene resulting in >2σ EGFP fluorescence above background as determined by Silencer Select Negative Control #1 siRNA.
Number of siRNA resulting in cytotoxicity with >80% inhibition relative to data median.
Number of siRNA resulting in knockdown using a threshold of 40% gene expression remaining.
Fig. 2.

Validation results and expression analysis with quantitative polymerase chain reaction (qPCR). Cells were tested against a focused panel of siRNA duplexes using reverse-transfection, then evaluated with image analysis and qPCR. (A) Representative images of cells treated with siRNA duplexes for EIF2C2, DGCR8, DROSHA, DICER1, XPO5, and PLK1. Images were acquired using the IN CELL Analyzer 3000 (INCA3000) with green channel for EGFP signal and blue channel for Hoechst-stained nuclei. (B) Gene expression analysis of cells treated with 5 nM of siRNA for 2 days. (C) miRNA expression analysis of cells treated with 50 nM of siRNA for 6 days.
To evaluate the mRNA levels of the tested genes after siRNA transfection, we performed qPCR gene expression analysis and scored those siRNA duplexes as active for gene knockdown using a threshold set at 60% gene knockdown. We find that all three siRNA duplexes per gene targeting DROSHA, TNRC6B, DICER1, TARBP2, EIF2C1, DHX9, XPO5, MOV10, SCPEP1, and PLK1 scored as active for gene knockdown as assessed by qPCR (Fig. 2B). However, those duplexes targeting EIF2C2, EIF2C4, TNRC6A, and TNRC6C had 2/3 knockdown activity, whereas DGCR8 had only one active duplex. Duplexes targeting EIF2C3 were found to be inactive for gene knockdown by qPCR. Furthermore, we assessed miR-21 levels following siRNA treatment on four genes, which constitute the core miRNA biogenesis regulators using qPCR miRNA expression analysis and scored siRNA duplexes as active for knockdown using a threshold of 40% miR-21 expression remanining. For EIF2C2, 3/3 siRNA duplexes were active for knockdown of miR-21. DGCR8 and DROSHA had 2/3 and 1/3 active siRNA duplexes, respectively; surprisingly, DICER1 had 0/3 active siRNA duplexes by miR-21 expression analysis (Fig. 2C). Taken together, these results demonstrate our assay's sensitivity and robustness; thus, providing a validated strategy to progress to the genome-wide screen.
Genome-Wide siRNA Screen for Modulators of the miR-21 Biogenesis Pathway
To identify genes and pathways that modulate miRNA biogenesis, the multiplexed-assay was screened against a genome-wide siRNA library containing 64,755 siRNA duplexes covering 21,565 genes with approximately three siRNA duplexes per gene target at a concentration of 50 nM. The screen was performed in duplicate to assess the assay robustness and performance. To monitor the assay's performance throughout the screen, Silencer Select Negative Control #1 siRNA was added to column 13 for negative control and EIF2C2 siRNA into column 14 for positive control. The controls worked as expected with box plot analysis of EGFP signal gain showing good separation from 1% to 100% for the negative and positive controls (Supplementary Fig. S1). However, the Z′ factor was <0 due to large standard deviations given the heterogeneity of gene silencing from cell to cell (Fig. 3A). As a gain-of-function assay, the Z′ factor had no impact on the performance and does not affect hit identification.
Fig. 3.

Assessment of the assay performance during screening. Cells (500 cells per well) were screened against Ambion Silencer Select v4.0 library covering the 21,565 genes at 50 nM using reverse transfection and evaluated at 6 days postseeding. (A) Frequency distribution plot of control wells for EGFP signal. Performance of negative control for % EGFP signal gain is 1%±20% and positive control is 100%±47%. (B) Distribution plot of siRNA duplex activity during screening for EGFP signal. Inset contains plot of active siRNA duplexes per gene for EGFP signal. (C) Distribution plot of siRNA duplex activity during screening for NUCL count. Inset contains plot of active siRNA duplexes per gene for NUCL count. (D) Distribution plot of siRNA duplex activity during screening for AB signal. Inset contains plot of active siRNA duplexes per gene for AB signal.
Hit Nomination Using the BDA Method
To identify modulators of the miR-21 biogenesis, we have implemented the BDA method to nominate active hits as nonessential or essential genes and further analyzed for their biological classification (Fig. 4). In the first step of active duplex identification for the EGFP signal readout, individual siRNA duplexes were scored using a threshold based on 2σ from the mean of the negative control that translates to an EGFP signal gain of 40% and 7,760 siRNA duplexes were identified as active (Fig. 3B and Supplementary Table S2). In the second step for active gene identification, we set an H score of ≥60 to identify active genes corresponding to at least 2/3 active siRNA duplexes. In total, 1,301 (21%) of the genes had an H score ≥60 and were nominated as modulators of miRNA biogenesis, while the remaining 4,981 (79%) of the genes were filtered out (Fig. 4).
Fig. 4.

The Bhinder–Djaballah analysis method workflow. Sequential systematic workflow consisting of five steps: (1) active siRNA duplex identification, (2) active gene identification, (3) off-target effect (OTE) filtering, (4) re-scoring, and (5) biological classifications.
In parallel for active duplex identification of NUCL count readout, individual siRNA duplexes were scored using a median-based method at 80% inhibition and 5,013 siRNA duplexes identified as cytotoxic (Fig. 3C). Using an H score ≥60, 755 (19%) of the genes were nominated as cytotoxic, while the remaining 3,215 (81%) of the genes were filtered out (Fig. 4). Similarly for AB signal readout, individual siRNA duplexes were scored using a median-based method at 80% inhibition and 930 siRNA duplexes were identified as cytotoxic (Fig. 3D). Using an H score of ≥60, 169 (24%) of the genes were nominated as cytotoxic, while the remaining 525 (76%) of the genes were filtered out (Fig. 4). The genes nominated using the AB signal had a complete overlap with the genes selected from the NUCL count, and we obtained a nominated list of 755 cytotoxic genes. We incorporated cytotoxicity data to classify the 1,301 nominated genes for EGFP signal into two categories: genes that were noncytotoxic, therefore nonessential, and genes that were cytotoxic, therefore essential. This classification resulted in 1,238 nonessential genes and 63 essential genes. The two categories were individually subjected to OTE filtering followed by re-scoring to obtain a final list of nominated hits.
Nomination and classification of the 1,216 nonessential gene candidates for the miR-21 biogenesis pathway
To nominate the final list of genes involved in the miR-21 biogenesis pathway, the third step of the BDA method consisted of OTE filtering to flag potential false positives due to enrichment of the 7-mer seed sequence (seed heptamer) in the antisense strand. The active siRNA duplexes from nonessential genes were tested for three characteristics of the seed heptamer in the antisense strand: over-representation in the active siRNA duplexes relative to the library with P<0.05 (Supplementary Fig. S2), >10% enrichment in the human 3′UTR sequences, and at least one perfect match with existing human miRNAs. We found 28 siRNA duplexes in nonessential genes qualifying in 3/3 criteria as high-confidence OTE that were, therefore, removed from subsequent analysis (Supplementary Table S3). Furthermore, 396 siRNA duplexes in nonessential genes qualifying in 2/3 criteria were flagged as low-confidence OTE and retained for subsequent analysis. As the fourth step, the remaining subset of the siRNA duplexes was re-scored with an H score ≥60 to obtain a final nominated list of 1,216 nonessential genes (Supplementary Table S4).
Among the 1,216 nonessential genes nominated in the screen, we have identified the four core components involved in miRNA biogenesis; namely, EIF2C2, DGCR8, DROSHA, and DICER1; consistent with our validation results. Similarly, the remaining genes from the focused panel did not score as active with the exception of EIF2C3; whereas 2/3 siRNA duplexes were active in the validation and only 1/3 siRNA duplexes was active in the screen. Among the nonessential genes, we also identified two genes previously known to have a regulatory role in miRNA biogenesis: HDAC1, and NHLH1 (HEN1). HDAC1 is a histone deacetylase, which has recently shown to be a regulator of DGCR8 activity.35 NHLH1, a methyltransferase that stabilizes miRNA during its processing on the 3′ end to prevent uridylation activity.36 Furthermore, RNA polymerase II has been described to be an initiator of the miRNA pathway as it is required for miRNA gene transcription.37 We identified GTF2A2, general transcription factor IIA, that is a transcription activator and component of the RNA polymerase II-mediated transcription machinery.
Focusing on the four core components (EIF2C2, DGCR8, DROSHA, and DICER1) as seed nodes in Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA), we constructed network maps to understand how our nominated genes interact with known modulators of miRNA biogenesis (Fig. 5A). Among the core components, we identified several upstream interactions, including RNASEH1 and NCK1, which directly interact with DROSHA, and HDAC1, which interacts with DGCR8. In addition, it has been previously shown that HSP90 stabilizes EIF2C2 and controls its localization to the P-bodies and stress granules38 and we identified 11 upstream interactions of HSP90: TGFBR1, NLRP2, CYP2E1, STUB1, ICK, CD28, CNTF, CD14, FGFR3, PRMT5, and BIRC5. Of the reported miRNA biogenesis components that were not active in our validation, network mapping revealed several known upstream regulators among the nominated genes, including SMAD4 and TNFRSF1B for XPO5, TRIM24 for MOV10, and GSN for DHX9. Furthermore, we performed network analysis on the entire list of nominated genes to identify highly interconnected gene clusters. Subsequently, we identified four high-scoring gene clusters (Fig. 5B). The most enriched cluster was found to be associated with the components of the exosome core complex. Additionally, we identified gene clusters involved in functions relating to transcription regulation, translation activity, and SMAD binding activity.
Fig. 5.

Biological classifications of nominated gene candidates. (A) The network map using ingenuity pathway analysis shows interactions between the core biogenesis proteins and the nominated genes. Identified core components of miRNA biogenesis are bolded in red. Reported miRNA biogenesis components that were inactive are bolded in black. The nonessential genes are depicted in blue and essential genes are depicted in green. (B) Top four core gene clusters found in nonessential genes. The ranking of each cluster is based on a statistical score from MCODE.
Next, we analyzed the 1,216 nonessential genes for enrichments in GO functional categories (Supplementary Fig. S3) and identified eight prominent functional clusters: ribonuclease activity (GO:0004540, GO:0004518, GO:0004527), ribonucleoprotein complex binding (GO:0043021), transcription factors and regulators (GO:0003700, GO:0030528), vesicular transport (GO:0016192), cell adhesion (GO:0007155), enzymatic activity (GO:0016407, GO:0004579, GO:0008080, GO:0016410, GO:0016765, and GO:0016769), metabolism, (GO:0032774, GO:0009100, GO:0046148, GO:0018196, GO:0006486, GO:0006487, and GO:0004576), and development (GO:0046530, GO:0030182, GO:0007423, GO:0048666, and GO:0043010). The protein domains enrichments in IPR classification revealed structural motifs, such as basic-leucine zipper (b-ZIP), bromodomain signatures, ATPase P-type proton pump, cadherin, and exoribonulease domains. The highest numbers of the active genes belonged to DNA/RNA binding and transcription regulators (P<0.002); especially 20 regulators of transcription were specifically associated with the RNA polymerase II activity. Genes involved in cell adhesion were identified and reported as activators of miRNA biogenesis.39 Also, we found multiple GO categories associated with the exonuclease activity (P<0.005) and these enrichment results suggest a closely knit circuit of miRNA biogenesis regulation primarily at the gene expression level. ATP2A2, ATP2A3, and ATP2C2, containing the ATPase P-type proton pump were found to be involved in translocation of Ca2+ from the cytosol to the sarcoplasmic reticulum lumen in muscle cells resulting in regulation of the Ca2+ levels. Modulation of Ca2+ levels are involved in multiple cellular processes, including signaling. In addition, we identified members involved in SMAD binding (P=0.03), including MEN1, TGFBR1, CREBBP, COL3A1, SMAD4, STUB1, and BMPR1A and its nuclear translocation are also members of the TGF-β pathway.
Furthermore, we analyzed the canonical pathway associations (Supplementary Fig. S4) for the 1,216 nonessential genes and have identified seven members of the BMP signaling with P=0.0007. The TGF-β/BMP signaling pathway is widely reported to have an established role in miRNA biogenesis through recruitment of SMADs on pri-miRNAs and enhancing processing activity of DROSHA.15 We also found enrichments in four other canonical pathways involved in signal transduction, including Activin A, PKA, cAMP, and SIP1. Activin belongs to the TGF-β superfamily and the signaling pathway is involved in SMAD2/SMAD3-SMAD4 complex-mediated regulation of transcription of certain genes. Similarly, the PKA signaling pathway is also involved in the SMAD4-mediated gene expression regulation. These findings not only reemphasizes the known role of SMAD2/3 in regulation of the microprocessor complex activity to further support the role of SMAD4 and multiple canonical SMAD pathways as modulators of miRNA biogenesis. We have identified eight nonessential genes, including clathrin as members of the clathrin-coated vesicle cycle pathway (CMT) with P=0.002 as a novel regulatory process in the miRNA processing.
Nomination and classification of the 57 essential gene candidates for the miR-21 biogenesis pathway
To nominate the final list of essential genes, the BDA method was employed and six siRNA duplexes qualifying in 3/3 criteria as high confidence-OTE and, therefore, removed from subsequent analysis (Supplementary Table S5). Forty-two siRNA duplexes qualifying in 2/3 criteria were flagged as low confidence-OTE and retained for subsequent analysis. The remaining subset of the siRNA duplexes was re-scored with an H score ≥60 to obtain a final nominated list of 57 essential genes (Supplementary Table S6).
Among the 57 essential genes, we identified PDCD6IP (ALIX), previously reported to be involved in miRNA biogenesis. PDCD6IP is associated with the late endosome/multivesicular bodies and was shown to be involved in GW182 sorting, which promotes RISC assembly.40 Next, we analyzed the 57 essential genes for enriched GO functional categories and found four main functional clusters (P<0.05): cell cycle, protein localization and transport, cytoskeleton organization, and ubiquitin-protein ligase activity (Supplementary Fig. S5). Based on the lowest P value of <6.0×10−5, cell cycle regulation and cytokinesis emerged as most prominent functions followed by protein transport and targeting. It has been reported that cell cycle is involved in mature miRNA activity during translation regulation41 and cytokinesis during mitosis regulates the activity of RNA polymerase II;42 therefore, it is plausible that these functions have an indirect role in miRNA biogenesis. Protein domains enrichments using IPR classification revealed several proteosome components. It has previously been reported that the activity of genes, such as TARBP and EIF2C2, are under the control of ubiquitination and proteosomal degradation.43,44 These findings indicate the possibility of ubiquitin-related post-transcription control on the core components that regulate miRNA biogenesis. Canonical pathway association of the essential genes revealed enriched processes, including cell cycle, metabolism, proteolysis, signaling, and translation among the top 10 pathways (Supplementary Fig. S6). In addition, we have identified Wnt signaling as an enriched pathway, which has been suggested to be involved in nuclear translocation of SMAD4 and might be interlinked with the TGF-β/BMP signaling pathway.45
Identification of Druggable Gene Targets
miRNA dysregulation has been described as oncogenic because of their emerging role in multiple types of cancer and, hence, has become an attractive therapeutic target.13,14 An alternate strategy instead of directly targeting miRNAs, is to target the genes involved in its biogenesis and accordingly targeting them with already known drugs can have a huge implication in accelerating the process of drug discovery. Our study reports 1,216 nonessential genes and 57 essential genes, with an implicated role in various stages of miRNA biogenesis and localization in specific, and thus provides a framework of various potential therapeutic targets. Based on the information obtained from the DrugBank, 11% of our 1,216 nominated nonessential genes are druggable targets out which 24 genes have FDA-approved drugs targeting them (Supplementary Table S4). Out of the 56 essential genes, 12% of the genes were identified as druggable targets out which three genes (PSMA4, PSMA7, and RRM2) have FDA-approved drugs targeting them (Supplementary Table S6). Among the targets, HDAC1, which regulates DGCR8 activity35 and whose enzymatic activity can be inhibited by an FDA-approved small molecule inhibitor vorinostat; currently used for treatment of cutaneous T-cell lymphoma. Perhaps, two interesting therapeutic targets would be AMHR2 and CREB1 because of their roles in the TGF-β signaling pathway and transcription regulation, respectively.
High-Confidence Gene Candidates for the miR-21 Biogenesis Pathway
Out of the total of 1,273 nominated gene candidates, 121 gene candidates were selected as high-confidence candidates as each gene had 3/3 siRNA duplexes active in the screen, and a thorough OTE analysis revealed no apparent and potential adverse effects. The list is summarized in Supplementary Table S7.
Discussion
Although the core components of the miRNA biogenesis pathway have been identified, yet the genes and pathways involved in its regulation remain largely unknown. For this purpose, we have developed a multiplexed high-content cell-based reporter assay to search for such modulators using RNAi technology. The assay was successfully validated against a focus panel of genes and identified previously known core components of the miRNA biogenesis pathway, including EIF2C2, DROSHA, DGCR8, and DICER1. Applying this assay approach, we report the first complete genome-wide siRNA screen scoring for modulators of miRNA biogenesis and cell death. We have successfully screened the Ambion Silencer Select v4.0 library and classified our nominated hits as nonessential and essential genes for miRNA biogenesis.
To nominate active genes in the genome-wide screen for modulators of miRNA biogenesis, we applied the BDA method for a standardized data processing workflow, while minimizing the false positives arising from off-target silencing. Target specificity is a major concern in RNAi screens and sequence-dependent off-target silencing is omnipresent.46 This implies that a single active siRNA duplex for a given gene might not be a true indicator of the gene to be nominated as a hit. Screening libraries are designed to allow combinatorial knockdowns using multiple siRNA duplexes per gene as such; a siRNA duplex library typically has three siRNA duplexes per gene, but this number may vary. In this case, a threshold based on the number of active siRNA duplexes for hit nomination is not very meaningful. We addressed this issue by assigning an activity ratio to each gene in the form of H score. Using a stringent H score of ≥60, only those genes are identified as hits, which have a reproducible phenotype across maximal targeting siRNA duplexes. After applying an H score, we filtered active siRNA duplexes for seed heptamer enrichment to eliminate putative false positives. Our OTE filtering analysis results revealed multiple cases of a seed heptamer mimic between an endogenous miRNA and an active siRNA duplex, therefore suggesting a potential false positive originating via an off-target silencing of a miRNA target (Table 3).
Table 3.
Off-Target Filtering Results of Matched microRNA Seed Sequences
| Antisense sequence | miRNA ID | miRNA sequence | ||
|---|---|---|---|---|
| Gene NCBI ID Supplier ID | BAHD1 NM_014952 s22606 | UAAGUGCUCAGGUCUGUAATA | hsa-miR-302a | UAAGUGCUUCCAUGUUUUGGUGA |
| hsa-miR-302b | UAAGUGCUUCCAUGUUUUAGUAG | |||
| hsa-miR-302c | UAAGUGCUUCCAUGUUUCAGUGG | |||
| hsa-miR-302d | UAAGUGCUUCCAUGUUUGAGUGU | |||
| hsa-miR-372 | AAAGUGCUGCGACAUUUGAGCGU | |||
| hsa-miR-373 | GAAGUGCUUCGAUUUUGGGGUGU | |||
| hsa-miR-520e | AAAGUGCUUCCUUUUUGAGGG | |||
| hsa-miR-520a-3p | AAAGUGCUUCCCUUUGGACUGU | |||
| hsa-miR-520b | AAAGUGCUUCCUUUUAGAGGG | |||
| hsa-miR-520c-3p | AAAGUGCUUCCUUUUAGAGGGU | |||
| hsa-miR-520d-3p | AAAGUGCUUCUCUUUGGUGGGU | |||
| hsa-miR-302e | UAAGUGCUUCCAUGCUU | |||
| Gene NCBI ID Supplier ID | TMEM14B NM_030969 s37836 | AAAAGUAACAGAUGUAGCGGC | hsa-miR-559 | UAAAGUAAAUAUGCACCAAAA |
| hsa-miR-548b-5p | AAAAGUAAUUGUGGUUUUGGCC | |||
| hsa-miR-548a-5p | AAAAGUAAUUGCGAGUUUUACC | |||
| hsa-miR-548c-5p | AAAAGUAAUUGCGGUUUUUGCC | |||
| hsa-miR-548d-5p | AAAAGUAAUUGUGGUUUUUGCC | |||
| hsa-miR-548j | AAAAGUAAUUGCGGUCUUUGGU | |||
| hsa-miR-548h | AAAAGUAAUCGCGGUUUUUGUC | |||
| hsa-miR-548i | AAAAGUAAUUGCGGAUUUUGCC | |||
| hsa-miR-548w | AAAAGUAACUGCGGUUUUUGCCU | |||
| hsa-miR-548y | AAAAGUAAUCACUGUUUUUGCC | |||
| hsa-miR-548ab | AAAAGUAAUUGUGGAUUUUGCU | |||
| hsa-miR-548ak | AAAAGUAACUGCGGUUUUUGA |
After implementing the five steps of the BDA method on the 21,565 genes screened, we nominated two classes of hits that affect miRNA biogenesis with 1,216 nonessential and 57 essential gene candidates for cell survival and viability. Among the nominated genes were four core components of the miRNA biogenesis pathway: DROSHA and DGCR8, components of the microprocessor complex; and EIF2C2 and DICER1, components of RISC. This result was found to be consistent with the validation of the focus panel, demonstrating the robustness of our assay and hit nomination methodology to efficiently identify key constituents of the miRNA pathway. However, we did not find several reported factors and regulators involved in miRNA biogenesis in both the validation and the genome-wide screen. Importantly, XPO5, a widely reported miRNA nuclear export protein, exhibited an overall EGFP signal marginally below our threshold settings in validation as well as genome-wide screen (Fig. 2A). Yi and coworkers had performed an RNAi study for involvement of XPO5 in miR-21 biogenesis, wherein a single siRNA duplex against XPO5 was found to significantly increase luciferase expression in 293T cells using the pCMV-luc-miR-21(P) reporter system.10 As eluded to earlier, a single active siRNA duplex might not be an efficient indicator of designated on-target silencing. Our data imply the stringency of the BDA method in identifying only those genes that associate with a consistently strong phenotype as determined by its maximal active siRNA duplexes. Of note, XPO5 was also not identified as a hit in the only other genome-wide miRNA biogenesis screen in C. elegans wherein the authors identified imb-4, an ortholog of XPO1.47 However, our network analysis revealed a number of nominated genes as upstream regulators of XPO5 and Ran-GTP to emphasize the merits of our biological classification in hit assessment.
The biological classification of our nominated genes revealed four major cellular control junctions in biogenesis pathway namely: epigenetic control, transcription regulation, RNA processing by the exosome complex, and vesicular transport via clathrin-mediated endocytosis. The epigenetic control is an important cellular process related to DNA methylation or histone post-translational modification and consequential control of gene expression in general. Histone acetylases and deacetylases are responsible for a transcription level control via the post-translational modifications of the lysine residues and, thus, generating binding sites for specific protein domains, such as bromodomains. In this study, we have identified HDAC1, histone deacetylase 1, an integral component of the microprocessor complex that regulates the activity of DGCR8 by deactelyation of its lysine residues.35 Interestingly, we have also found six genes (ELP3, TAF10, TAF5, CREBBP, MGEA5, and CDY2A) with histone acetyltransferase activity, which has an antagonistic effect to that of histone deacetylases. All six genes with acetyl transferase activity are recruited by estrogen receptor for transcriptional activation; a recent study has reported a role of the estrogen receptor in dissociation of the microprocessor complex.48 Additionally, the proteins containing bromodomains bind to the acetylated lysine residues and further exert a control on gene expression. We have identified CREBBP, the CREB binding protein, the bromodomains of which have been shown to bind with the lysine acetylated p53 and consequentially leading to a cell cycle arrest mediated via activation of p21. Taken together, the above observations provide evidence toward an orchestrated epigenetic control of miRNA biogenesis at a gene expression level.
For the second control junction, we report an enriched cluster of five transcription regulation factors with previously unknown association with miRNA biogenesis: TAF4, TAF4B, TAF5, TAF10, and GTF2A2. These factors are involved in the transcription initiation activity of RNA polymerase II, the key enzyme driving the transcription of miRNAs. In total, we have found 66 genes with a transcription regulation activity and out of these report 11 genes (MAF, MAFG, ATF4, FOSL2, CREB3, CEBPD, CREB1, ATF7, TEF, FOSB, and NFIL3) are b-ZIP transcription factors. As a third control junction, we report six core components of the exosome complex; namely, EXOSC1, EXOSC3, EXOSC5, EXOSC6, EXOSC9, and DIS3. The proteins constituting the exosome complex are primarily enriched in RNase domain and have a 3′-5′ exoribonuclease activity. The exact mechanism of miRNA processing via the argonaute proteins during miRNA biogenesis is not known. Recently, Xue and coworkers used a Northern blot analysis to demonstrate that the rrp44 (DISC3) and mtr3 (EXOSC6) knockdown of neurospora strains resulted in a decrease in mature miRNA like small RNAs (milR) and an increase in pre-milR levels.49 As a result, the exosome complex is required in milR maturation due to its role in trimming of pre-milR into a smaller length and generation of single stranding pre-milR. Furthermore, the 3′-end trimming activity of exosomes in drosophila has also been demonstrated in miR-1017, and intron-derived miRNAs referred to as mirtrons through knockdown experiments.50 Flynt and coworkers have shown depletion of four exosome proteins: dis3, rrp6 (EXOSC10), mtr3, and rrp45 (EXOSC9) of which leads to reduction in mature miRNA levels. These observations extend strong support to our findings of enrichments in DISC3, EXOSC6, and EXOSC9 along with three additional core components of the exosome complex, further emphasizing the importance of this complex in the miRNA biogenesis pathway and their processing into the mature form.
Previously, it has been suggested that late endosome/multivesicular bodies are the sites for miRNA-loaded RISC accumulation and are enriched in GW182, a regulator of EIF2C2 activity.40 Additionally, exosomes have been shown to contain mature miRNAs and have been suggested to be possibly involved in their export to the neighboring cells.51 Altogether, these findings by other groups indicate involvement of a vesicular transport system for miRNA localization to its target sites. Similarly, we have not only identified genes involved in vesicular transport and trafficking in general, but also report eight genes in specific, which are involved in all stages of the CMT pathway from initiation of vesicle formation to recycling endosome (Fig. 6). The CMT pathway is novel with regard to its role in miRNA biogenesis and the exact mechanism of its function in the biogenesis pathway still needs to be elucidated. We hypothesize that this might be an essential step in miRNA sorting and targeting post the miRNA processing stage, and thus accounting for the fourth control junction.
Fig. 6.

Network map of nominated genes associated with vesicular transport and different stages of clathrin-mediated endocytosis.
We report the first genome-wide siRNA screen for modulators of miRNA biogenesis in mammalian cells. Parry and coworkers reported the only other genome-wide screen for miRNA biogenesis modulators in C. elegans.47 Out of the 213 gene hits reported, 152 human orthologs were identified using OrthoList,52 and a comparison identified 12 hits overlapping with active genes in our screen: 9 nonessential genes (DICER1, BCL11A, DCTN1, NCBP1, PGGT1B, POU2F1, RPN2, TUBA1C, and ZW10) and three essential genes (CUL1, PRC1, and PSMA4). Of the active genes, four genes (DCTN1, PRC1, TUBA1C, and ZW10) are constituents of cytoskeleton, 2 genes (BCL11A, POU2F1) are regulators of transcription by RNA polymerase II, two genes (PSMA4, CUL1) are involved in proteolysis, and two others (DICER1, NCBP1) participate in RNA catabolic processes. Out of the remaining genes, PGGT1B has acetyl transferase activity, and RPN2 is involved in protein modification. Additionally, R-SMADs have been hypothesized to play an important role during the processing of pre-miRNA to mature miRNAs after translocation regulated by the members of the TGF-β and the BMP family.15 We have identified genes with SMAD binding and translocation activity to further demonstrate our assay's robustness. In addition to the already-known association of the TGF-β/BMP signaling pathways, we also found five other pathways involved in SMAD-mediated gene expression regulation: Wnt signaling, Activin A, PKA, cAMP, and SIP1.
In conclusion, we report on the successful implementation of a multiplexed high-content assay and the execution of the first genome scale screen to identify modulators of miRNA biogenesis using stringent thresholds for hit scoring and nomination. We validated this multiplexed high-content assay strategy as a novel way to score for both nonessential and essential gene function with four core component genes of the biogenesis pathway identified. Our overall gene function analysis reveals four novel control junction points together, with several druggable genes as potential modulators of the miRNA biogenesis pathway. We report on 121 gene candidates as high-confidence hits. These newly identified genes and pathways may provide novel insights into the biogenesis machinery, with the ultimate goal of the development of small molecule therapeutics for controlling miRNA function in disease state.
Supplementary Material
Abbreviations
- AB
alamar blue
- BDA
Bhinder–Djaballah analysis
- BMP
bone morphogenetic protein
- DMEM
Dulbecco's modified Eagle's medium
- EGFP
enhanced green fluorescent protein
- FBS
fetal bovine serum
- GO
gene ontology
- H score
hit rate per gene
- INCA3000
IN CELL Analyzer 3000
- miRNAs
microRNAs
- NUCL
nuclei count
- OTE
off-target effect
- pre-miRNA
precursor-miRNA
- pri-miRNA
primary miRNA
- RISC
RNA-induced silencing complex
- RNAi
RNA interference
- siRNA
short interfering RNA
- TGF-β
transforming growth factor beta
- TRBP
tar-RNA binding protein
- UTR
untranslated region
- XPO5
Exportin-5
Acknowledgments
The authors wish to thank members of the High-Throughput Screening Core Facility for their help during the course of this study and Terry Helms (Medical Graphics, MSKCC) for her contribution to the artwork in this article. The HTS Core Facility is partially supported by Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Experimental Therapeutics Center of MSKCC, the William Randolph Hearst Fund in Experimental Therapeutics, the Lillian S. Wells Foundation, and by an NIH/NCI Cancer Center Support Grant 5 P30 CA008748-44.
Disclosure Statement
The authors declare no competing financial interests.
References
- 1.Lee RC. Feinbaum RL. Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 2.Reinhart BJ. Slack FJ. Basson M, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 3.Calin GA. Dumitru CD. Shimizu M, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Winter J. Jung S. Keller S. Gregory RI. Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 6.Gregory RI. Yan KP. Amuthan G, et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 7.Lee Y. Ahn C. Han J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 8.Lee Y. Han J. Yeom KH, et al. Drosha in primary microRNA processing. Cold Spring Harb Symp Quant Biol. 2006;71:51–57. doi: 10.1101/sqb.2006.71.041. [DOI] [PubMed] [Google Scholar]
- 9.Han J. Lee Y. Yeom KH, et al. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yi R. Qin Y. Macara IG. Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chendrimada TP. Gregory RI. Kumaraswamy E, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okamura K. Ishizuka A. Siomi H. Siomi MC. Distinct roles for Argonaute proteins in small RNA-directed RNA cleavage pathways. Genes Dev. 2004;18:1655–1666. doi: 10.1101/gad.1210204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kloosterman WP. Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 14.Garzon R. Calin GA. Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167–179. doi: 10.1146/annurev.med.59.053006.104707. [DOI] [PubMed] [Google Scholar]
- 15.Davis BN. Hilyard AC. Lagna G. Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukuda T. Yamagata K. Fujiyama S, et al. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat Cell Biol. 2007;9:604–611. doi: 10.1038/ncb1577. [DOI] [PubMed] [Google Scholar]
- 17.Adamson B. Smogorzewska A. Sigoillot FD, et al. A genome-wide homologous recombination screen identifies the RNA- binding protein RBMX as a component of the DNA-damage response. Nat Cell Biol. 2012;14:318–328. doi: 10.1038/ncb2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie L. Kassner M. Munoz RM, et al. Kinome-wide siRNA screening identifies molecular targets mediating the sensitivity of pancreatic cancer cells to Aurora kinase inhibitors. Biochem Pharmacol. 2012;83:452–461. doi: 10.1016/j.bcp.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shum D. Bhinder B. Radu C, et al. An image-based biosensor assay strategy to screen for modulators of the microRNA 21 biogenesis pathway. Comb Chem High Throughput Screen. 2012;15:529–541. doi: 10.2174/138620712801619131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhinder B. Djaballah H. A simple method for analyzing actives in random RNAi screens: introducing the “H score” for gene nomination and prioritization. Comb Chem High Throughput Screen. 2012;15:686–704. doi: 10.2174/138620712803519671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng A. Magdaleno S. Vlassov AV. Optimization of transfection conditions and analysis of siRNA potency using real-time PCR. Methods Mol Biol. 2011;764:199–213. doi: 10.1007/978-1-61779-188-8_13. [DOI] [PubMed] [Google Scholar]
- 22.Jacobsen N. Andreasen D. Mouritzen P. Profiling microRNAs by real-time PCR. Methods Mol Biol. 2011;732:39–54. doi: 10.1007/978-1-61779-083-6_4. [DOI] [PubMed] [Google Scholar]
- 23.Wang X. Wang X. Varma RK, et al. Selection of hyperfunctional siRNAs with improved potency and specificity. Nucleic Acids Res. 2009;37:e152. doi: 10.1093/nar/gkp864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karolchik D. Hinrichs AS. Furey TS, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D4966. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kozomara A. Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wishart DS. Knox C. Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robb GB. Rana TM. RNA helicase A interacts with RISC in human cells and function in RISC loading. Mol Cell. 2007;26:523–537. doi: 10.1016/j.molcel.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 28.Doi N. Zenno S. Ueda R, et al. Short-interfering-RNA-mediated gene silencing in mammalian cells requires Dicer and eIF2C translation initiation factors. Curr Biol. 2003;13:41–46. doi: 10.1016/s0960-9822(02)01394-5. [DOI] [PubMed] [Google Scholar]
- 29.Peters L. Meister G. Argonaute proteins: mediators of RNA silencing. Mol Cell. 2007;26:611–623. doi: 10.1016/j.molcel.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Daniels SM. Melendez-Pena CE. Scarborough RJ, et al. Characterization of the TRBP domain required for dicer interaction and function in RNA interference. BMC Mol Biol. 2009;10:38. doi: 10.1186/1471-2199-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lazzaretti D. Tournier I. Izaurralde E. The C-terminal domains of human TNRC6A, TNRC6B, and TNRC6C silence bound transcripts independently of Argonaute proteins. RNA. 2009;15:1059–1066. doi: 10.1261/rna.1606309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pshezhetsky AV. Hinek A. Serine carboxypeptidases in regulation of vasoconstriction and elastogenesis. Trends Cardiovasc Med. 2009;19:11–17. doi: 10.1016/j.tcm.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 33.Marcus AI. Peters U. Thomas SL, et al. Mitotic kinesin inhibitors induce mitotic arrest and cell death in Taxol-resistant and -sensitive cancer cells. J Biol Chem. 2005;280:11569–11577. doi: 10.1074/jbc.M413471200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reagan-Shaw S. Ahmad N. Silencing of polo-like kinase (Plk) 1 via siRNA causes induction of apoptosis and impairment of mitosis machinery in human prostate cancer cells: implications for the treatment of prostate cancer. FASEB J. 2005;19:611–613. doi: 10.1096/fj.04-2910fje. [DOI] [PubMed] [Google Scholar]
- 35.Wada T. Kikuchi J. Furukawa Y. Histone deacetylase 1 enhances microRNA processing via deacetylation of DGCR8. EMBO Rep. 2012;13:142–149. doi: 10.1038/embor.2011.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J. Yang Z. Liu J. Chen X. Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabidopsis. Curr Biol. 2005;15:1501–1507. doi: 10.1016/j.cub.2005.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee Y. Kim M. Han J, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pare JM. Tahbaz N. Lopez-Orozco J, et al. Hsp90 regulates the function of argonaute 2 and its recruitment to stress granules and P-bodies. Mol Biol Cell. 2009;20:3273–3284. doi: 10.1091/mbc.E09-01-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hwang HW. Wentzel EA. Mendell JT. Cell-cell contact globally activates microRNA biogenesis. Proc Natl Acad Sci USA. 2009;106:7016–7021. doi: 10.1073/pnas.0811523106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibbings DJ. Ciaudo C. Erhardt M. Voinnet O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol. 2009;11:1143–1149. doi: 10.1038/ncb1929. [DOI] [PubMed] [Google Scholar]
- 41.Vasudevan S. Tong Y. Steitz JA. Cell-cycle control of microRNA-mediated translation regulation. Cell Cycle. 2008;7:1545–1549. doi: 10.4161/cc.7.11.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parsons GG. Spencer CA. Mitotic repression of RNA polymerase II transcription is accompanied by release of transcription elongation complexes. Mol Cell Biol. 1997;17:5791–5802. doi: 10.1128/mcb.17.10.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JY. Moon HJ. Lee WK, et al. Merlin facilitates ubiquitination and degradation of transactivation-responsize RNA-binding protein. Oncogene. 2006;25:1143–1152. doi: 10.1038/sj.onc.1209150. [DOI] [PubMed] [Google Scholar]
- 44.Dueck A. Meister G. TRIMming microRNA function in mouse stem cells. Nat Cell Biol. 2009;11:1392–1393. doi: 10.1038/ncb1209-1392. [DOI] [PubMed] [Google Scholar]
- 45.Nishita M. Hashimoto MK. Ogata S, et al. Interaction between Wnt and TGF-beta signaling pathways during formation of Spemann's Organizer. Nature. 2000;403:781–785. doi: 10.1038/35001602. [DOI] [PubMed] [Google Scholar]
- 46.Sudbery I. Enright AJ. Fraser AG. Dunham I. Systematic analysis of off-target effects in an RNAi screen reveals microRNAs affecting sensitivity to TRAIL-induced apoptosis. BMC Genomics. 2010;11:175. doi: 10.1186/1471-2164-11-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parry DH. Xu J. Ruvkun G. A whole-genome RNAi Screen for C. elegans miRNA pathway genes. Curr Biol. 2007;17:2013–2022. doi: 10.1016/j.cub.2007.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamagata K. Fujiyama S. Ito S, et al. Maturation of microRNA is hormonally regulated by a nuclear receptor. Mol Cell. 2009;36:340–347. doi: 10.1016/j.molcel.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 49.Xue Z. Yuan H. Guo J. Liu Y. Reconstitution of an arognaute-dependent small RNA biogenesis pathway reveals a handover machanism involving the RNA exosome and the exonuclease QIP. Mol Cell. 2012;46:299–310. doi: 10.1016/j.molcel.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flynt AS. Greimann JC. Chung WJ, et al. MicroRNA biogenesis via splicing and exosome-mediated trimming in Drosophila. Mol Cell. 2010;38:900–907. doi: 10.1016/j.molcel.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Valadi H. Ekström K. Bossios A, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 52.Shaye DD. Greenwald I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 2011;6:e20085. doi: 10.1371/journal.pone.0020085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.