

Significance
Triple-negative breast cancers lack targeted therapies and are subdivided into molecular subtypes, including basal and claudin-low. Preclinical models representing these subtypes are limited. We have developed a murine model in which mammary gland expression of a receptor tyrosine kinase (MET) and loss of tumor suppressor gene p53 (Trp53), synergize to promote tumors with pathological and molecular features of claudin-low breast cancer. These tumors require MET signaling for proliferation, as well as mesenchymal characteristics, which are key features of claudin-low biology. This work associates MET expression and p53 loss with claudin-low breast cancers and highly proliferative breast cancers of poor outcome.
Keywords: Met RTK, EMT, mouse model, gene expression
Abstract
Triple-negative breast cancer (TNBC) accounts for ∼20% of cases and contributes to basal and claudin-low molecular subclasses of the disease. TNBCs have poor prognosis, display frequent mutations in tumor suppressor gene p53 (TP53), and lack targeted therapies. The MET receptor tyrosine kinase is elevated in TNBC and transgenic Met models (Metmt) develop basal-like tumors. To investigate collaborating events in the genesis of TNBC, we generated Metmt mice with conditional loss of murine p53 (Trp53) in mammary epithelia. Somatic Trp53 loss, in combination with Metmt, significantly increased tumor penetrance over Metmt or Trp53 loss alone. Unlike Metmt tumors, which are histologically diverse and enriched in a basal-like molecular signature, the majority of Metmt tumors with Trp53 loss displayed a spindloid pathology with a distinct molecular signature that resembles the human claudin-low subtype of TNBC, including diminished claudins, an epithelial-to-mesenchymal transition signature, and decreased expression of the microRNA-200 family. Moreover, although mammary specific loss of Trp53 promotes tumors with diverse pathologies, those with spindloid pathology and claudin-low signature display genomic Met amplification. In both models, MET activity is required for maintenance of the claudin-low morphological phenotype, in which MET inhibitors restore cell-cell junctions, rescue claudin 1 expression, and abrogate growth and dissemination of cells in vivo. Among human breast cancers, elevated levels of MET and stabilized TP53, indicative of mutation, correlate with highly proliferative TNBCs of poor outcome. This work shows synergy between MET and TP53 loss for claudin-low breast cancer, identifies a restricted claudin-low gene signature, and provides a rationale for anti-MET therapies in TNBC.
Despite recent improvements in breast cancer mortality, this disease remains the second leading cause of cancer-related deaths for women worldwide (1). Gene expression profiling and molecular pathology have revealed that breast cancers naturally divide into luminal A and B, human epidermal growth factor receptor 2 (HER2)-enriched, basal-like, and the recently identified claudin-low subtypes (2, 3). Targeted therapies that rely on tumor cell expression of estrogen and v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 (ErbB2) receptors can be effective in the treatment of luminal and HER2-positive breast cancers (4). However, basal-like and claudin-low breast cancers are predominately negative for these receptors, referred to as triple negative (TN), and are associated with poor prognosis. TN breast cancers account for up to 20% of breast cancer cases (5), emphasizing the need to identify molecular targets for their treatment.
Claudin-low tumors were originally distinguished from other subtypes on the basis of gene expression profiling (3) and have subsequently been correlated with tumors of metaplastic and medullary pathology (6). These tumors are characterized by loss of tight junction markers (notably claudins) and high expression of markers of epithelial-to-mesenchymal transition (EMT), in addition to being enriched for markers of mammary stem cells (6).
Signaling through MET, the receptor tyrosine kinase (RTK) for hepatocyte growth factor (HGF) influences diverse cellular processes during both developmental and cancer progression (7, 8). MET is expressed in the epithelium of numerous tissues, including breast, and regulates cell proliferation, migration, and invasion, as well as EMT (7, 8). Increased expression of MET is associated with TN breast cancers and correlates with poor outcome (8–11). In normal breast, activation of MET in ductal epithelium can occur through paracrine signaling, as a result of the secretion of HGF by stromal fibroblasts, and increased amounts of HGF are detected in serum of patients with breast cancer who have high-grade disease (12, 13).
Transgenic mice expressing a weakly oncogenic variant of Met under the control of the murine mammary tumor virus (MMTV) promoter (MMTV-Metmt), or knock-in of Metmt into its endogenous promoter, develop mammary tumors that are histologically diverse (14, 15). Consistent with elevated MET in TN breast cancer, 50% of MMTV-Metmt tumors exhibit a molecular signature of the basal-like subclass of human breast cancer and are positive for basal cytokeratins (14, 15). However, the long latency of the MMTV-Metmt model supports the requirement for cooperating oncogenic events. Loss-of-function mutations in the tumor suppressor gene TP53 (tumor protein p53) are detected in ∼80% of TN breast cancers (2). Interplay between TP53 and MET is supported by the observation that in a mouse model of mammary tumorigenesis involving Trp53 (murine p53) deletion, 73% of tumors carry amplification of Met (16). Moreover, Met mRNA levels are regulated by the p53-regulated microRNA (miRNA) miR34a (17). However, synergy between MET and Trp53 loss during mammary tumor formation has not been tested.
To study the consequences of Trp53 loss during MET-induced mammary tumorigenesis, we generated a conditional mouse model in which mammary gland–specific expression of Met (MMTV-Metmt) is combined with Cre-recombinase (MMTV-Cre)–mediated deletion of floxed Trp53 alleles in the mammary gland. We document a significant reduction in tumor latency coupled with a dramatic increase in tumor penetrance in MMTV-Metmt;Trp53fl/+;Cre mice compared with MMTV-Metmt and a significant increase in penetrance compared with Trp53fl/+;Cre mice. The majority of mammary tumors that arise in MMTV-Metmt;Trp53fl/+;Cre mice and Trp53fl/+;Cre mice possess a distinctive spindloid pathology, and a comparison of gene expression data with human breast cancer datasets reveals a significant correlation between these mammary tumors and human claudin-low breast cancer. In both cases, the claudin-low phenotype is correlated with amplification of Met and requires continuous MET signaling. This work highlights the fact that MET and TP53 loss act synergistically in promoting breast tumors and provides a model to study the claudin-low subtype.
Results
MMTV-Metmt;Trp53fl/+;Cre Tumors Exhibit a Predominately Spindloid Pathology.
To investigate the consequence of elevated MET in the absence of functional TP53, we generated a transgenic mouse model in which mammary gland expression of a weakly oncogenic MET receptor (MMTV-Metmt) is combined with conditional deletion of Trp53 in the mammary glands of FVB/N [Friend Leukaemia virus type B (susceptibility)-NIH] mice (MMTV-Metmt;Trp53fl/+;MMTV-Cre-recombinase). Compared with MMTV-Metmt or Trp53fl/+;Cre control mice, we observed a dramatic increase in tumor penetrance, going from 31% and 24%, respectively, to 70% for MMTV-Metmt;Trp53fl/+;Cre mice (Table 1 and Fig. 1A). Moreover, although the MMTV-Metmt model required multiple rounds of pregnancy to stimulate tumor development, 71% of virgin MMTV-Metmt;Trp53fl/+;Cre mice developed tumors (Table 1). Unlike the MMTV-Metmt model, in which a spectrum of tumor pathologies was observed (14), the majority of mammary tumors that arose in MMTV-Metmt;Trp53fl/+;Cre mice (80%) and, to a lesser extent, in Trp53fl/+;Cre mice (63%) displayed a spindloid pathology, with the remaining tumors being poorly differentiated adenocarcinomas (Fig. 1B).
Table 1.
Tumor penetrance and latency values for mammary tumor development in MMTV-Metmt;Trp53fl/+;Cre, MMTV-Metmt and Trp53fl/+;Cre mice
| Parity | Genotype | Tumor-bearing mice/total mice | Penetrance, % | Latency, d |
| Nulliparous | MMTV-Metmt;Trp53fl/+;Cre | 15/21 | 71.4 | 278 |
| Trp53fl/+;Cre | 4/12 | 33.3 | 305 | |
| Multiparous | MMTV-Metmt;Trp53fl/+;Cre | 13/19 | 68.4 | 280 |
| Trp53fl/+;Cre | 2/13 | 15 | 276 | |
| MMTV-Metmt | 16/52 | 31 | 430 |
Loss of mammary gland expression of Trp53 in the MMTV-Metmt model led to an increase in tumor penetrance and shortened latency, in addition to abrogating the requirement for parity for tumor development. Compared with Trp53fl/+;Cre control mice, MMTV-Met;Trp53fl/+;Cre mice developed tumors with a similar latency but at a significantly higher penetrance, indicating Met expression as an important event in tumor initiation.
Fig. 1.

MMTV-Metmt;Trp53fl/+;Cre mammary tumors are highly penetrant, have a spindloid pathology, and selectively amplify the endogenous Met locus. A Kaplan-Meier plot illustrates that MMTV-Metmt;Trp53fl/+;Cre (n = 35) and Trp53fl/+;Cre mice (n = 25) have similar tumor onsets (∼300 d), occurring earlier than tumors in MMTV-Metmt mice (n = 52) (∼400 d) (A). However, MMTV-Metmt;Trp53;Cre mice are associated with a significantly higher tumor penetrance (∼70%) compared with Trp53fl/+;Cre mice (∼24%), resulting in a steeper curve (A). Tumor pathology was similar between MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre mice, ranging from spindloid to poorly differentiated adenocarcinomas (B). Cells with enlarged nuclei (arrow in B, iv) and large areas of necrosis (outlined in B, iii) were common. Spindloid tumors often contained ducts with atypical morphology (Inset, B, ii). All MMTV-Metmt;Trp53fl/+;Cre tumors contained genomic amplification of Met and adjacent loci, as determined by array-CGH (C), a phenomenon also observed in Trp53fl/+Cre tumors of spindloid pathology but not in Trp53fl/+;Cre adenocarcinomas (Fig. S2). High expression and activation (phosphorylation) of endogenous MET in MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre tumors was confirmed by immunostaining (D). A Trp53fl/+;Cre adenocarcinoma without amplification of Met and little activated MET is shown as a comparison (D). (Scale bars, 50 µm.)
Cytokeratin (CK) expression can be used to infer the differentiation status of breast tumors (17, 18). Interestingly, although nonspindloid MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre adenocarcinomas expressed basal (CK14) and luminal (CK8/18) cytokeratins, as well as CK5 (associated with progenitor cells), spindloid tumors showed only weak and sporadic expression of all CKs tested (CK14, 8/18, 5/6) (Fig. S1A). Spindloid tumor cells stained strongly for the mesenchymal marker vimentin and were negative for the epithelial marker E-cadherin (Fig. S1), which is supportive of an EMT (20). Interestingly, coexpression of both cytokeratins and vimentin was detected by immunofluorescence in spindloid tumor cells as well as hyperplastic glands (Fig S1B), thus capturing EMTs. Together, these data support the idea that expression of activated MET in combination with the loss of Trp53 in the mouse mammary gland promotes the formation of tumors with high penetrance and pronounced features that are typical of EMT.
MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre Tumors Undergo Loss of Heterozygosity for Trp53 and Selectively Amplify the Endogenous Met Locus.
Models of mammary tumorigenesis involving loss of a single allele of a tumor suppressor gene frequently undergo loss of heterozygosity during tumor progression, resulting in loss of the second allele (21). Consistent with this, all MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre mammary tumors tested showed Cre-mediated deletion of the conditional Trp53 allele as well as loss of the wild-type (unfloxed) Trp53 allele (Fig. S2). As loss of TP53 is associated with genomic instability (22), we used array-based comparative genomic hybridization (aCGH) to investigate whether consistent chromosomal alterations were associated with the MMTV-Metmt;Trp53fl/+;Cre and/or Trp53fl/+;Cre tumors. In addition to validating loss of the Trp53 locus (Fig. S3C), array-CGH data also showed copy number changes consistent with human breast cancer. For example, three of seven MMTV-Metmt;Trp53fl/+;Cre spindloid tumors (but not Trp53fl/+;Cre spindloid tumors) showed gain of the locus encoding myelocytomatosis oncogene (Myc) (MsChr15:61.8Mb) (Fig. S3), which is amplified in 46.7% of human TN breast cancers of the claudin-low subclass (23). Although Myc amplification was not detected in Trp53fl/+;Cre spindloid tumors, both MMTV-Metmt;Trp53fl/+;Cre tumors and Trp53fl/+;Cre tumors with a spindloid component contained genomic amplification of the endogenous Met locus (Chr6 17.4–17.5Mb) (Fig. 1C and Fig. S3). Although variable, tumors contained a broad region of amplification at this locus (Chr6 16.7–18.2Mb), which included not only Met but also other genes adjacent to Met; including Cav1 (caveolin 1), Cav2 (caveolin 2), Wnt2 (wingless-related MMTV-integration site 2) and Cftr (cystic fibrosis transmembrane conductance regulator) (Fig. S3). Notably, amplification of Met was absent in all Trp53fl/+;Cre tumors of adenocarcinoma pathology. The association between Met amplification and Trp53-null mammary tumors of spindloid but not adenocarcinoma-type pathology is highly significant (P = 0.01786), supporting an association between Met amplification and Trp53-deficient tumors with spindle-cell pathology.
Consistent with Met amplification, MMTV-Metmt;Trp53fl/+;Cre tumors showed strong immunohistochemical staining for the endogenous murine MET protein (Fig. 1D). In tumors as well as tumor lysates, the murine MET protein was highly phosphorylated on tyrosines 1234/5 (within the activation loop), consistent with its amplification and constitutive activation (Fig. 1D and Fig. S4) (6). This supports a possible “addiction” of the tumors to MET signaling. Endogenous Met amplification in MMTV-Metmt;Trp53fl/+;Cre tumors correlated with repression of the MMTV-Metmt transgene (Fig. 1D and Fig. S4) and is consistent with suppression of the MMTV promoter after EMT, as shown previously (24). Notably, Trp53fl/+;Cre spindloid tumors, but not adenocarcinomas, also expressed elevated levels of endogenous murine MET at similar levels of activity to that of MMTV-Metmt;Trp53fl/+;Cre tumors (Fig. S4). Thus, genomic amplification of Met leads to constitutive activation of the MET RTK in the absence of its ligand HGF, supporting a potential dependency of these Trp53-deficient mammary tumors on MET signaling.
MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre Spindloid Tumors Are Characterized by a Strong EMT, Met Signaling Axis, and Significant Immune Infiltrate.
To gain insight into the contribution of Trp53 loss to Met-induced mammary tumorigenesis, gene expression profiles were generated from 14 MMTV-Metmt;Trp53fl/+;Cre, 8 Trp53fl/+;Cre tumors, 8 MMTV-Metmt tumors, and 11 whole mammary gland (mammary fat pad, MFP) controls. Unsupervised hierarchical clustering with those genes that have an interquartile range greater than or equal to 2 over all samples identified three distinct clusters (Fig. 2A). The clusters were associated with tumor pathology in which all MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors clustered together and tumors with an adenocarcinoma pathology clustered together, regardless of genotype. Normal mammary gland controls formed a distinct cluster away from the tumor samples. Genes differentially expressed between clusters are indicated in Dataset S1, Tables S1–S3.
Fig. 2.

MMTV-Metmt;Trp53fl/+;Cre spindloid tumors display elevated expression of genes associated with a mesenchymal, migratory phenotype and are distinct from MMTV-Metmt mammary tumors. Unsupervised hierarchical clustering identifies three distinct groups. In the first group, 12 MMTV-Metmt;Trp53fl/+;Cre tumors (blue) form a cluster with six Trp53fl/+;Cre tumors (yellow) and one MMTV-Metmt tumor (purple); this cluster represents tumors of predominantly spindloid pathology and with genomic amplification of Met. In the next cluster, poorly differentiated adenocarcinomas (two MMTV-Metmt;Trp53fl/+;Cre and two Trp53fl/+;Cre tumors) cluster with tumors of the MMTV-Metmt model. MMTV-Metmt tumors further segregate into solid and mixed subtypes in accordance with their pathology (14). Normal mammary gland controls (green) form the third cluster. Tumor characterizations below the heat map are represented in white for negative, black for positive, and gray for unknown. “Other_All” refers to tumors of various pathology types; for example, tumor A899 contained regions of spindloid and adenocarcinoma-type pathologies. Genes highly expressed in MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors are associated with cell migration and invasion, signaling through the MET receptor, and EMT (B). Low expression of cell-cell junction markers and moderate expression of epithelial cytokeratins is also observed (B). A number of these genes were validated by qRT-PCR (n = 5 MMTV-Metmt;Trp53fl/+;Cre, 5 Trp53f/+;Cre tumors, 3 MMTV-Metmt mixed tumors, and 3 MMTV-Metmt solid tumors); expression relative to wild-type mammary gland is shown. Error bars, SEM (C).
Compared with MMTV-Metmt tumors or normal MFP (Dataset S1, Tables S1–S3), a striking feature of MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors was high expression of several markers of the previously determined EMT core signature (Snai1/2, Twist1/2, and Zeb1/2) (Fig. 2 B and C) (25), weak expression of cytokeratins as observed by immunohistochemical (IHC) analysis (Fig. S1 and Fig. 2B), and decreased representation of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) processes such as cell-cell junction organization, tight junction, and cell junction maintenance (Fig. 2B and Dataset S1, Tables S4–S7).
Analysis of the genes differentially expressed between MMTV-Metmt;Trp53fl/+;Cre spindloid and MMTV-Metmt tumors also identified enrichment for GO and KEGG categories such as actin filament–based movement and regulation of cell projection organization (Dataset S1, Table S4 and Fig. 2B), as well as inflammatory response, positive regulation of macrophage chemotaxis, regulation of lymphocyte-mediated immunity, cytokine-cytokine receptor interaction, and chemokine signaling pathway (Dataset S1, Table S5). Consistent with this, high expression of several chemokines and chemokine receptors associated with monocyte and lymphocytic infiltration (Ccr1, Cxcl10, and Cxcl1) (Dataset S1, Table S2) (26, 27) suggested a strong inflammatory response in MMTV-Metmt;Trp53fl/+;Cre tumors. Immunostaining for the T- and B-lymphocyte markers CD3 and CD20 (Fig. S5 A and B) and the macrophage marker F4/80 (Fig. S5C) revealed elevated lymphocytic and macrophage content in MMTV-Metmt;Trp53fl/+;Cre spindloid tumors compared with in MMTV-Metmt tumors.
In addition, the GO analysis included the category HGF receptor signaling pathway, reflecting a strong MET signaling axis within MMTV-Metmt;Trp53fl/+;Cre tumors (Dataset S1, Table S4). Consistent with Met amplification and activation, both MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors show elevated expression of the Met gene, in addition to high expression of the MET receptor ligand Hgf, Cd44 (a potential coreceptor for MET) (28), Ets1, and Ybx1 (proposed transcriptional activators of Met) (Fig. 2B and Dataset S1, Tables S1–S3) (29, 30).
MMTV-Metmt;Trp53fl/+;Cre Tumors and Trp53fl/+;Cre Spindloid Tumors Cluster with the Claudin-Low Subtype of TN Breast Cancers.
To determine whether MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre tumors were representative of a subtype of human breast cancer, gene expression profiles were compared with those of Herschkowitz and colleagues (3). Notably, all MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre tumors of spindloid, but not adenocarcinoma, pathology clustered with the claudin-low subclass of human breast cancers (Fig. 3A). The human claudin-low subclass signature reflects high expression of transcriptional drivers of EMT and low expression of markers of adherens and tight junctions, such as E-cadherin and claudins 1, 3, 4, and 7 (6). As validated by quantitative RT-PCR, MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors showed similar expression of genes within this signature, expressing high levels of Snai1/2, Twist1/2, and Zeb1/2 (Fig. 2C) and low levels of claudins such as Cldn1,3,4 and 7 and E-cadherin (Fig. 2C). Importantly, application of a claudin-low subclass gene signature derived from human tumors (6) identified MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+Cre spindloid tumors as strongly correlative (P < 0.0001) (Fig. 3B). Conversely, application of the differentially expressed gene signature from MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors to human breast cancer subtypes induced a cluster of claudin-low subjects, and this human subtype was found to be highly associated with the signature derived from the murine spindloid tumors (P < 0.0001) (Fig. S6).
Fig. 3.

Gene and miRNA expression profiles of MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors correlate with those of human claudin-low breast cancer. A cross-species comparison with human breast cancer subtypes reveals that a large proportion of MMTV-Metmt;Trp53fl/+;Cre tumors and Trp53fl/+;Cre tumors cluster with the claudin-low molecular subclass at the level of gene expression (A). Application of a published claudin-low breast cancer gene expression signature to the mouse model data confirmed this association (P < 0.0001) (B) and showed that tumors of spindloid pathology were those that correlated with the signature. Similarly, a significant association in miRNA expression was identified through the application of a human claudin-low miRNA signature to MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre tumor data (P = 4 × 10−4) (C). To further identify genes associated with claudin-low tumor cell biology and to remove genes expressed by cells in the tumor microenvironment, an intersect of genes highly expressed in human claudin-low breast cancers, MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors (compared with MMTV-Metmt tumors) and human basal B (claudin-low) breast cancer cell lines, was generated (D). This comprised 36 genes (E), a selection of which was validated by qRT-PCR (n = 5 MMTV-Metmt;Trp53fl/+;Cre, 5 Trp53f/+;Cre tumors, 3 MMTV-Metmt mixed tumors, and 3 MMTV-Metmt solid tumors), data were normalized to wild-type mammary gland. Error bars, SEM (F).
MicroRNA expression profiles are also associated with human breast cancer pathological features and molecular subtypes (31–33). Using a signature of ∼50 significantly differentially expressed miRNAs that distinguish claudin-low tumors from other human breast cancer subtypes (33), we identified a near-homogeneous clustering of MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors that were highly associated with the signature (P = 0.0004) (Fig. 3C and Dataset S1, Table S8). Notably, consistent with a strong EMT gene expression signature, MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors showed a significant decrease in expression of miR-200 family members, whose targets include the transcription factors Zeb1/2 and are known inhibitors of EMT and stemness (34–36). Together, these analyses indicate that MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors, but not adenocarcinomas, share multiple features in common with human claudin-low breast cancers.
Identification of a Core Claudin-Low Gene Signature.
The human claudin-low gene signature constitutes 777 genes (6). To establish whether a restricted, core claudin-low signature could be identified and whether MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors share common features with human claudin-low tumors, we compared genes systematically highly expressed in MMTV-Metmt;Trp53fl/+;Cre spindloid tumors, Trp53fl/+;Cre spindloid tumors, human claudin-low tumors, and human basal B breast cancer cell lines (Fig. 3D). This analysis highlighted more than 700 genes that are expressed at elevated levels in either just MMTV-Metmt;Trp53fl/+;Cre or just Trp53fl/+;Cre tumors, but not the other, a proportional difference that was significantly higher than expected (P = 0.009). When overall gene variance was measured, Trp53fl/+;Cre spindloid tumors were significantly more heterogeneous than MMTV-Metmt;Trp53fl/+;Cre spindloid tumors (P < 2.2 × 10−16) (Fig. S7). It is possible that the higher degree of homogeneity observed among MMTV-Met;Trp53fl/+;Cre tumors may result from expression of the MMTV-Met transgene at the point of tumor initiation, whereas Trp53-null-alone tumors arise as a result of more stochastic tumorigenic events subsequent to Trp53 loss. Elevated genes in common between MMTV-Metmt;Trp53fl/+;Cre spindloid tumors, human claudin-low tumors, and basal B-cell lines were enriched for signatures related to EMT, HGF signaling, and immune infiltration (Dataset S1, Table S10). In contrast, genes uniquely elevated in Trp53fl/+;Cre spindloid tumors, human claudin-low tumors, and basal B-cell lines (but not MMTV-Metmt;Trp53fl/+;Cre spindloid tumors) had enrichment for signatures related to p53 function such as MDM2 and AURKB targets, in addition to apoptosis and chemotherapy response (Dataset S1, Table S10). Hence, although MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors are more similar to one another than to MMTV-Metmt tumors (Fig. 2A), these tumors are not identical.
In addition to differences, this analysis generated an intersect containing 36 genes in common among MMTV-Metmt;Trp53fl/+;Cre spindloid tumors, Trp53fl/+;Cre spindloid tumors, human claudin-low tumors, and human basal B breast cancer cell lines (Fig. 3D). Consistent with the highly mesenchymal phenotype of our murine as well as human claudin-low tumors, the core 36-gene intersect includes genes linked to EMT (Twist1, Zeb2, and Vim) in addition to actin cytoskeleton dynamics (Fscn1) (37), extracellular matrix interaction, and cell migration (Msn, lamb1, and Itga5) (38, 39) (Fig. 3 D and E). The 36-gene intersect also included the proinflammatory cytokine Il-18 and genes associated with poor-outcome breast cancers [Vegfc (40) and Ybx1 (41)]. To test whether the 36-gene intersect alone could identify human claudin-low tumors, we applied it to a human breast cancer dataset containing claudin-low patients (6). Compared with the published claudin-low predictor of Prat et al. (6), which includes 426 genes with elevated expression and 351 genes with decreased, the 36-gene intersect, which represents a small subset, identified claudin-low patients with an equivalent degree of accuracy as the published predictor (Fig. S8) (P < 0.0001). Thus, our 36-gene set is functionally equivalent at identifying human claudin-low tumors while elucidating core aspects of claudin-low biology, including potential biomarkers.
Claudin-Low EMT Phenotype Is Dependent on MET Kinase.
Met was identified within the intersect of MMTV-Metmt;Trp53fl/+;Cre tumors, Trp53fl/+;Cre tumors, and basal B-cell lines (Dataset S1, Table S9) and is also retained as part of the published claudin-low predictor (6). To establish whether MET is involved in the maintenance of claudin-low characteristics, primary cells from MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors, which amplify the endogenous Met locus and maintain a strong EMT morphology in culture, were treated with two small-molecule MET-kinase inhibitors (PHA665752 and Crizotinib) (Fig. S9). On inhibition of MET kinase activity, a striking change in cell morphology was observed in both MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre tumor cells. Cells lost their elongated mesenchymal morphology, formed cell-cell junctions positive for the tight junction marker zona occludens protein 1 (ZO-1), and remodeled their actin cytoskeleton with enhanced appearance of cortical actin (Fig. 4A). Consistent with the formation of cell-cell junctions and the loss of the EMT morphological phenotype, elevated levels of Claudin 1 protein (CLDN1) were observed (Fig. 4B), as well as an elevation in Cldn1 (Claudin 1) and Cdh1 (E-cadherin) mRNA (Fig. 4C). In contrast, and surprisingly, mRNA levels of EMT transcriptional drivers Snail, Twist, and Zeb were not significantly reduced (Fig. 4D). This demonstrates that continued MET signaling has an important role in regulating cell-cell junction disassembly, even in the presence of high levels of key EMT regulators, a characteristic of claudin-low tumor pathology.
Fig. 4.

Treatment of spindloid tumor cells with pharmacological MET inhibitors leads to reversal of the claudin-low phenotype. MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumor cells were treated in vitro with small-molecule inhibitors of MET kinase (PHA665752 [1 μM] or Crizotinib [1 μM]) for 48–72 h. On treatment, cells underwent a distinct morphological change from a mesenchymal to an epithelial-like state (A), which included the formation of cell-cell junctions, as demonstrated by the appearance of cortical actin and localization of ZO-1 at sites of cell-cell contact (A). (Scale bars, 20 μm.) This was also accompanied by elevated levels of Claudin1 protein, as shown by Western blotting (B). Although we also observed an increase in mRNA levels of Claudin1 (Cldn1) and E-cadherin (Cdh1) on Met inhibition (C), there was no corresponding decrease in genes that are well-established as transcriptional drivers of EMT (Twist1/2, Zeb1/2, and Snai1/2) (D). Averaged PCR data for four spindloid tumor cell lines (two MMTV-Metmt;Trp53fl/+;Cre and two Trp53fl/+;Cre lines) are presented. Error bars, SEM.
In addition to restoring tight junctions and reverting the mesenchymal cell morphology, MET inhibition resulted in significantly impaired proliferation of both MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumor cells, both under normal (adherent) growth conditions and in soft agar (Fig. 5 A–C). In addition, Annexin V and propidium iodide labeling revealed a significant decrease in the viability of cells that had been treated for 48 h with either PHA665752 or Crizotinib (Fig. 5 D and E). Together, these data support that both MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumor cells are dependent on MET activity for their proliferation and survival.
Fig. 5.
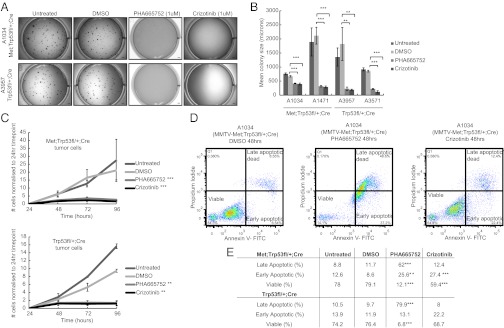
Inactivation of MET kinase inhibits the proliferation and survival of Met-amplified spindloid tumor cells. Tumor cells isolated from two MMTV-Metmt;Trp53fl/+;Cre and two Trp53fl/+;Cre spindloid mammary tumors formed smaller colonies in soft agar during a 10-d assay in the presence of MET kinase inhibitors (PHA665752 [1 μM] and Crizotinib [1 μM]); representative images for two cell lines are shown (A). (Scale bars, 1,000 μm.) Reduction in colony size was highly significant in all four cell lines (B). Error bars, SEM. Significantly impaired proliferation resulting from MET inhibition was also demonstrated in a 4-d proliferation assay in which the same cell lines were grown on tissue culture plastic and counted every 24 h (C). Error bars, SEM. To assess any effect on cell viability, cells treated with MET inhibitors for 48 h were stained with Annexin-V and propidium iodide and analyzed by flow cytometry. Representative plots for one MMTV-Metmt;Trp53fl/+;Cre cell line are shown (D), and averaged data for two MMTV-Metmt;Trp53fl/+;Cre and two Trp53fl/+ cell lines are tabulated (E). All four cell lines responded similarly and showed a dramatic increase in the proportion of cells in late-stage apoptosis after treatment with PHA665752 (e.g., 11.7% of MMTV-Metmt;Trp53fl/+;Cre cells were in late apoptosis in the DMSO control vs. 62% in the PHA667572 treatment). The effect of Crizotinib on cell viability was more moderate (only 12.4% of MMTV-Metmt;Trp53fl/+;Cre cells treated with Crizotinib were in late apoptosis). ***P < 0.01; **P < 0.05 (E).
MET Inhibition in Vivo Results in Decreased Metastatic Burden.
Despite the apparently aggressive phenotype of MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors, overt lung metastases were not observed. This may be because of the rapid proliferation of the primary tumors, which reach biological endpoint within 2 wk postpalpation. Alternatively, metastasis may be limited by an antitumor immune response, as could be suggested from the gene expression and immune profiling of these tumors (Fig. S5). To establish whether these cells are capable of invasive growth and metastatic spread, as is associated with MET signaling (7), we used a tail vein injection assay to determine whether MMTV-Metmt;Trp53fl/+;Cre spindloid tumor cells could grow in the lung microenvironment of immunocompromised mice. Introduction of a firefly luciferase gene allowed visualization of growth in vivo by bioluminescent imaging. MMTV-Metmt;Trp53fl/+;Cre spindloid tumor cells were highly aggressive, and by 3 wk postinjection were detected in both the lungs and liver of injected mice, in addition to other sites such as the lymph nodes and peritoneal cavity (Fig. 6). Examination of the lung and liver samples confirmed that MMTV-Metmt;Trp53fl/+;Cre tumor cells extravasate and proliferate as lesions external to the blood vessels (Fig. S10), indicating an invasive phenotype. The identification of cells at a variety of anatomical sites in this assay is unusual, as cells introduced via the tail vein bypass the normal metastatic cascade and are delivered directly to the lung, only rarely being detected in other organs (42–44). Notably, daily treatment of injected mice with the orally available MET inhibitor Crizotinib (45 mg⋅kg−1⋅d−1) significantly reduced metastatic growth both in the lungs and livers of the mice (Fig. 6), showing that the metastatic growth of these EMT mammary tumor cells is highly dependent on MET activity.
Fig. 6.

MET inhibition impairs the metastatic potential of spindloid mammary tumor cells. An MMTV-Metmt;Trp53fl/+;Cre spindloid tumor cell line expressing firefly luciferase was injected i.v. by the tail vein into 35 nude mice (0.5 × 106 cells/mouse). Mice were imaged on the day of injection (A) and twice per week thereafter to monitor the development of metastases. A control group of 15 mice was gavaged daily with water and compared with 20 mice receiving a daily gavage of Crizotinib (45 mg/kg/d). By day 24, control mice showed extensive metastatic burden compared with Crizotinib-treated mice (A). Lungs and livers were harvested from all animals at day 24 and scored histologically for metastatic lesions. Mice treated with Crizotinib showed a significant reduction in the number of lesions detected in both the lungs and liver (B). Representative histology from three control and three Crizotinib-treated mice is shown (C and D).
Elevated MET and TP53 Protein Correlates with Hormone Receptor-Negative Status and Poor Prognosis in Human Breast Cancer.
Alterations in TP53 are typically associated with the basal subtype of TN breast cancer (2). Missense mutations are associated with increased stability of the TP53 protein and can be detected by IHC analysis, as significantly higher tumor tissue staining is observed compared with tumors with TP53 truncating mutations or wild-type TP53 (45). Overexpression of MET and expression of mutant TP53 proteins have both been shown to have prognostic value individually; however, the significance of their coexistence in the same tumor has not been examined. The examination of MET and TP53 protein in a cohort of 618 axillary lymph node–negative human breast cancer cases (46) revealed that tumor epithelium was positive for MET immunostaining and/or TP53 staining, with an absence of staining in the stroma (Fig. 7A). Tumors that stained strongly for MET were more likely to be TP53 positive than those negative for MET, as 13.9% of all 618 tumors studied were MET+/TP53+ compared with 9.1% that were MET−/TP53+ (Fig. 7B) (P < 0.0001).
Fig. 7.

Elevated MET expression in human breast cancer is associated with TP53 mutation and combining MET with TP53 positive IHC identifies patients with poor prognosis. A human breast cancer tissue microarray comprising 618 node-negative patients was stained for MET and TP53 (A). Analysis showed that MET-positive tumors were more likely to stain positively for TP53 (indicative of mutated TP53) than MET-negative tumors (B) and that patients with MET-positive–TP53-positive tumors had a significantly worse outcome than patients with either MET or TP53 positivity alone (P = 0.0012) (C). Within TN patients specifically (n = 93), there was a trend toward MET-TP53 copositivity correlating with a poorer outcome (P = 0.3774), with a clear separation from patients with other combinations of MET and TP53 IHC within the first 36 mo after diagnosis.
Tumors that scored for both high MET and TP53 were observed in all histological subtypes, but a significantly greater proportion of MET/TP53 copositive tumors were estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and CK5-positive (61%, 71%, and 44%, respectively) than tumors with other combinations of MET and TP53 (24%, 38%, and 14%, respectively; P < 0.0001) (Dataset S1, Table S11). Basal, TN phenotype (TNP)-nonbasal, Her2, and luminal subtypes were determined as previously described (47). MET/P53 copositive tumors were found to correlate most significantly with the basal (P < 0.0001) and TNP-nonbasal (P = 0.0211) subtypes (Table 2). More precise identification of claudin-low patients would require an examination of a claudin-low gene expression signature within this set and/or the use of a positive IHC marker for claudin-low, which is currently not known. However, on the basis of the available information for this cohort, both of these subtypes could include patients of claudin-low pathology.
Table 2.
Association of MET-positive-, TP53-positive breast tumors with the basal and TNP-nonbasal subtypes
| MET+/TP53+ (n = 86) |
Other combinations of MET and TP53 (n = 532) |
||||
| Subgroup | No. | % | No. | % | P |
| Basal | |||||
| Yes | 26 | 30.2 | 42 | 7.9 | <0.0001 |
| No | 60 | 69.8 | 490 | 92.1 | |
| TNP-nonbasal | |||||
| Yes | 6 | 7.0 | 11 | 2.0 | 0.0211 |
| No | 80 | 93.0 | 521 | 98.0 | |
Scoring for MET and TP53 IHC on a human breast cancer tissue microarray was correlated with subtype. Breast cancers that stained positively for both MET and TP53 were more likely to be classified as basal, than breast cancers with other combinations of MET and TP53 staining (30.2% vs. 7.9%). Likewise, more MET/TP53 copositive breast cancers were classified as TNP-nonbasal, than breast cancers positive to MET or TP53 alone (7.0% vs. 2.0%).
The majority of MET/TP53-positive tumors (94%) scored high for cell proliferation marker KI67 compared with 57% for other combinations of MET and TP53 (P < 0.0001) (Dataset S1, Table S11). Consistent with this, combined MET/TP53-positive tumor status correlates with poor disease-free survival among lymph node–negative patients (Fig. 7C; log rank P = 0.0012) compared with patients with other combinations of MET/TP53 status, demonstrating that the combination of elevated MET with positive TP53 IHC is a strong predictor of poor outcome. This association persisted in multivariate analysis after adjustment for traditional histopathological prognostic factors (Dataset S1, Tables S11 and S12). Finally, MET/TP53 copositivity can also identify poor-outcome patients within the TN group alone (Fig. 7D). Together, these results strongly support a role for MET/TP53 signaling in human ER/PR-negative, CK5-positive breast cancers and in breast cancers with high KI67 staining and poor outcome.
Discussion
One of the challenges for the effective treatment of breast cancer is the heterogeneity of the disease (48). TN breast cancers alone encompass at least 2 (and potentially 6, some of which are more recently identified) (49) molecular subtypes referred to as basal-like and claudin-low (3, 6), for which there are a lack of known therapeutic targets and suitable animal models. Evidence supports that the MET RTK is elevated in human TN breast cancers (8). This, together with the observation that murine models expressing a weakly activated Met in the mammary epithelium develop tumors with basal-like characteristics, supports a role for MET in the development of basal-like mammary tumors (14, 15). However, the involvement of MET in other subtypes within TN or the ability of MET to synergize with known alterations in TN breast cancer has not been addressed. To create a more accurate model for human TN breast cancer, we have exploited the frequent occurrence of TP53 mutations in TN breast cancer and generated a model combining expression of a weakly oncogenic MET receptor (MMTV-Metmt) (14) with conditional deletion of Trp53 in the mammary glands of FVB/N mice (MMTV-Metmt;Trp53fl/+;MMTV-Cre-recombinase). The resulting MMTV-Metmt;Trp53fl/+;Cre mouse model shows effective cooperation of Met with Trp53 loss in mammary tumorigenesis, manifested as a significant increase in tumor penetrance over both MMTV-Metmt and Trp53fl/+;Cre control groups.
Notably, the majority of mammary tumors that form in the MMTV-Metmt;Trp53fl/+;Cre model (80%) share molecular features and histological markers of the claudin-low subtype of human TN breast cancer (6). Key aspects include enrichment for a claudin-low gene expression signature (P < 0.0001) (6) and miRNA signature, including loss of Claudin gene expression (e.g., Cldn1, Cldn3, Cldn4, and Cldn7), expression of the core EMT gene signature (Snai1/2, Twist1/2, and Zeb1/2), and lymphocytic infiltration (6, 23). This phenotype is shared by 5/8 Trp53fl/+;Cre tumors, which, in addition to loss of Trp53, show amplification of Met and a similar claudin-low gene expression signature to MMTV-Metmt;Trp53fl/+;Cre spindloid tumors. In contrast, MMTV-Metmt tumors clustered with basal and luminal subtypes (14), and only a single MMTV-Metmt tumor with a spontaneous Trp53 mutation, expressed a claudin-low signature (Fig. 3B). One important difference within Trp53fl/+;Cre tumors is that Met amplification was not detected in Trp53fl/+;Cre tumors of adenocarcinoma pathology. This indicates that loss of Trp53 alone, as evident in Trp53fl/+;Cre adenocarcinomas, is insufficient for spindloid pathology and a penetrant claudin-low phenotype and supports a synergistic role for Met, together with Trp53 loss, in promoting tumors with a spindloid pathology and claudin-low molecular subtype in the FVB background. This is consistent with the enhanced penetrance (70%) and high incidence of spindloid (80%), claudin-low-type tumors in MMTV-Metmt;Trp53fl/+;Cre mice.
Compared with other mouse mammary tumor models, MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre tumors of spindloid pathology clustered together and in close proximity to tumors belonging to models such as p53-null transplants, in addition to 7,12-Dimethylbenz(a)anthracene (DMBA), MMTV-CreBrca1co/co, and whey acidic protein (WAP)-Myc (Fig. S11). Interestingly, the WAP-Myc model can also induce tumors of spindloid pathology (3), and amplification of the Myc locus is observed in 3 of 7 of the MMTV-Metmt;Trp53fl/+;Cre spindloid tumors and 47% of human claudin-low tumors (23). However, although 80% of the MMTV-Metmt;Trp53fl/+;Cre tumors described here are spindloid or contain a spindle-cell component, only a fraction of tumors in the aforementioned models display this phenotype (3). Hence, MMTV-Metmt;Trp53fl/+;Cre tumors represent a robust model for efficient induction of claudin-low breast cancer. Similarly, only 10% of tumors arising in a transplant model of Trp53-null mammary epithelium display a claudin-low phenotype (50), providing further evidence that loss of Trp53 may be insufficient for this phenotype. Consistent with this, all Trp53fl/+;Cre tumors of spindloid pathology, correlating with a claudin-low subtype, contained amplification of the Met locus and variable adjacent genes. This links MET and P53 synergistically in promoting spindloid pathology and claudin-low like tumors in the FVB genetic background, especially as Trp53fl/+;Cre tumors of adenocarcinoma pathology did not amplify Met (Fig. S3A).
The mechanism selecting for Met amplification in the Trp53fl/+;Cre FVB model is unclear. A similar amplification of Met is observed in 73% of mammary tumors involving germ-line loss of Trp53 in combination with a conditional breast cancer 1 (Brca1) mutation (Brca1Δ11/co;MMTV-Cre;Trp53+/−) (16). However, although Met amplification in cell lines established from Brca1Δ11/co;MMTV-Cre;Trp53+/− tumors was carried on double minutes and lost from cells in culture (16), Met amplification in cell lines derived from MMTV-Met;Trp53fl/+;Cre and Trp53fl/+;Cre tumors is stable and retained during serial passage (Fig. S12). Moreover, these cell lines are continuously dependent on MET signaling for their EMT phenotype, as well as for their proliferation and survival both in culture and in vivo. Thus, Met amplification with consequent constitutive activation of the kinase is required to maintain the claudin-low mesenchymal phenotype of these cells. The unstable nature of the Met amplicon in the Brca1Δ11/co;MMTV-Cre;Trp53+/− model may reflect loss of function of Brca1, which contributes to chromosomal instability, whereas we observe no decrease in Brca1 or Brca2 expression in MMTV-Met;Trp53fl/+;Cre tumors compared with normal mammary gland (Dataset S1, Table S2). Interestingly, an amplicon containing Met was also recently detected in murine mammary tumors that arise as a result of potentiated Notch signaling and that also model both basal-like and claudin-low breast cancers (51). Although the stability of this amplicon was not addressed in this study, this lends further support for a specific role for MET signaling in murine models of claudin-low breast cancer.
Cell explants derived from MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid claudin-low-like tumors retain a mesenchymal phenotype that is highly dependent on continued MET signaling. When treated with two pharmacological MET inhibitors, a reversal of the EMT morphological phenotype was observed, with elevated levels of Claudin 1 and reformation of ZO-1 positive cell-cell junctions, which are claudin-dependent (52). Although the effect of MET signaling on tight junction disassembly is clear, we observed no changes in the mRNA levels of the core transcriptional drivers of EMT (Snai1/2, Twist1/2, and Zeb1/2) on MET inhibition (Fig. 4), demonstrating that continued MET activation is essential to maintain the EMT morphological phenotype and the loss of claudin gene expression, a hallmark of human claudin-low tumors (6).
Although MET can promote elevated expression of Zeb1 and Snail to initiate EMT (14), the core EMT signature is elevated in Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre spindloid tumors compared with MMTV-Metmt basal subtype tumors. This likely reflects the role for wild-type Trp53 in promoting an epithelial phenotype through transcriptional activation of the miR-200 family (underexpressed within the human claudin-low miRNA signature) that negatively regulates the key regulators of EMT (34). Consistent with this, after loss of Trp53 in MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre tumors, we observe a decrease in the miR-200 family and correspondingly high levels of EMT transcriptional drivers that are not altered after MET inhibition.
Accumulating evidence supports a role for MET and MET-dependent signals in human claudin-low breast cancer. MET contributes to a published claudin-low predictor (6). A strong MET signaling network is present in both MMTV-Metmt;Trp53fl/+;Cre and Trp53l/+;Cre tumors [Hgf, Cd44, Plaur (plasminogen activator, urokinase receptor), Plau (plasminogen activator, urokinase), Ets1 and Ybx1] (28–30, 53, 54), elements of which are also represented in the 36-gene intersect formed with human claudin-low tumors and basal B-cell lines (Cd44 and Ybx1) (Fig. 3E). The selection for amplification of the Met locus in Trp53- null tumors of spindloid pathology is striking and highlights an emerging concept in cancer whereby genes that function synergistically to enhance signaling will frequently be coselected during tumor formation or progression.
We propose that Met synergizes in this context with loss of function of Trp53 but may also synergize with other regulators of this phenotype such as Notch (51). The observed amplification of genes also amplified in human basal and claudin-low breast cancer such as Caveolin 1 and Myc in the MMTV-Metmt;Trp53fl/+;Cre model provides a valuable tool to understand the molecular events and signaling pathways that drive TN breast cancers. This model also presents an opportunity to study the tumor microenvironment of claudin-low breast cancer, as demonstrated by the evidence for robust leukocyte infiltration. Because human claudin-low breast cancer is especially difficult to treat due to the lack of biomarkers, determining molecular targets that can be used in drug therapy is of utmost importance. In addition, because small-molecule MET inhibitors are presently in clinical trials for multiple cancers, this raises the possibility that TP53 status may be important for patient selection.
Materials and Methods
Transgenic Mice.
MMTV-Metmt mice were described previously (14). MMTV-Cre mice were generated in the laboratory of W.J. Muller (55). Mice with floxed-Trp53 alleles are described elsewhere (21), were obtained from the National Cancer Institute mouse repository, and were bred onto a pure FVB background. Mice were housed in accordance with McGill University Animal Ethics Committee guidelines.
Immunohistochemical and Immunofluorescent Analyses of Mouse Tissue and Cell Lines.
Cells were fixed and histology samples prepared as described in SI Materials and Methods. Primary and secondary antibodies are detailed in Dataset S1, Table S13.
Microarray Data.
Gene expression profiles were generated using Agilent 4 × 44K whole-mouse genome gene expression microarrays. Copy number gains and losses were assessed using Agilent 4 × 44K whole-mouse genome CGH arrays. miRNA profiling was performed using the Agilent 8 × 15K mouse miRNA platform. Raw and normalized microarray data have been deposited in the Gene Expression Omnibus database under accession no. GSE41748. All analyses are detailed in the SI Materials and Methods.
Isolation and Culture of Mouse Mammary Tumor Cells.
Primary cells were isolated from mouse mammary tumors as described (56). Cells were cultured in DMEM supplemented with 5% (vol/vol) serum, epidermal growth factor (5 ng/mL), insulin (5 μg/mL), bovine pituitary extract (35 μg/mL), and hydrocortisone (1 μg/mL).
Met Inhibition.
MMTV-Metmt;Trp53fl/+;Cre and Trp53fl/+;Cre tumor cell lines were treated with PHA665752 (Pfizer) or Crizotinib (LC Laboratories) at a final concentration of 1 μM. Control cells were incubated with an equivalent concentration of DMSO alone for the same amount of time.
Supplementary Material
Acknowledgments
We thank Anie Monast for maintenance of our transgenic mouse colony and genotyping. We also thank Dr. Peter Siegel and Dr. Josie Ursini-Siegel for helpful discussions, the Goodman Cancer Research Centre Histology service, Ken McDonald and Diane Ethier for assistance with flow cytometry, and Dr. Pierre Lepage at the Genome Quebec Innovation Centre for loss-of-heterozygosity analysis. The Met inhibitor PHA665752 was a kind gift from Pfizer. We acknowledge infrastructure support and technical assistance from the Breast Cancer Functional Genomics Group, which is partially supported by funds from the Terry Fox New Frontiers Program. This research was supported with funds from a Terry Fox New Frontier grant (to M.P). in addition to funds from the Canadian Institutes for Health Research (to I.L.A. and M.P.)., a Susan G. Komen for the Cure postdoctoral fellowship (to J.F.K.), a McGill Faculty of Medicine Studentship (to S.M.S.), and a Canadian Institutes for Health Research studentship (to R.L.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE41748).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1210353110/-/DCSupplemental.
References
- 1.Ferlay, et al. Globocan 2008 v2.0, Cancer Incidence and Mortality Worldwide. IARC CancerBase 10. 2008. Available at https://globocan.iarc.fr.
- 2.Sørlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herschkowitz JI, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8(5):R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brenton JD, Carey LA, Ahmed AA, Caldas C. Molecular classification and molecular forecasting of breast cancer: Ready for clinical application? J Clin Oncol. 2005;23(29):7350–7360. doi: 10.1200/JCO.2005.03.3845. [DOI] [PubMed] [Google Scholar]
- 5.Sorlie T, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100(14):8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prat A, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4(12):915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 8.Ponzo MG, Park M. The Met receptor tyrosine kinase and basal breast cancer. Cell Cycle. 2010;9(6):1043–1050. doi: 10.4161/cc.9.6.11033. [DOI] [PubMed] [Google Scholar]
- 9.Camp RL, Rimm EB, Rimm DL. Met expression is associated with poor outcome in patients with axillary lymph node negative breast carcinoma. Cancer. 1999;86(11):2259–2265. doi: 10.1002/(sici)1097-0142(19991201)86:11<2259::aid-cncr13>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 10.Garcia S, et al. Poor prognosis in breast carcinomas correlates with increased expression of targetable CD146 and c-Met and with proteomic basal-like phenotype. Hum Pathol. 2007;38(6):830–841. doi: 10.1016/j.humpath.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 11.Ghoussoub RA, et al. Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer. 1998;82(8):1513–1520. doi: 10.1002/(sici)1097-0142(19980415)82:8<1513::aid-cncr13>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 12.Taniguchi T, et al. Serum concentrations of hepatocyte growth factor in breast cancer patients. Clin Cancer Res. 1995;1(9):1031–1034. [PubMed] [Google Scholar]
- 13.Tyan SW, et al. Breast cancer cells induce cancer-associated fibroblasts to secrete hepatocyte growth factor to enhance breast tumorigenesis. PLoS ONE. 2011;6(1):e15313. doi: 10.1371/journal.pone.0015313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ponzo MG, et al. Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc Natl Acad Sci USA. 2009;106(31):12903–12908. doi: 10.1073/pnas.0810402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graveel CR, et al. Met induces diverse mammary carcinomas in mice and is associated with human basal breast cancer. Proc Natl Acad Sci USA. 2009;106(31):12909–12914. doi: 10.1073/pnas.0810403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smolen GA, et al. Frequent met oncogene amplification in a Brca1/Trp53 mouse model of mammary tumorigenesis. Cancer Res. 2006;66(7):3452–3455. doi: 10.1158/0008-5472.CAN-05-4181. [DOI] [PubMed] [Google Scholar]
- 17.He L, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447(7148):1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 19.Lim E, et al. kConFab Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15(8):907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 20.Cardiff RD. The pathology of EMT in mouse mammary tumorigenesis. J Mammary Gland Biol Neoplasia. 2010;15(2):225–233. doi: 10.1007/s10911-010-9184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jonkers J, et al. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29(4):418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 22.Junttila MR, Evan GI. p53—a Jack of all trades but master of none. Nat Rev Cancer. 2009;9(11):821–829. doi: 10.1038/nrc2728. [DOI] [PubMed] [Google Scholar]
- 23.Weigman VJ, et al. Basal-like Breast cancer DNA copy number losses identify genes involved in genomic instability, response to therapy, and patient survival. Breast Cancer Res Treat. 2012;133(3):865–880. doi: 10.1007/s10549-011-1846-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White DE, Cardiff RD, Dedhar S, Muller WJ. Mammary epithelial-specific expression of the integrin-linked kinase (ILK) results in the induction of mammary gland hyperplasias and tumors in transgenic mice. Oncogene. 2001;20(48):7064–7072. doi: 10.1038/sj.onc.1204910. [DOI] [PubMed] [Google Scholar]
- 25.Taube JH, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA. 2010;107(35):15449–15454. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitamura T, Taketo MM. Keeping out the bad guys: Gateway to cellular target therapy. Cancer Res. 2007;67(21):10099–10102. doi: 10.1158/0008-5472.CAN-07-2100. [DOI] [PubMed] [Google Scholar]
- 27.Groom JR, Luster AD. CXCR3 ligands: Redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89(2):207–215. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matzke A, et al. Haploinsufficiency of c-Met in cd44-/- mice identifies a collaboration of CD44 and c-Met in vivo. Mol Cell Biol. 2007;27(24):8797–8806. doi: 10.1128/MCB.01355-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gambarotta G, et al. Ets up-regulates MET transcription. Oncogene. 1996;13(9):1911–1917. [PubMed] [Google Scholar]
- 30.Finkbeiner MR, et al. Profiling YB-1 target genes uncovers a new mechanism for MET receptor regulation in normal and malignant human mammary cells. Oncogene. 2009;28(11):1421–1431. doi: 10.1038/onc.2008.485. [DOI] [PubMed] [Google Scholar]
- 31.Blenkiron C, et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007;8(10):R214. doi: 10.1186/gb-2007-8-10-r214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iorio MV, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65(16):7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 33.Buffa FM, et al. microRNA-associated progression pathways and potential therapeutic targets identified by integrated mRNA and microRNA expression profiling in breast cancer. Cancer Res. 2011;71(17):5635–5645. doi: 10.1158/0008-5472.CAN-11-0489. [DOI] [PubMed] [Google Scholar]
- 34.Chang CJ, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13(3):317–323. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim T, et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208(5):875–883. doi: 10.1084/jem.20110235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gregory PA, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 37.Sedeh RS, et al. Structure, evolutionary conservation, and conformational dynamics of Homo sapiens fascin-1, an F-actin crosslinking protein. J Mol Biol. 2010;400(3):589–604. doi: 10.1016/j.jmb.2010.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nürnberg A, Kitzing T, Grosse R. Nucleating actin for invasion. Nat Rev Cancer. 2011;11(3):177–187. doi: 10.1038/nrc3003. [DOI] [PubMed] [Google Scholar]
- 39.Valastyan S, Benaich N, Chang A, Reinhardt F, Weinberg RA. Concomitant suppression of three target genes can explain the impact of a microRNA on metastasis. Genes Dev. 2009;23(22):2592–2597. doi: 10.1101/gad.1832709. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Mohammed RA, et al. Prognostic significance of vascular endothelial cell growth factors -A, -C and -D in breast cancer and their relationship with angio- and lymphangiogenesis. Br J Cancer. 2007;96(7):1092–1100. doi: 10.1038/sj.bjc.6603678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stratford AL, et al. Epidermal growth factor receptor (EGFR) is transcriptionally induced by the Y-box binding protein-1 (YB-1) and can be inhibited with Iressa in basal-like breast cancer, providing a potential target for therapy. Breast Cancer Res. 2007;9(5):R61. doi: 10.1186/bcr1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodale D, Phay C, Postenka CO, Keeney M, Allan AL. Characterization of tumor cell dissemination patterns in preclinical models of cancer metastasis using flow cytometry and laser scanning cytometry. Cytometry A. 2009;75(4):344–355. doi: 10.1002/cyto.a.20657. [DOI] [PubMed] [Google Scholar]
- 43.Francia G, Cruz-Munoz W, Man S, Xu P, Kerbel RS. Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat Rev Cancer. 2011;11(2):135–141. doi: 10.1038/nrc3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du YC, Chou CK, Klimstra DS, Varmus H. Receptor for hyaluronan-mediated motility isoform B promotes liver metastasis in a mouse model of multistep tumorigenesis and a tail vein assay for metastasis. Proc Natl Acad Sci USA. 2011;108(40):16753–16758. doi: 10.1073/pnas.1114022108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ozcelik H, Pinnaduwage D, Bull SB, Andrulis IL. Type of TP53 mutation and ERBB2 amplification affects survival in node-negative breast cancer. Breast Cancer Res Treat. 2007;105(3):255–265. doi: 10.1007/s10549-006-9452-0. [DOI] [PubMed] [Google Scholar]
- 46.Mulligan AM, Pinnaduwage D, Bull SB, O’Malley FP, Andrulis IL. Prognostic effect of basal-like breast cancers is time dependent: Evidence from tissue microarray studies on a lymph node-negative cohort. Clin Cancer Res. 2008;14(13):4168–4174. doi: 10.1158/1078-0432.CCR-07-4543. [DOI] [PubMed] [Google Scholar]
- 47.Voduc KD, et al. Breast cancer subtypes and the risk of local and regional relapse. J Clin Oncol. 2010;28(10):1684–1691. doi: 10.1200/JCO.2009.24.9284. [DOI] [PubMed] [Google Scholar]
- 48.Curtis C, et al. METABRIC Group The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lehmann BD, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herschkowitz JI, et al. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci USA. 2012;109(8):2778–2783. doi: 10.1073/pnas.1018862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu K, et al. Lunatic fringe deficiency cooperates with the Met/Caveolin gene amplicon to induce basal-like breast cancer. Cancer Cell. 2012;21(5):626–641. doi: 10.1016/j.ccr.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Furuse M. Molecular basis of the core structure of tight junctions. Cold Spring Harb Perspect Biol. 2010;2(1):a002907. doi: 10.1101/cshperspect.a002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zöller M. CD44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11(4):254–267. doi: 10.1038/nrc3023. [DOI] [PubMed] [Google Scholar]
- 54.Jeffers M, Rong S, Vande Woude GF. Enhanced tumorigenicity and invasion-metastasis by hepatocyte growth factor/scatter factor-met signalling in human cells concomitant with induction of the urokinase proteolysis network. Mol Cell Biol. 1996;16(3):1115–1125. doi: 10.1128/mcb.16.3.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andrechek ER, et al. Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc Natl Acad Sci USA. 2000;97(7):3444–3449. doi: 10.1073/pnas.050408497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ling C, Zuo D, Xue B, Muthuswamy S, Muller WJ. A novel role for 14-3-3sigma in regulating epithelial cell polarity. Genes Dev. 2010;24(9):947–956. doi: 10.1101/gad.1896810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.