

Abstract
The unfolded protein response (UPR) is a cellular response highly conserved in eukaryotes to obviate accumulation of misfolded proteins in the endoplasmic reticulum (ER). Inositol-requiring enzyme 1 (IRE1) catalyzes the cytoplasmic splicing of mRNA encoding bZIP transcription factors to activate the UPR signaling pathway. Arabidopsis IRE1 was recently shown to be involved in the cytoplasmic splicing of bZIP60 mRNA. In the present study, we demonstrated that an Arabidopsis mutant with defects in two IRE1 paralogs showed enhanced cell death upon ER stress compared with a mutant with defects in bZIP60 and wild type, suggesting an alternative function of IRE1 in the UPR. Analysis of our previous microarray data and subsequent quantitative PCR indicated degradation of mRNAs encoding secretory pathway proteins by tunicamycin, DTT, and heat in an IRE1-dependent manner. The degradation of mRNAs localized to the ER during the UPR was considered analogous to a molecular mechanism referred to as the regulated IRE1-dependent decay of mRNAs reported in metazoans. Another microarray analysis conducted in the condition repressing transcription with actinomycin D and a subsequent Gene Set Enrichment Analysis revealed the regulated IRE1-dependent decay of mRNAs-mediated degradation of a significant portion of mRNAs encoding the secretory pathway proteins. In the mutant with defects in IRE1, genes involved in the cytosolic protein response such as heat shock factor A2 were up-regulated by tunicamycin, indicating the connection between the UPR and the cytosolic protein response.
Keywords: heat stress, protein folding, bioinformatics
The unfolded protein response (UPR) or the endoplasmic reticulum (ER) stress response is a cellular response that is highly conserved in eukaryotes to obviate accumulation of misfolded proteins and to alleviate protein overload in the ER (1–3). Inositol-requiring enzyme 1 (IRE1), which is the primary transducer of the UPR in various organisms, catalyzes the unconventional or cytoplasmic splicing of mRNAs encoding bZIP transcription factors to up-regulate the UPR-related genes, such as genes for the ER-resident molecular chaperones, through its ribonuclease domain. The cytoplasmic splicing by IRE1 activates the bZIP transcription factors HAC1, XBP1, and bZIP60 in yeast, animals, and plants, respectively, by producing active proteins. Although the fundamental mechanism of the cytoplasmic splicing by IRE1 appears to be highly conserved, the mechanism of transcription factor activation is diverse among organisms (4).
In addition to the cytoplasmic splicing of mRNAs for transcription factors, other functions of metazoan IRE1 have been reported. One such function is the degradation of mRNA-encoding proteins in the secretory pathway referred to as regulated IRE1-dependent decay (RIDD) (5–7). RIDD is considered to contribute to reducing the amount of proteins entering the ER in the UPR. The metazoan UPR has an alternative mechanism to reduce the amount of protein entering the ER, and this mechanism is regulated by PKR-like ER kinase (PERK) (8). PERK attenuates protein translation through eIF2α phosphorylation, also resulting in the reduction of the protein load in the ER. Thus, the metazoan UPR has two roles: increasing the protein-folding capacity in the ER and decreasing the protein load in the ER (1). In plants, the IRE1–bZIP60 pathway, which increases protein-folding capacity, was recently discovered (9, 10); however, whether plants also have machinery to decrease protein load in the ER is yet to be elucidated. In addition to cytoplasmic splicing and RIDD, IRE1α in mammalian cells activates the apoptotic pathway under prolonged ER stress (1, 11). Although ER stress-mediated programmed cell death (PCD) has also been reported in Arabidopsis (12), the contribution of IRE1 to this process has not yet been examined.
Results
Enhanced Cell Death in an Arabidopsis Mutant with Defects in IRE1 During Acute ER Stress.
Arabidopsis contains two IRE1 paralogs, IRE1A and IRE1B. Our previous study showed distinct inhibition of germination in a mutant with a knockout of both genes (hereafter referred to as ire1a/b) by tunicamycin (Tm) compared with the wild-type (WT) and single-knockout mutants (ire1a and ire1b) (10). Inhibition of germination by Tm was more severe in ire1a/b than in WT and in bzip60, a bZIP60-knockout mutant, as shown in Fig. 1A. Similarly, germination was more severely inhibited by another ER stress inducer, DTT, in ire1a/b than in WT, bzip60, and ire1 single mutants (Fig. S1 A–C). Seedlings of ire1a/b were also more severely damaged by Tm (Fig. 1B), indicating enhanced cell death in ire1a/b. In addition, ion leakage, which indicates cell death, was prominent in ire1a/b after Tm treatment (Fig. 1C). Evans blue staining indicated enhanced cell death in ire1a/b (Fig. S1D). DNA fragmentation was observed in ire1a/b treated with Tm, but not in WT, suggesting that Tm-induced PCD was accelerated by defects in IRE1 (Fig. S1 E and F).
Fig. 1.
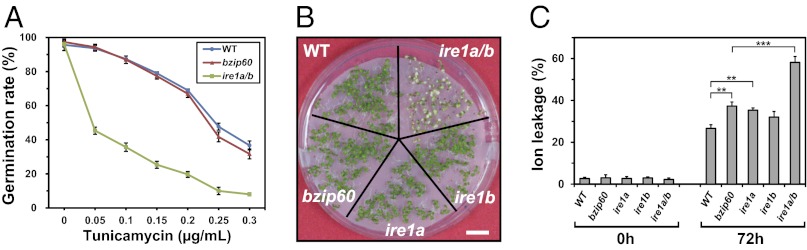
ire1a/b exhibited enhanced Tm-induced cell death. (A) The effects of Tm concentration on the germination of WT, bzip60, and ire1a/b. The germination rate was determined as described (10) from three independent experiments (n = 100). (B) Tm sensitivity of the mutants. Five-day-old seedlings of the indicated lines were treated with Tm (0.3 µg/mL) and grown for an additional 10 d to capture images as described in Methods. (C) Ion leakage after Tm treatment. Ten-day-old seedlings were treated with Tm (0.3 µg/mL) for 72 h, and ion leakage was measured as described in Methods. Data are means ± SD of at least three independent experiments. **P < 0.01; ***P < 0.001. P values were calculated by using the Student t test.
Degradation of mRNAs Encoding Secretory Pathway Proteins Induced by Tm Was Not Observed in ire1a/b.
The difference in sensitivity to Tm and DTT between ire1a/b and bzip60 suggested an alternative function of IRE1 other than activating bZIP60 in the UPR. To elucidate this function, we examined the result of our previous microarray (10) and found that Tm treatment down-regulated a batch of transcripts encoding secretory pathway proteins in WT, but not in ire1a/b; that is, among 125 genes down-regulated (fold change of <0.5, P < 0.05, and signal intensity of >30) in WT, including 118 genes (94.4%) encoding predicted signal sequences and/or transmembrane domains (SS/TM), the transcripts for 120 genes (96%) were unchanged or up-regulated in ire1a/b (Fig. 2A and Dataset S1). This observation suggested occurrence of RIDD in Arabidopsis as well as Drosophila and mammals (5–7).
Fig. 2.

IRE1-dependent down-regulation of transcripts by Tm. (A) Transcript level ratios (Tm/mock) of 125 genes considerably down-regulated (log2 < −1) by Tm (5 µg/mL) in WT (open circle) are shown on a log2 scale according to the degree of down-regulation. Ratios in ire1a/b are denoted by filled circles. Genes encoding predicted SS/TM are denoted by the gray bars. See Dataset S1 for detailed information. (B) Relative transcript abundance after Tm treatment of three selected genes and BiP1 as a control as presented on a log2 scale. RNA was prepared from WT, bzip60, and ire1a/b seedlings treated with DMSO (mock; open circle) or Tm (5 µg/mL) (filled circle) for the indicated periods and subjected to qPCR. Seedlings were pretreated with ActD (75 µM) for 2 h before Tm treatment (triangles). (C) The relative transcript abundance of PR-4 and PRX34 as presented on a log2 scale. RNA was prepared from WT and ire1a/b seedlings treated with various concentrations of Tm (black bars) or 2 mM DTT (gray bars) for 5 h and subjected to qPCR. Data are means ± SD of four independent experiments. Data for AT1G78850 are presented in Fig. S4B because of space limitations.
Transcripts of several genes after Tm treatment were monitored by quantitative real-time PCR (qPCR) in WT and the mutants. As shown in Fig. S2, induction of BiP3 and BiP1 observed in WT was severely and moderately reduced, respectively, in both bzip60 and ire1a/b, consistent with previous studies (10, 13). Their induction profiles in ire1a and ire1b were similar to that of WT, also as reported (10). Unspliced bZIP60 (bZIP60u) was up-regulated in WT and all of the mutants excluding bzip60, whereas up-regulation of spliced bZIP60 (bZIP60s) was not detected in ire1a/b, as reported (10). Transcripts for IRE1A and IRE1B were rather constant, as reported (14). To verify RIDD using these RNAs, genes for PR-4 (AT3G04720), the peroxidase PRX34 (AT3G49120), and a curculin-like (mannose-binding) lectin family protein (AT1G78850) were selected from the down-regulated genes in Fig. 2A. The transcripts for these three genes decreased after Tm treatment in WT, bzip60, ire1a, and ire1b (Fig. S3). However, in ire1a/b, the transcripts for PRX34 and AT1G78850 were unchanged or slightly increased after Tm treatment, and PR-4 transcripts were gradually decreased in both mock- and Tm-treated samples. Subsequently, seedlings were treated with actinomycin D (ActD) to prevent transcription. Up-regulation of BiP1 was clearly inhibited by ActD, indicating that transcription was inhibited (Fig. 2B). In contrast, the down-regulation of PR-4, PRX34, and AT1G78850 observed in WT and bzip60 was not affected by ActD. Thus, the decrease in mRNA abundance was considered to be due to mRNA degradation rather than transcriptional attenuation. Together, we concluded that both IRE1A and IRE1B contribute to RIDD in addition to the cytoplasmic splicing of bZIP60 mRNA.
Modulated Regulation of Cytoplasmic Splicing and RIDD in Different Stress Conditions.
Cytoplasmic splicing of bZIP60 and the subsequent induction of BiP3 were monitored in WT and ire1a/b after treatment with various concentrations of Tm and DTT. As shown in Fig. S4A, clear induction of bZIP60s and BiP3 was observed under each condition in WT, but not in ire1a/b, indicating the occurrence of IRE1-dependent UPR. The down-regulation of PR-4, PRX34, and AT1G78850 transcripts was also observed under each condition in an IRE1-dependent manner (Fig. 2C and Fig. S4B). Although we used different concentrations of Tm for cell death (0.3 μg/mL) and microarray (5 μg/mL) analyses, cytoplasmic splicing and RIDD did not appear to be largely affected by the Tm concentration. DTT (2 mM) prominently down-regulated (fold reduction of >80) PR-4 in WT, but not PRX34 and AT1G78850. This difference may have occurred due to induction of two latter genes by DTT.
In addition to Tm and DTT, heat is known to induce the cytoplasmic splicing of bZIP60 (9). Therefore, the profiles of BiP3, bZIP60s, PR-4, PRX34, and AT1G78850 after Tm, DTT, and heat treatment were monitored by qPCR in WT and ire1a/b. As shown in Fig. S4C, all treatments induced bZIP60s substantially within 30 min in WT, but only slightly in ire1a/b, whereas the effect of heat was rather small compared with that of Tm and DTT. Despite its lower effect on bZIP60s and BiP3 induction, heat clearly down-regulated the transcripts for PR-4, PRX34, and AT1G78850, indicating the occurrence of RIDD under heat stress. Although the down-regulation of these transcripts was detected within 30 min of DTT and heat treatment in WT, it was observed after 2 h in the case of Tm treatment. Down-regulation of transcripts was completely abolished in ire1a/b. In WT, transcripts for PRX34, which were rapidly down-regulated by DTT, increased to the basal level after 5 h. This increase is considered to be due to transcriptional activation by DTT because the transcripts were apparently up-regulated in ire1a/b. A similar expression profile was observed for AT1G78850. These observations were consistent with the results in Fig. 2C and Fig. S4B.
The drastic reduction in PR-4 transcripts in response to DTT was confirmed by Northern blotting (Fig. S5A). A 5′ RACE analysis of the PR-4 transcripts was performed by using a procedure independent of the cap structure of mRNA (15) (cRACE; see Methods for details) to detect degradation intermediates. As shown in Fig. S5B, seven independent RT-PCRs were performed by using mRNA prepared from WT and ire1a/b, which were untreated or treated with DTT for 0.5 and 1 h. Various sizes of amplicons, possibly derived from truncated PR-4, were detected in DTT-treated WT, whereas amplicons of identical size produced from full-length transcripts were obtained from untreated WT and DTT-treated ire1a/b. These results confirmed the degradation of PR-4 mRNA under DTT-induced UPR in WT, but not in ire1a/b.
Significant Portion of mRNAs Encoding Secretory Pathway Proteins Was Possibly Degraded by RIDD.
To identify possible targets of RIDD, microarray analysis was performed by using mock- and Tm-treated (5 µg/mL) WT and ire1a/b seedlings. All samples were treated with ActD (75 µM) for 2 h before Tm treatment to reduce the de novo transcription. Among 22,746 probes on an Affymetrix ATH1 chip, 9,413 probes with reliable signals (detection P < 0.05 and signal intensity of >200) were used for Gene Set Enrichment Analysis (GSEA). GSEA computationally determines the statistical significance between a list of ranked genes from transcriptome analysis and a gene set of any biological state by calculating the enrichment score (ES), which reflects the degree to which a gene set is overrepresented at the extremes (top or bottom) of a ranked gene list (16). We ordered the 9,413 genes according to the signal-to-noise ratio (mean divided by the SD) of Tm-treated WT and Tm-treated ire1a/b as a ranked gene list. A gene set containing 2,921 of the 9,413 genes encoding predicted SS/TM was created, and the analysis was performed by using GSEA software (Version 2.07). Fig. 3 shows the apparent correlation between down-regulated genes in Tm-treated WT compared with Tm-treated ire1a/b and genes for SS/TM, i.e., the negative enrichment of genes for the SS/TM gene set. GESA can define the core of a gene set that accounts for ES as the leading-edge subset. In the present study, 1,505 genes from the SS/TM gene set consisting of 2,921 genes comprised the leading-edge subset, as shown in the right part of the ES peak (−0.76) in Fig. 3 Upper. To evaluate this result in a different manner, we calculated the frequency of the appearance of genes in the SS/TM gene set in the ranked gene list (Fig. 3 Lower). The frequency of appearance of SS/TM genes increased from approximately the 6,000th ranked gene, and it was almost saturated at approximately the 8,000th gene. Although a similar correlation between Tm–down-regulated genes and SS/TM genes was observed in WT, this correlation was completely disordered in ire1a/b (Fig. S6A).
Fig. 3.

GSEA showing the correlation between RIDD targets and genes encoding secretory pathway proteins. (Upper) Enrichment plot of SS/TM genes (black bars) in the ranked gene list ordered by signal-to-noise scores, which are shown as a ranked list metric, of 9,413 genes derived from a microarray comparing WT and ire1a/b seedlings treated with Tm (5 µg/mL) in the presence of ActD (75 µM). The green line indicates ES calculated by GSEA. (Lower) Frequency of SS/TM gene appearance for every 20 genes in the ranked gene list was plotted.
Because RIDD was indicated to be induced by heat, as described , the effects of heat on the down-regulation of transcripts was estimated genome-wide by using data deposited in the Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo). Four microarray datasets related to heat stress (GSE12619, GSE18666, GSE19603, and GSE26266) obtained from the GEO database were used to create a ranked gene list consisting of 22,746 genes in descending order of fold change after heat treatment (Fig. S6B Upper). The frequency of appearance of SS/TM genes was also plotted (Fig. S6B Lower) to identify a significant correlation between the down-regulation of transcripts by heat and the prediction of SS/TM.
Independent of the aforementioned analysis, 12 gene sets presumably encoding proteins translocated in the ER were selected according to the cellular component aspect in the Gene Ontology (GO) database (www.geneontology.org/), and GSEA was performed for each gene set. In each gene set, down-regulated genes in Tm-treated WT were significantly [false discovery rate (FDR) < 0.01] enriched compared with Tm-treated ire1a/b (Fig. S6C). In this analysis, 1,213 of 2,849 genes (42.6%) were included in the leading edge subset.
Up-Regulation of Cytosolic Protein Response Genes in ire1a/b by ER Stress.
The enhanced cell death by Tm in ire1a/b observed in Fig. 1 and Fig. S1 may be due to defects in RIDD; i.e., in ire1a/b that cannot degrade ER-localized mRNAs, overloading of secretory pathway proteins may occur in the UPR, resulting in trigger of cell death pathway. To evaluate this prediction, the effects of ActD on Tm-induced cell death were examined. Although ion leakage was considerably higher in ire1a/b after Tm treatment, it was reduced by cotreatment of ActD (Fig.4A). We considered that transcriptional attenuation by ActD reduced the amount of mRNAs for secretory pathway proteins mimicking degradation of mRNA by IRE1 and thus alleviated Tm-induced cell death in ire1a/b. To further explore the physiological function of RIDD in Arabidopsis, we examined genes specifically up-regulated in ire1a/b in the UPR.
Fig. 4.

Effects of RIDD deficiency on cell death and CPR in the UPR. (A) Effects of ActD treatment on ion leakage after Tm treatment. Ten-day-old seedlings of the indicated lines were treated with Tm (0.3 µg/mL) and/or ActD (25 µM) for 72 h. When both TM and ActD were used, seedlings were treated with ActD for 2 h before Tm treatment. Data are means ± SD of six independent experiments. ***P < 0.001; n.s., not significant. P values were calculated by using the Student t test. (B) The relative transcript abundance of CPR genes in WT and mutant lines after 5 or 10 h of mock or Tm (5 µg/mL) treatment was quantified by qPCR. All qPCR data were normalized to the corresponding 5-h mock-treated sample. Data are means ± SD of four independent experiments. (C) Relative transcript abundance of HSP90.1 and HSFA2 in WT and ire1a/b seedlings treated with various concentrations of Tm for 5 h. The same RNA samples used in Fig. 2C were subjected to qPCR. Data are means ± SD of four independent experiments.
Dataset S2 represents 190 genes that were up-regulated (fold change of >4, P < 0.05 and signal intensity of >30) in ire1a/b but not in WT (fold increase of <2) by Tm; these genes were retrieved from our previous microarray data (10). Classification of these genes according to the biological process aspect in the GO database revealed that genes responding to stress or stimuli (GO: 0006950, 0009628, and 0009607) were enriched in the gene set (38.5% of the up-regulated 190 genes vs. 9.9% of all Arabidopsis genes) (Fig. S7A). In particular, genes for defense response (49 genes; GO: 0006952), response to heat (29 genes; GO: 0009408), oxidative stress (27 genes; GO: 0006979), and wounding (23 genes; GO: 0009611) were prominent.
Among these processes, we focused on heat stress because it can promote the accumulation of unfolded proteins in the cytosol and the ER. It is well known that a cellular response different from the UPR occurs in the cytosol if protein folding in the cytosol is disturbed. This response is referred to as the cytosolic protein response (CPR) (17), and genes induced in the CPR in Arabidopsis were identified by microarray-excluding genes induced in the UPR (18). However, 30 of 190 genes up-regulated by Tm in ire1a/b were overlapped with the CPR genes, similar to the heat stress-responsive genes (Fig. S7B).
Subsequently, we examined whether the CPR occurred specifically in ire1a/b after Tm treatment. The expression profiles of HSP90.1 and two heat shock factors, HSFA2 and HSFA7a, reported to be specifically induced during the CPR were monitored by qPCR. Distinct increases of the HSFs and HSP90.1 transcripts after 5 or 10 h of treatment with Tm (5 µg/mL) were observed in ire1a/b, but not in WT, bzip60, and ire1 single mutants (Fig. 4B). The induction of HSFA2-II, an alternative splicing variant of HSFA2 reported to indicate the CPR (18), was also specifically observed in ire1a/b. HSP90.1 and HSFA2 induction was not observed after treatment with 15 µg/mL Tm in WT, whereas they were induced by 0.3 µg/mL Tm in ire1a/b (Fig. 4C), indicating that the CPR in ire1a/b was not due to an overdose of the ER stress inducer.
Discussion
Prolongation of the UPR is believed to induce apoptosis or PCD in animals (1). The UPR-dependent apoptosis is considered to be mediated by IRE1α (7, 11), which is negatively regulated by BAX inhibitor-1 (BI-1) through a direct interaction (19). Cell death after prolonged treatment by Tm was also observed in cultured plant cells (20, 21), and ER stress-mediated PCD modulated by BI-1 was reported in Arabidopsis (12). Thus, the signaling machinery inducing PCD in the UPR is anticipated in plants, as in animals. However, the present study revealed that defects in IRE1 enhanced Tm-mediated PCD, implying a negative contribution of IRE1 to PCD in Arabidopsis. Given that the machinery of PCD is not highly conserved between animals and plants (22, 23), this inconsistency at a glance may not necessarily be controversial. Detailed characterization of the enhancement of cell death in ire1a/b is needed in the future to understand the function of IRE1 in ER stress-mediated PCD in plants.
Because the enhancement of cell death found in ire1a/b was not observed in bzip60, consistent with a previous report (24), an alternative function of IRE1A and IRE1B other than cytoplasmic splicing of bZIP60 mRNA was assumed. Analysis of our previous microarray data and subsequent qPCR analysis comparing transcripts levels between WT and ire1a/b clearly demonstrated the occurrence of RIDD in Arabidopsis in addition to Drosophila (5) and mammalian cells (6, 7, 25); that is, most genes (94.4%) down-regulated by Tm in WT, but not in ire1a/b, encoded the secretory pathway proteins, namely proteins with predicted SS/TM. Consistent with our previous study indicating the functional redundancy of IRE1A and IRE1B for the cytoplasmic splicing of bZIP60 (10), both IRE1A and IRE1B were revealed to contribute to the degradation of PR-4, PRX34, and AT1G78850 mRNAs. Because the down-regulation of transcripts by Tm was slightly retarded in ire1b (Fig. S3), IRE1B may play a more primary role in RIDD. Previous transcriptome studies revealed the down-regulation of transcripts for secretory pathway proteins in the UPR in Arabidopsis (26, 27), and the present study demonstrated involvement of IRE1 in the degradation of mRNAs. IRE1-dependent reduction of transcripts for some secretory pathway proteins was also recently reported in rice (28, 29). In addition to the cytoplasmic splicing of bZIP60 (9), RIDD was activated by heat. Analysis of transcriptomic data retrieved from four independent experiments deposited in the GEO database indicated that RIDD could be widely induced by heat stress.
Another microarray analysis comparing transcripts between WT and ire1a/b after Tm treatment was performed under the condition of ActD-mediated suppression of transcription, and 9,413 probes with reliable signals were subjected to GSEA to evaluate the contribution of RIDD. Among 9,413 genes, 2,921 genes described as SS/TM genes were predicted to encode proteins in the secretory pathway, consistent with the general description that one-third of proteins are synthesized in the ER (30). The gene set defined as the “leading-edge subset” by computational calculation that contributed to the negative enrichment of transcripts by Tm in WT, but not in ire1a/b, contained 1,505 of the 2,921 SS/TM genes. Thus, more than half (51.5%) of transcripts encoding proteins in the secretory pathway could be estimated to be targets of RIDD. The frequencies of the appearance of SS/TM genes among the 9,413 genes were also plotted, and the frequency started to increase at approximately the 6,000th gene (Fig. 3 Lower). If genes beyond this point (genes ranked 6,000th or later) were counted as being considerably down-regulated in WT, but not in ire1a/b, 66.3% of SS/TM gene transcripts could be regarded as targets of RIDD. Although these genome-wide analyses may have limitations because of the possible inaccurate prediction of SS/TM in the database and incomplete suppression of transcription by ActD, it is assumed that considerable portions of mRNAs encoding secretory pathway proteins could be degraded in an IRE1-dependent manner. In mammalian cells, certain secretory proteins are considered to be degraded before reaching the ER membrane in the UPR (31). This substrate-specific translocational attenuation during ER stress was defined as a “pre-emptive quality control,” and similar regulation was also suggested in plants (32). Thus, although a large segment of ER-localized mRNAs is considered to be degraded by IRE1 in the UPR in Arabidopsis, it may not necessarily be the case that all of the transcripts for SS/TM genes are RIDD targets.
Studies in mammalian cells indicated that the splicing of XBP1 could be artificially induced by mutated IRE1 without ER stress, whereas RIDD required IRE1 activity and ER stress (6, 7). Thus, the distinct regulation of two IRE1 outputs, cytoplasmic splicing and RIDD, was suggested in mammalian cells. In the present study, we also observed modulated outputs of the two IRE1 functions, splicing of bZIP60 and RIDD, in response to different ER stresses; that is, DTT and heat stress induced RIDD within 30 min, whereas Tm needed a longer period to induce its effects, even though bZIP60s appeared within 30 min of Tm treatment. The induction of bZIP60s by heat was less obvious than that by Tm and DTT, whereas RIDD induction by heat was apparent (Fig. S4C). These findings implied that the plant IRE1 may provide different catalytic modes for bZIP60 splicing and RIDD, specific and promiscuous cleavage of mRNA, as suggested for XBP1 splicing and RIDD in mammals (1). We monitored the IRE1-dependent endonucleolytic cleavage of PR-4 mRNA in vivo using cRACE, which amplifies the 5′ end of mRNA independent of the CAP structure (15). As shown in Fig. S5B, the size and sequence of amplified fragments were not identical, suggesting promiscuous cleavage by IRE1. Alternatively, IRE1 may cleave mRNA by recognizing partly conserved sequences, and then translationally inactive mRNA would be degraded by other RNases as suggested (5).
As described above, Arabidopsis IRE1s participate in RIDD in addition to the cytoplasmic splicing of bZIP60. Because ActD-mediated suppression of transcription alleviated Tm-induced cell death in ire1a/b, but not in WT and bzip60 (Fig. 4A), defects in RIDD in ire1a/b were assumed to enhance cell death under ER stress. However, given the proposed multifunction of IRE1 in mammals (33), we cannot rule out other possibilities for the enhanced cell death observed in ire1a/b. For instance, IRE1-mediated autophagy recently reported in Arabidopsis (34) may be involved in PCD in the UPR. In any case, the alternative function of IRE1 other than bZIP60 splicing is apparently crucial for ER homeostasis under serious ER stress.
A considerable number of the CPR genes were found among the stress-responsive genes that were up-regulated in ire1a/b under ER stress. The CPR is the cellular response corresponding to the accumulation of unfolded proteins in the cytoplasm, but not in the ER, and the UPR genes induced by Tm were excluded from the CPR genes (18). This observation can be interpreted as follows. The ribosome-nascent chain complex (RNC), which synthesizes proteins in the secretory pathway, is attached to the translocon in the ER membrane (35) to deliver the mRNA for secretory pathway proteins to the RNase domain of IRE1. In the UPR, IRE1 can thus randomly or specifically cleave mRNAs in RNC to reduce the polypeptides entering the ER lumen through the translocon. Degradation of mRNAs is considered to result in the dislocation of RNC from the ER membrane. It has been proposed that ER stress induces the disruption of the RNC–translocon seal, resulting in the exposure of nascent polypeptides that enter the ER lumen under a no-stress condition to the cytoplasm, where they may interact with cytosolic chaperones (36). Thus, in the absence of IRE1, RNC may be stuck on the ER membrane, and nascent polypeptides requiring cytosolic chaperones may overflow into the cytoplasm.
Inhibition of protein secretion into the culture medium by Tm has been reported (37–39). Incorporation of radioactive amino acids into microsomal fraction was not largely inhibited by Tm in these studies, and then RIDD may not seem to be involved in inhibition of protein secretion. Contribution of RIDD to protein synthesis needs to be carefully considered with further experimental data. Considering the lack of evidence of PERK homologs in plants (4), RIDD may have a more important function in the UPR in plants than in animals. Very recently, RIDD was reported in fission yeast, although it has not been reported in budding yeast (40). The primary function of IRE1 in evolution may be RIDD rather than cytoplasmic splicing of mRNA of transcription factors. The physiological importance and evolutionary conservation of RIDD in plants need to be elucidated in the near future.
Methods
Plant Lines and Stress Treatments.
T-DNA insertion mutants of Arabidopsis thaliana and the method of determining the germination rate have been described (10). To test the sensitivity of seedlings to Tm, 5-d-old seedlings grown on a nylon mesh placed on a 1/2 Murashige and Skoog agar plate containing 1% sucrose were transferred to a plate containing Tm (0.3 µg/mL) with a nylon mesh, grown for 4 d, retransferred to the plate without Tm, and further grown for 10 d to capture images. Other stress treatments were applied to 10-d-old seedlings in liquid medium as described (10). To inhibit de novo transcription, seedlings were treated with ActD (75 µM) for 2 h before stress treatments.
Evans Blue Staining.
Seedlings were treated with Tm (5 µg/mL) for 0–3 h, washed five times with culture medium without Tm, and further cultured for 24 h without Tm. Staining was conducted as described (41).
Ion Leakage.
Seedlings were washed five times with an excess of Milli-Q water. For Tm treatment, seedlings were incubated in Milli-Q water containing Tm (0.3 µg/mL) for 72 h. For ActD pretreatment, seedlings were incubated with 25 µM ActD for 2 h before Tm treatment. The conductivity of the bathing solution was measured with an electrical conductivity meter (B-173; Horiba). After autoclaving samples for 20 min, the conductivity was remeasured to obtain the total amount of ions in the cell. The ion leakage was presented as a percentage of the ratio of the conductivity before autoclaving to that after autoclaving.
DNA Fragmentation Analysis.
Seedlings were treated either with mock (0.1% DMSO) or Tm. DNA was extracted by using Nucleon PhytoPure DNA extraction kits (GE Healthcare), electrophoresed in a 2% (wt/vol) agarose gel, and subjected to Southern transfer. Hybridization was performed with a digoxigenin-labeled HaeIII/MspI-digested Arabidopsis genomic DNA probe at 44 °C.
RNA Preparation and Analyses.
For qPCR and cRACE (15), RNA was extracted by using a NucleoSpin RNA Plant kit (Takara) according to the manufacturer’s protocol. For qPCR, 500 ng of RNA was subjected to reverse transcription with random primers using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer’s protocol. Real-time PCR was performed in an ABI 7300 Real-Time PCR System (Applied Biosystems) using Thunderbird SYBR qPCR Mix (Toyobo), normalizing the transcript abundance to that of ACT8. cRACE was performed by using the 5′-Full RACE Core Set (Takara) according to the manufacturer’s protocol. The primers used for qPCR and cRACE are listed in Table S1. For RNA gel blot analysis, RNA was extracted according to the method described by Chomczynski and Sacchi (42). RNA gel blot analysis was conducted by using the PCR DIG Probe Synthesis Kit (Roche) as described (10). Primers used to prepare the probe are shown in Table S1.
Microarray.
WT and ire1a/b seedlings grown for 10 d were treated with ActD (75 µM) for 2 h followed by treatment with mock (0.1% DMSO) or Tm (5 µg/mL) for 5 h. The experiment was conducted with three biological replicates. RNAs prepared using an RNeasy Plant Mini Kit (Qiagen) according to the manufacturer’s instructions were subjected to microarray analysis using GeneChip (Affymetrix Arabidopsis ATH1 Genome Array) as described (10). The data were transformed to a log scale, and then statistical analyses were conducted by using R as described (43). The FDR and q value were calculated according to a report (44). Microarray data can be found in the GEO database (www.ncbi.nlm.nih.gov/geo) under accession no. GSE39690.
Bioinformatics.
Genes down-regulated by Tm in WT were extracted from our previous microarray data deposited in the ArrayExpress database (www.ebi.ac.uk/arrayexpress/) under accession no. E-MEXP-3186. Four public prediction programs, TargetP (www.cbs.dtu.dk/services/TargetP/) (45), SOSUI (http://bp.nuap.nagoya-u.ac.jp/sosui/) (46), TMHMM (www.cbs.dtu.dk/services/TMHMM/) (47), and HMMTOP (www.enzim.hu/hmmtop/) (48), were used to predict SS/TM in the derived amino acid sequences. If one of these programs detected SS/TM, then the corresponding gene was regarded as an SS/TM gene.
GSEA was performed by using GSEA software (Version 2.07) obtained from the Broad Institute according to a report (16). Gene sets comprising predicted SS/TM were built according to the TargetP information for SSs and TMHMM information for TMs. Gene sets of intracellular locations according to representative GO terms (www.geneontology.org/) were built by using the KAGIANA tool (49). The gene sets were screened against the GSEA-ranked microarray datasets to calculate ES for each gene set. Because of the small number of array samples (n = 3), we used “gene set” permutation (1,000 times) to assess the significance of ES. The distribution of GO terms of the biological process aspect assigned to the genes up-regulated in ire1a/b was compared with that for all Arabidopsis genes by using a gene-to-GO term relationship dataset (ATH_GO_GOSLIM.txt; downloaded July 5, 2012) obtained from The Arabidopsis Information Resource database (www.arabidopsis.org/).
Supplementary Material
Acknowledgments
We thank Ms. Chitose Takahashi, Akiko Sato, and Sachiko Oyama for technical assistance in GeneChip analysis. This work was supported by Ministry of Education, Culture, Sports, Science, and Technology of Japan Grant-in-Aid for Scientific Research 22020031 (to N.K.). Y.N. is a Research Fellow of the Japan Society for the Promotion of Science.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. A.V. is a guest editor invited by the Editorial Board.
Data deposition: The microarray data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE39690).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1219047110/-/DCSupplemental.
References
- 1.Walter P, Ron D. The unfolded protein response: From stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 2.Cao SS, Kaufman RJ. Unfolded protein response. Curr Biol. 2012;22(16):R622–R626. doi: 10.1016/j.cub.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Moore KA, Hollien J. The unfolded protein response in secretory cell function. Annu Rev Genet. 2012;46:165–183. doi: 10.1146/annurev-genet-110711-155644. [DOI] [PubMed] [Google Scholar]
- 4.Iwata Y, Koizumi N. Plant transducers of the endoplasmic reticulum unfolded protein response. Trends Plant Sci. 2012;17(12):720–727. doi: 10.1016/j.tplants.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 5.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313(5783):104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 6.Hollien J, et al. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186(3):323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han D, et al. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138(3):562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5(5):897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 9.Deng Y, et al. Heat induces the splicing by IRE1 of a mRNA encoding a transcription factor involved in the unfolded protein response in Arabidopsis. Proc Natl Acad Sci USA. 2011;108(17):7247–7252. doi: 10.1073/pnas.1102117108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagashima Y, et al. Arabidopsis IRE1 catalyses unconventional splicing of bZIP60 mRNA to produce the active transcription factor. Sci Rep. 2011;1:29. doi: 10.1038/srep00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe N, Lam E. BAX inhibitor-1 modulates endoplasmic reticulum stress-mediated programmed cell death in Arabidopsis. J Biol Chem. 2008;283(6):3200–3210. doi: 10.1074/jbc.M706659200. [DOI] [PubMed] [Google Scholar]
- 13.Iwata Y, Fedoroff NV, Koizumi N. Arabidopsis bZIP60 is a proteolysis-activated transcription factor involved in the endoplasmic reticulum stress response. Plant Cell. 2008;20(11):3107–3121. doi: 10.1105/tpc.108.061002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koizumi N, et al. Molecular characterization of two Arabidopsis Ire1 homologs, endoplasmic reticulum-located transmembrane protein kinases. Plant Physiol. 2001;127(3):949–962. [PMC free article] [PubMed] [Google Scholar]
- 15.Maruyama IN, Rakow TL, Maruyama HI. cRACE: A simple method for identification of the 5′ end of mRNAs. Nucleic Acids Res. 1995;23(18):3796–3797. doi: 10.1093/nar/23.18.3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subramanian A, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aparicio F, et al. Virus induction of heat shock protein 70 reflects a general response to protein accumulation in the plant cytosol. Plant Physiol. 2005;138(1):529–536. doi: 10.1104/pp.104.058958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sugio A, Dreos R, Aparicio F, Maule AJ. The cytosolic protein response as a subcomponent of the wider heat shock response in Arabidopsis. Plant Cell. 2009;21(2):642–654. doi: 10.1105/tpc.108.062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lisbona F, et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1α. Mol Cell. 2009;33(6):679–691. doi: 10.1016/j.molcel.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crosti P, Malerba M, Bianchetti R. Tunicamycin and Brefeldin A induce in plant cells a programmed cell death showing apoptotic features. Protoplasma. 2001;216(1-2):31–38. doi: 10.1007/BF02680128. [DOI] [PubMed] [Google Scholar]
- 21.Iwata Y, Koizumi N. Unfolded protein response followed by induction of cell death in cultured tobacco cells treated with tunicamycin. Planta. 2005;220(5):804–807. doi: 10.1007/s00425-004-1479-z. [DOI] [PubMed] [Google Scholar]
- 22.Hoeberichts FA, Woltering EJ. Multiple mediators of plant programmed cell death: Interplay of conserved cell death mechanisms and plant-specific regulators. Bioessays. 2003;25(1):47–57. doi: 10.1002/bies.10175. [DOI] [PubMed] [Google Scholar]
- 23.van Doorn WG, et al. Morphological classification of plant cell deaths. Cell Death Differ. 2011;18(8):1241–1246. doi: 10.1038/cdd.2011.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, Brandizzi F. AtIRE1A/AtIRE1B and AGB1 independently control two essential unfolded protein response pathways in Arabidopsis. Plant J. 2012;69(2):266–277. doi: 10.1111/j.1365-313X.2011.04788.x. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura D, et al. Mammalian ER stress sensor IRE1β specifically down-regulates the synthesis of secretory pathway proteins. FEBS Lett. 2011;585(1):133–138. doi: 10.1016/j.febslet.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 26.Martínez IM, Chrispeels MJ. Genomic analysis of the unfolded protein response in Arabidopsis shows its connection to important cellular processes. Plant Cell. 2003;15(2):561–576. doi: 10.1105/tpc.007609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwata Y, Sakiyama M, Lee MH, Koizumi N. Transcriptomic response of Arabidopsis thaliana to tunicamycin-induced endoplasmic reticulum stress. Plant Biotechnol. 2010;27(2):161–171. [Google Scholar]
- 28.Hayashi S, Wakasa Y, Takaiwa F. Functional integration between defence and IRE1-mediated ER stress response in rice. Sci Rep. 2012;2:670. doi: 10.1038/srep00670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wakasa Y, Hayashi S, Ozawa K, Takaiwa F. Multiple roles of the ER stress sensor IRE1 demonstrated by gene targeting in rice. Sci Rep. 2012;2:944. doi: 10.1038/srep00944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999;13(10):1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 31.Kang SW, et al. Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell. 2006;127(5):999–1013. doi: 10.1016/j.cell.2006.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall RS, et al. Signal peptide-regulated toxicity of a plant ribosome-inactivating protein during cell stress. Plant J. 2011;65(2):218–229. doi: 10.1111/j.1365-313X.2010.04413.x. [DOI] [PubMed] [Google Scholar]
- 33.Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: Assembling the IRE1α interactome. Mol Cell. 2009;35(5):551–561. doi: 10.1016/j.molcel.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, et al. Degradation of the endoplasmic reticulum by autophagy during endoplasmic reticulum stress in Arabidopsis. Plant Cell. 2012;24(11):4635–4651. doi: 10.1105/tpc.112.101535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walter P, Blobel G. Translocation of proteins across the endoplasmic reticulum III. Signal recognition protein (SRP) causes signal sequence-dependent and site-specific arrest of chain elongation that is released by microsomal membranes. J Cell Biol. 1981;91(2 Pt 1):557–561. doi: 10.1083/jcb.91.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oyadomari S, et al. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell. 2006;126(4):727–739. doi: 10.1016/j.cell.2006.06.051. [DOI] [PubMed] [Google Scholar]
- 37.Faye L, Chrispeels MJ. Apparent inhibition of β-fructosidase secretion by tunicamycin may be explained by breakdown of the unglycosylated protein during secretion. Plant Physiol. 1989;89(3):845–851. doi: 10.1104/pp.89.3.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Driouich A, Gonnet P, Makkie M, Laine AC, Faye L. The role of high-mannose and complex asparagine-linked glycans in the secretion and stability of glycoproteins. Planta. 1989;180(1):96–104. doi: 10.1007/BF02411415. [DOI] [PubMed] [Google Scholar]
- 39.Okushima Y, Koizumi N, Sano H. Glycosylation and its adequate processing is critical for protein secretion in tobacco BY2 cells. J Plant Physiol. 1999;154(5–6):623–627. [Google Scholar]
- 40.Kimmig P, et al. The unfolded protein response in fission yeast modulates stability of select mRNAs to maintain protein homeostasis. eLIFE. 2012;1:e00048. doi: 10.7554/eLife.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mergemann H, Sauter M. Ethylene induces epidermal cell death at the site of adventitious root emergence in rice. Plant Physiol. 2000;124(2):609–614. doi: 10.1104/pp.124.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chomczynski P, Sacchi N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: Twenty-something years on. Nat Protoc. 2006;1(2):581–585. doi: 10.1038/nprot.2006.83. [DOI] [PubMed] [Google Scholar]
- 43.Goda H, et al. The AtGenExpress hormone and chemical treatment data set: Experimental design, data evaluation, model data analysis and data access. Plant J. 2008;55(3):526–542. doi: 10.1111/j.0960-7412.2008.03510.x. [DOI] [PubMed] [Google Scholar]
- 44.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2(4):953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 46.Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14(4):378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 47.Möller S, Croning MDR, Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 2001;17(7):646–653. doi: 10.1093/bioinformatics/17.7.646. [DOI] [PubMed] [Google Scholar]
- 48.Tusnády GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17(9):849–850. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- 49.Ogata Y, et al. KAGIANA: An excel-based tool for retrieving summary information on Arabidopsis genes. Plant Cell Physiol. 2009;50(1):173–177. doi: 10.1093/pcp/pcn179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.