

The evolutionary pressure to populate rewarding niches can require organisms to survive high-risk environments. For bacteria that inhabit the nutrient-rich human gut, whether they are our friends or foes, the trip through the stomach requires clever strategies to survive harsh, low-pH conditions. Gut-resident Escherichia coli strains deploy a complex set of responses to counter the impact of the low pH they experience as they travel through the stomach (1). Some of their responses, such as the amino acid decarboxylases, act to keep the cytoplasmic pH above a dangerous level. However, the permeability of the outer membrane leaves the periplasmic space unprotected from the perilously low pH of the external medium. Consequently, these bacteria had to develop strategies to protect periplasmic proteins from irreversible pH denaturation. A network of chaperones participates in this protection; the key pH-responsive members are the small, abundant HdeA and HdeB proteins, the activity of which is triggered by low pH. In a recent study reported in PNAS, Foit et al. (2) apply a multipronged approach to learn how E. coli HdeA uses low pH-induced protonation of a small number of acidic residues to shift from an inactive, stably folded dimeric state to a partially folded monomer that is capable of reversibly binding unfolded substrates.
The mystery underlying HdeA function is how pH could change its properties in such a way that turns on its chaperone activity. Given the pH shift that this protein would experience between the stomach (pH 2) and the small intestine (pH 7), the most likely titratable residues involved in the modulation of activity are aspartate or glutamate. Foit et al. (2) identify several potential “pH switches” based on conservation of Asp and Glu residues in the HdeA family, the location of the conserved Asp and Glu residues on the structure of the inactive dimeric form of HdeA, and a powerful computational method called constant pH molecular dynamics (CpHmd) calculations (3). The CpHmd calculations are particularly informative, because they provide estimates of the pKa shifts each Asp and Glu residue would experience between the low-pH stable dimer (4) and models of the monomeric active state of HdeA built from the dimer. Strikingly, the authors find that mutation of the two aspartates predicted to experience the largest pKa shifts (D20 and D51), which were also among the most highly conserved acidic residues, to alanines created a variant HdeA that is constitutively active at neutral pH. Using thermodynamic coupling relationships, the pKa shifts were related to the expected extent of destabilization of the dimer by the Asp to Ala mutations, and, indeed, the two resulting HdeA variants showed a substantial reduction in apparent dimer-melting temperature. Mutation of other acidic residues, which were not predicted to shift pKa to as great an extent as D20 and D51, hardly perturbed the apparent dimer melting temperature. Notably, the pH-dependent ability of the D20, D51 HdeA variant to bind a fluorescent dye, used as a measure of the exposure of the hydrophobic surface, was shifted significantly toward higher pH relative to that of wild-type HdeA. Also, circular dichroism showed that the constitutively active double mutant had significantly reduced secondary structure at neutral pH than the wild-type protein, and the loss was similar to that triggered in wild-type HdeA by lowering pH. Most importantly, the double mutant completely blocked aggregation of unfolded malate dehydrogenase at pH 5, and substantially inhibited it at neutral pH, substantiating the identification of D20 and D51 as major pH-switch residues. Analytical ultracentrifugation analysis confirmed that the exposure of hydrophobic surface, loss of secondary structure, and increase in chaperone activity were all coincident with a shift in the wild-type HdeA dimer–monomer equilibrium toward monomer for the D20A, D51A variant.
How does HdeA function as a chaperone? Major classes of bacterial cytoplasmic chaperones like GroEL and DnaK use ATP binding and hydrolysis to switch between high- and low-affinity states and to set the timing of a cycle of binding and release of unfolded substrates. These mechanistic steps simultaneously optimize folding assistance and minimize accumulation of unfolded substrates, thus lowering the risk of aggregation. In the case of HdeA, which must function in the periplasm where there is no ATP, pH gradients appear to play a role comparable to ATP. Upon exposure to low pH, the HdeA dimer rapidly dissociates, and the monomer binds an array of different acid-denatured, periplasmic proteins (4, 5) (Fig. 1). Among them, intriguingly, are DegP and SurA, themselves periplasmic chaperones. The shift back to higher pH after the bacterium’s traversal of the stomach leads to a relatively slow release of bound clients from HdeA, apparently enabling refolding to occur with minimal risk of aggregation. It will be interesting to determine the rate of release and refolding of the DegP and SurA chaperones to see whether they might be available to facilitate the refolding of other HdeA clients. Similarly, it will be of interest to determine the role of HdeB, which shares many properties with HdeA but, from in vitro studies, has a different optimal pH range for its chaperone function (6). It seems likely that these two similar small periplasmic chaperones act as a synergistic team.
Fig. 1.
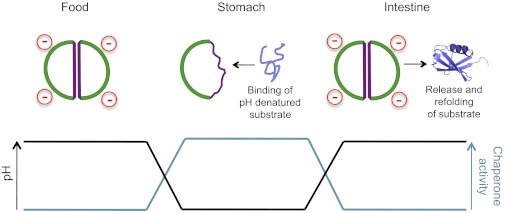
pH-dependent activity of HdeA (in green and purple outline) allows it to have maximal chaperone activity in the low-pH environment of the stomach. Here, periplasmic proteins (example in blue) will be destabilized by the harsh acidic conditions. Binding to HdeA protects them such that they may be released for refolding upon arrival of the bacterium in the small intestine. The work of Foit et al. (2), reported in PNAS, reveals how protonation of two key aspartic acid residues shifts HdeA to a partially unfolded, chaperone-active monomeric state.
The study by Foit et al. (2), combined with previous work from this group (7, 8), makes a compelling case for a direct link between partial unfolding in HdeA and its enhanced chaperone activity. Intrinsically disordered proteins (IDPs) and intrinsically disordered regions of proteins (IDRs) have recently entered the limelight of the protein science world (9, 10). Defying the dogma that 3D structure is required for a protein to function, IDPs and IDRs exist as ensembles of highly dynamic states. IDPs and IDRs are implicated in molecular recognition, signaling, interdomain linkages, etc., and their structural plasticity is implicated in their promiscuous binding (10, 11). Although the molecular details are not yet clear, the fact that HdeA chaperone activity is associated with its partially folded monomeric state suggests a role for conditionally disordered regions in HdeA in binding a wide array of unfolded substrates. These regions may work together with the intersubunit hydrophobic surface that is exposed in the monomer and sequestered in the stable HdeA dimer to mediate binding of unfolded substrates. Provocatively, IDRs have been suggested to have evolved as folding assistants (12), and, indeed, they are quite prevalent in chaperones (13): the mobile loop of GroES is a classic IDR (14); it serves as the GroEL-interactive site of GroES, shares its binding site with GroEL clients, and mimics the properties of clients. GroEL itself has unstructured C-terminal sequences that have been proposed both to interact with bound substrates (15) and to serve as malleable space fillers in reducing the GroEL cavity size (16). A disordered interdomain linker in heat shock protein (Hsp) 90 has been postulated to regulate substrate binding (17, 18). DnaK has two IDRs, the interdomain linker functions in allostery (19, 20), and the C-terminal disordered region has been proposed to bind protein substrates (21). Hsp33 has a disordered region that is exposed for substrate binding in a redox switchable manner (22). HdeA, thus, joins a growing number of chaperones in using disorder in its function (13).
The study by Foit et al. (2) elegantly illustrates the power of combining powerful computational modeling with well-designed experiments. The successful creation of an HdeA mutant that is active as a chaperone at neutral pH provides a promising system to elucidate its mechanism in greater depth. How disordered is the active form of HdeA? Do some parts of the protein retain structure, whereas others become unstructured, or is the active state globally dynamic? We eagerly await further details about the mode of substrate binding and the intrinsic dynamics of the monomeric state of HdeA, both of which will be greatly aided by the availability of the constitutively active mutant HdeA. The resulting insights will shed light on how disorder is exploited in this fascinating minimalist chaperone machine.
Acknowledgments
Work on in-cell protein folding and chaperone mechanisms in the laboratory of L.M.G. is supported by National Institutes of Health Grants GM027616 and OD00945. K.S.H. is supported in part by a fellowship from the University of Massachusetts, Amherst, as a part of the Chemistry-Biology Interface Program (funded by NIH Grant T32 GM08515).
Footnotes
The authors declare no conflict of interest.
See companion article on page E1254.
References
- 1.Hong W, Wu YE, Fu X, Chang Z. Chaperone-dependent mechanisms for acid resistance in enteric bacteria. Trends Microbiol. 2012;20(7):328–335. doi: 10.1016/j.tim.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Foit L, George JS, Zhang BW, Brooks CL, III, Bardwell JC. Chaperone activation by unfolding. Proc Natl Acad Sci USA. 2013;110:E1254–E1262. doi: 10.1073/pnas.1222458110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee MS, Salsbury FR, Jr, Brooks CL., 3rd Constant-pH molecular dynamics using continuous titration coordinates. Proteins. 2004;56(4):738–752. doi: 10.1002/prot.20128. [DOI] [PubMed] [Google Scholar]
- 4.Yang F, Gustafson KR, Boyd MR, Wlodawer A. Crystal structure of Escherichia coli HdeA. Nat Struct Biol. 1998;5(9):763–764. doi: 10.1038/1796. [DOI] [PubMed] [Google Scholar]
- 5.Zhang M, et al. A genetically incorporated crosslinker reveals chaperone cooperation in acid resistance. Nat Chem Biol. 2011;7(10):671–677. doi: 10.1038/nchembio.644. [DOI] [PubMed] [Google Scholar]
- 6.Abdallah J, Caldas T, Kthiri F, Kern R, Richarme G. YhbO protects cells against multiple stresses. J Bacteriol. 2007;189(24):9140–9144. doi: 10.1128/JB.01208-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tapley TL, Franzmann TM, Chakraborty S, Jakob U, Bardwell JC. Protein refolding by pH-triggered chaperone binding and release. Proc Natl Acad Sci USA. 2010;107(3):1071–1076. doi: 10.1073/pnas.0911610107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tapley TL, et al. Structural plasticity of an acid-activated chaperone allows promiscuous substrate binding. Proc Natl Acad Sci USA. 2009;106(14):5557–5562. doi: 10.1073/pnas.0811811106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turoverov KK, Kuznetsova IM, Uversky VN. The protein kingdom extended: Ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog Biophys Mol Biol. 2010;102(2-3):73–84. doi: 10.1016/j.pbiomolbio.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dyson HJ. Expanding the proteome: Disordered and alternatively folded proteins. Q Rev Biophys. 2011;44(4):467–518. doi: 10.1017/S0033583511000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown CJ, Johnson AK, Dunker AK, Daughdrill GW. Evolution and disorder. Curr Opin Struct Biol. 2011;21(3):441–446. doi: 10.1016/j.sbi.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kovacs D, Szabo B, Pancsa R, Tompa P. Intrinsically disordered proteins undergo and assist folding transitions in the proteome. Arch Biochem Biophys. 2013;531(1-2):80–89. doi: 10.1016/j.abb.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 13.Bardwell JC, Jakob U. Conditional disorder in chaperone action. Trends Biochem Sci. 2012;37(12):517–525. doi: 10.1016/j.tibs.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Landry SJ, Zeilstra-Ryalls J, Fayet O, Georgopoulos C, Gierasch LM. Characterization of a functionally important mobile domain of GroES. Nature. 1993;364(6434):255–258. doi: 10.1038/364255a0. [DOI] [PubMed] [Google Scholar]
- 15.Elad N, et al. Topologies of a substrate protein bound to the chaperonin GroEL. Mol Cell. 2007;26(3):415–426. doi: 10.1016/j.molcel.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang YC, et al. Structural features of the GroEL-GroES nano-cage required for rapid folding of encapsulated protein. Cell. 2006;125(5):903–914. doi: 10.1016/j.cell.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 17.Hainzl O, Lapina MC, Buchner J, Richter K. The charged linker region is an important regulator of Hsp90 function. J Biol Chem. 2009;284(34):22559–22567. doi: 10.1074/jbc.M109.031658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsutsumi S, et al. Charged linker sequence modulates eukaryotic heat shock protein 90 (Hsp90) chaperone activity. Proc Natl Acad Sci USA. 2012;109(8):2937–2942. doi: 10.1073/pnas.1114414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swain JF, et al. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell. 2007;26(1):27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogel M, Mayer MP, Bukau B. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J Biol Chem. 2006;281(50):38705–38711. doi: 10.1074/jbc.M609020200. [DOI] [PubMed] [Google Scholar]
- 21.Smock RG, Blackburn ME, Gierasch LM. Conserved, disordered C terminus of DnaK enhances cellular survival upon stress and DnaK in vitro chaperone activity. J Biol Chem. 2011;286(36):31821–31829. doi: 10.1074/jbc.M111.265835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reichmann D, et al. Order out of disorder: Working cycle of an intrinsically unfolded chaperone. Cell. 2012;148(5):947–957. doi: 10.1016/j.cell.2012.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]