

Abstract
Breakage-fusion-bridge (BFB) is a mechanism of genomic instability characterized by the joining and subsequent tearing apart of sister chromatids. When this process is repeated during multiple rounds of cell division, it leads to patterns of copy number increases of chromosomal segments as well as fold-back inversions where duplicated segments are arranged head-to-head. These structural variations can then drive tumorigenesis. BFB can be observed in progress using cytogenetic techniques, but generally BFB must be inferred from data such as microarrays or sequencing collected after BFB has ceased. Making correct inferences from this data is not straightforward, particularly given the complexity of some cancer genomes and BFB’s ability to generate a wide range of rearrangement patterns. Here we present algorithms to aid the interpretation of evidence for BFB. We first pose the BFB count-vector problem: given a chromosome segmentation and segment copy numbers, decide whether BFB can yield a chromosome with the given segment counts. We present a linear time algorithm for the problem, in contrast to a previous exponential time algorithm. We then combine this algorithm with fold-back inversions to develop tests for BFB. We show that, contingent on assumptions about cancer genome evolution, count vectors and fold-back inversions are sufficient evidence for detecting BFB. We apply the presented techniques to paired-end sequencing data from pancreatic tumors and confirm a previous finding of BFB as well as identify a chromosomal region likely rearranged by BFB cycles, demonstrating the practicality of our approach.
Keywords: bioinformatics, cancer genomics, combinatorial pattern matching, gene amplification
Genomic instability allows cells to acquire the functional capabilities needed to become cancerous (1), so understanding the origin and operation of genomic instability is crucial to finding effective treatments for cancer. Numerous mechanisms of genomic instability have been proposed (2), including the faulty repair of double-stranded DNA breaks by recombination or end-joining and polymerase hopping caused by replication fork collapse (3). These mechanisms are generally not directly observable, so their elucidation requires the deciphering of often subtle clues after genomic instability has ceased.
In contrast, the breakage-fusion-bridge (BFB) mechanism creates gross chromosomal abnormalities that can be seen in progress using methods that have been available for decades (4). BFB begins when a chromosome loses a telomere (Fig. 1 A and B). Then, during replication, the two sister chromatids of the telomere-lacking chromosome fuse together (Fig. 1 C and D). During anaphase, as the centromeres of the chromosome migrate to opposite ends of the cell (Fig. 1E), the fused chromatids are torn apart (Fig. 1F). Each daughter cell receives a chromosome missing a telomere, and the cycle can begin again. As this process repeats, it can lead to the rapid accumulation of amplifications and rearrangements that facilitate the transition to malignancy (5).
Fig. 1.
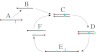
Schematic BFB process.
This process produces several identifiable cytogenetic signatures such as anaphase bridges and dicentric chromosomes. However, as cancer genomics has shifted to high-throughput techniques, the signatures of BFB have become less clear. Methods such as microarrays and sequencing do not allow for direct observation of BFB, and similar to other mechanisms, BFB must be inferred from its footprint in complex data.
Multiple groups have begun to address the problem of finding evidence for BFB in high-throughput data. For example, Bignell et al. found a pattern of inversions and exponentially increasing copy numbers “[bearing] all the architectural hallmarks at the sequence level” of BFB (6). Kitada and Yamasaki found a pattern of copy counts and segment organization consistent with a particular set of BFB cycles (7). Hillmer et al. used paired-end sequencing to find patterns of inversions and amplification explainable by BFB (8).
The procedures of these investigators, among others (9–11), share an element in common: They determine whether a particular observation is consistent with or could be explained by BFB. Although this is helpful, it does not on its own allow one to infer whether or not BFB occurred. Indeed, in a previous work (12) we examined short patterns of copy number increases consisting of five or six chromosome segments. We found that many such patterns, whether produced by BFB or not, were consistent or nearly consistent with BFB. Thus, finding that such a pattern was consistent with BFB would only be weak evidence that it had been produced by BFB. This finding highlights the need for a rigorous and systematic approach to the interpretation of modern data for BFB to avoid being misled by the complexity of cancer genomes and the BFB mechanism itself.
Here we present a framework for interpreting high-throughput data for signatures of BFB. We incorporate observations of breakpoints as well as copy numbers to create a scoring scheme for chromosomes. Through simulations, we find appropriate threshold scores for labeling a chromosome as having undergone BFB based on varying models of cancer genome evolution and tolerances for error. This framework complements the work of previous groups by not only finding breakpoint and copy number patterns consistent with BFB but also showing under what assumptions they are more likely to be observed if BFB occurred than if it did not.
The key technical contribution that underlies our scoring scheme is a fast algorithm for determining if a given pattern of copy counts is consistent with BFB. This algorithm is related to a previously described algorithm (12) in that it takes advantage of a distinctive feature of BFB: When fused chromatids are torn apart, they may not tear at the site of fusion. This yields chromosomes with either a terminal deletion or a terminal inverted duplication. When a chromosome undergoes this process repeatedly, it results in particular patterns of copy number increases. The running time of the earlier algorithm grew exponentially with the amount of amplification and the number of segments in a copy number pattern. This greatly narrowed the scope of copy number patterns that could be investigated. This was particularly limiting as it appeared that copy number patterns with more segments would be more useful for identifying BFB, but these patterns could not be evaluated in a reasonable amount of time with the previous method. The algorithm presented here is linear time and therefore allows complex copy number patterns to be checked in a trivial amount of time.
We begin by describing the kinds of high-throughput data that can provide evidence for BFB. We then lay out some formalization needed to precisely describe scoring methods of samples based on BFB evidence implied from such data. Next, we define related computational problems, followed by an outline of algorithms for these problems. In Results, we detail the simulations used to measure the performance of our scoring system. Based on simulation parameters, we find false and true positive rates for different BFB signatures. We apply our methods to two datasets. The first is copy number data from 746 cancer cell lines (13). We find three chromosomes that have long copy number patterns consistent with BFB, but the false positive rates from our simulations suggest that these may be false discoveries. We also examine paired-end sequencing data from pancreatic cancers. We find two chromosomes that likely have undergone BFB, one that was also identified in ref. 14 and one additional finding.
High-Throughput Evidence for BFB
We consider two experimental sources for evidence for BFB: microarrays and sequencing. Microarrays allow for the estimation of the copy number of segments of a chromosome by measuring probe intensities (15). Sequencing also yields copy number estimates by measuring depth of sequence coverage (16). In addition, sequencing can reveal genomic breakpoints where different portions of the genome are unexpectedly adjacent. This is generally the extent of evidence available from either technique. Sequencing does not allow for a full reconstruction of a rearranged chromosome, as the repetitive nature of the genome leads to multiple alternative assemblies. Neither method can resolve segment copy numbers by orientation, so copy numbers from both forward and reversed chromosome segments are summed. Nevertheless, BFB should leave its signature both in breakpoints and copy counts, and we examine each in turn.
Breakpoints.
During BFB, the telomere-lacking sister chromatids are fused together. This causes the ends of the sister chromatids to become adjacent but in opposite orientations (Fig. 1D). This adjacency is unlikely to be disrupted by subsequent BFB cycles and will remain in the final sequence as two duplicated segments arranged head-to-head. If the chromosome is paired-end sequenced, the rearrangement will appear as two ends that map very near each other but in opposite orientations. This type of rearrangement has been termed a “fold-back inversion” (14), and regions of a chromosome rearranged by BFB should have an enrichment of these fold-back inversions. Reliable indications for fold-back inversions may or may not be available, depending on the type of experiment and its intensity.
Copy Counts.
Each BFB cycle duplicates some telomeric portion of the chromosome undergoing BFB. These repeated duplications lead to certain characteristic copy number patterns. We would like to evaluate copy numbers observed from microarrays or sequencing and determine if the copy numbers contain the footprint of BFB. Previous groups have searched for such a footprint by manually inspecting copy number data and searching for a set of BFB cycles that could produce the observed copy numbers (6, 7). This approach is challenging and labor intensive, but developing a more general approach turns out to be rather difficult. A key technical contribution of this paper is the development of efficient algorithms to evaluate copy counts for consistency with BFB.
Formalizing BFB.
Creating an efficient method for evaluating copy numbers requires some formalization, so we begin with some definitions and basic results.
We represent a chromosome as a string ABC…, where each letter corresponds to a contiguous segment of the chromosome. For example, the string ABCD would symbolize a chromosome arm composed of four segments, where A is the segment nearest the centromere. More generally, we use σl for the lth segment in a chromosome. So, ABCD could be written σ1σ2σ3σ4. A bar notation 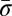 is used to signify that a segment is reversed. Greek letters α, β, γ, ρ denote concatenations of chromosomal segments, and a bar will again mean that the concatenation is reversed. For example, if
is used to signify that a segment is reversed. Greek letters α, β, γ, ρ denote concatenations of chromosomal segments, and a bar will again mean that the concatenation is reversed. For example, if 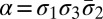 ,
, 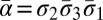 . An empty string is denoted by ɛ.
. An empty string is denoted by ɛ.
Consider the following BFB cycle on a chromosome X • ABCD, where “•” represents the centromere, X is one chromosomal arm, and ABCD is the four-segmented other chromosomal arm which has lost a telomere. The cycle starts with the duplication of the chromosome into two sister chromatids and their fusion at the ends of the “D” segments, generating the dicentric chromosome 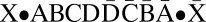 . During anaphase, the two centromeres migrate to opposite poles of the cell and a breakage of the dicentric chromosome occurs between the centromeres, say between
. During anaphase, the two centromeres migrate to opposite poles of the cell and a breakage of the dicentric chromosome occurs between the centromeres, say between 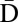 and
and 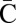 , providing one daughter cell with the chromosome
, providing one daughter cell with the chromosome 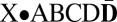 , and another daughter cell with the chromosome X•ABC (chromosomes
, and another daughter cell with the chromosome X•ABC (chromosomes 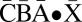 and X•ABC are equivalent). The now-amplified segment D in the first daughter cell may confer some proliferative advantage, causing its descendants to increase in frequency. The daughter cells also lack a telomere on one chromosome arm and may undergo additional BFB cycles. One possible subsequent cycle could, for example, cause an inverted duplication of the suffix
and X•ABC are equivalent). The now-amplified segment D in the first daughter cell may confer some proliferative advantage, causing its descendants to increase in frequency. The daughter cells also lack a telomere on one chromosome arm and may undergo additional BFB cycles. One possible subsequent cycle could, for example, cause an inverted duplication of the suffix 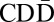 , yielding the chromosome
, yielding the chromosome 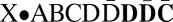 . As these BFB cycles continue, the count of segments on the modified chromosome can increase significantly.
. As these BFB cycles continue, the count of segments on the modified chromosome can increase significantly.
The notation 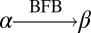 will be used to indicate that the string β can be obtained by applying 0 or more BFB cycles over the string α, as formally described in Definition 1.
will be used to indicate that the string β can be obtained by applying 0 or more BFB cycles over the string α, as formally described in Definition 1.
Definition 1:
For two strings α, β, say that
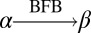 if β = α, or α = ργ for some strings ρ,γ such that γ ≠ ɛ, and
if β = α, or α = ργ for some strings ρ,γ such that γ ≠ ɛ, and 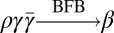 .
.
We say that β is an l-BFB string if for some consecutive chromosomal region α = σlσl+1… starting at the lth segment σl, 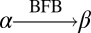 . Say that β is a BFB string if it is an l-BFB string for some l. As examples, CDE = σ3σ4σ5 is a 3-BFB string, and so are CDE
. Say that β is a BFB string if it is an l-BFB string for some l. As examples, CDE = σ3σ4σ5 is a 3-BFB string, and so are CDE and CDE
and CDE E
E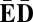 . The empty string ɛ is considered an l-BFB string for every integer l > 0.
. The empty string ɛ is considered an l-BFB string for every integer l > 0.
Denote by 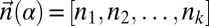 the count vector of α, where α represents a chromosomal arm σ1σ2…σk with k segments, and nl is the copy number of σl and
the count vector of α, where α represents a chromosomal arm σ1σ2…σk with k segments, and nl is the copy number of σl and 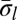 in α. For example, for
in α. For example, for 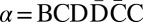 ,
, 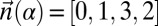 . Say that a vector
. Say that a vector 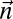 is a BFB count vector if there exists some 1-BFB string α such that
is a BFB count vector if there exists some 1-BFB string α such that 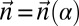 .
.
Handling Experimental Imprecision.
Experimental methods do not provide the accurate copy number of a given chromosome segment. Instead, some measurement error is expected. Moreover, in a cancer genome, it is plausible that a region undergoing BFB may also be rearranged by other mechanisms. So, when we evaluate a count vector for consistency with BFB, we must also consider whether the count vector is “nearly” consistent with BFB. For this, we define a distance measure δ between count vectors, where 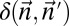 reflects a penalty for assuming that the real copy counts are
reflects a penalty for assuming that the real copy counts are 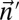 whereas the measured counts are
whereas the measured counts are 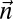 . We implement such a distance measure based on the Poisson likelihood of the observation, as follows: Let
. We implement such a distance measure based on the Poisson likelihood of the observation, as follows: Let 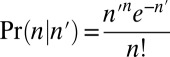 be the Poisson probability of measuring a copy number n, given that the segment’s true copy number is n′. Assuming measurement errors are independent, the probability for measuring a count vector
be the Poisson probability of measuring a copy number n, given that the segment’s true copy number is n′. Assuming measurement errors are independent, the probability for measuring a count vector 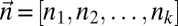 , where the true counts are
, where the true counts are 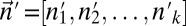 , is given by
, is given by 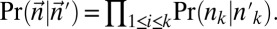 Define the distance of
Define the distance of 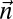 from
from 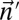 by
by
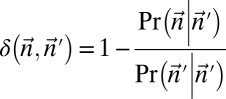 |
For every pair of count vectors 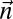 and
and 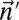 of the same length
of the same length 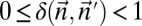 , the closer to 0 the greater is the similarity between
, the closer to 0 the greater is the similarity between 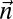 and
and 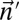 .
.
BFB Count-Vector Problem.
With these definitions, we can now precisely pose a set of problems that needs to be solved to evaluate copy number patterns for consistency with BFB.
BFB Count-Vector Problem Variants.
• Input: A count vector
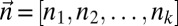 .
.• The decision variant: decide if
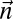 is a BFB count vector.
is a BFB count vector.• The search variant: if
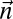 is a BFB count vector, find a BFB string α such that
is a BFB count vector, find a BFB string α such that 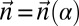 .
.• The distance variant: Identify a BFB count vector
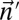 such that
such that 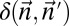 is minimized. Output δ.
is minimized. Output δ.
Outline of the BFB Count-Vector Algorithms
We defer the full details of the algorithms we have developed to the accompanying SI Text, presenting here only essential properties of BFB strings and some intuition of how to incorporate these properties in algorithms for BFB count-vector problems. We focus on the search variant of the problem, where the goal of the algorithm is to output a BFB string α consistent with the input counts, if such a string exists.
Properties of BFB Palindromes.
Call an l-BFB string β of the form 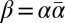 an l-BFB palindrome.† For an l-BFB string α, the string
an l-BFB palindrome.† For an l-BFB string α, the string 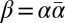 is an l-BFB palindrome by definition (choosing ρ = ɛ and γ = α in Definition 1). In ref. 12 it was shown that every prefix of a BFB string is itself a BFB string; thus, for an l-BFB palindrome
is an l-BFB palindrome by definition (choosing ρ = ɛ and γ = α in Definition 1). In ref. 12 it was shown that every prefix of a BFB string is itself a BFB string; thus, for an l-BFB palindrome 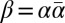 , the prefix α of β is also an l-BFB string. Hence, it follows that α is an l-BFB string if and only if
, the prefix α of β is also an l-BFB string. Hence, it follows that α is an l-BFB string if and only if 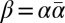 is an l-BFB palindrome. For a BFB string α with
is an l-BFB palindrome. For a BFB string α with 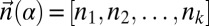 and a corresponding BFB palindrome
and a corresponding BFB palindrome 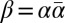 , we have that
, we have that 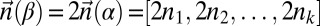 . Thus, a count vector
. Thus, a count vector 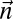 is a BFB count vector if and only if there is a 1-BFB palindrome β such that
is a BFB count vector if and only if there is a 1-BFB palindrome β such that 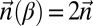 . Considering BFB palindromes instead of BFB strings will facilitate the algorithm description.
. Considering BFB palindromes instead of BFB strings will facilitate the algorithm description.
Define an l-block as a palindrome of the form 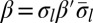 , where β′ is an (l + 1)-BFB palindrome. For example, from the 4-BFB palindromes
, where β′ is an (l + 1)-BFB palindrome. For example, from the 4-BFB palindromes 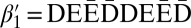 and
and 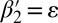 , we can produce the 3-blocks
, we can produce the 3-blocks 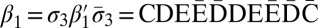 and
and 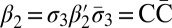 . It may be asserted that an l-block is a special case of an l-BFB palindrome. Next, we show how l-BFB palindromes may be decomposed into l-block substrings.
. It may be asserted that an l-block is a special case of an l-BFB palindrome. Next, we show how l-BFB palindromes may be decomposed into l-block substrings.
For a string α ≠ ɛ, denote by top(α) the maximum integer t such that σt or 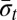 occur in α, and define top(ɛ) = 0. For two strings α and β, say that α ≤t
β if top(α) ≤ top(β), and that α <t
β if top(α) ≤ top(β). For example, for α = AB and
occur in α, and define top(ɛ) = 0. For two strings α and β, say that α ≤t
β if top(α) ≤ top(β), and that α <t
β if top(α) ≤ top(β). For example, for α = AB and 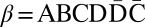 , top(α) = 2 and top(β) = 4, therefore α <t
β.
, top(α) = 2 and top(β) = 4, therefore α <t
β.
Definition 2:
A string α is a convexed l-palindrome if α = ɛ, or α = γβγ such that γ is a convexed l-palindrome, β is an l-BFB palindrome, and γ <t β.
Although every l-BFB palindrome α is also a convexed l-palindrome (because α = ɛαɛ), not every convexed l-palindrome is a valid BFB string. For example, 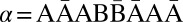 is a convexed 1-palindrome (choosing
is a convexed 1-palindrome (choosing 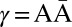 ,
,  ), yet it is not a 1-BFB string. Instead, we have the following claim, proven in SI Text:
), yet it is not a 1-BFB string. Instead, we have the following claim, proven in SI Text:
Claim 1.
A string α is an l-BFB palindrome if and only if α = ɛ, α is an l-block, or α = βγβ, such that β is an l-BFB palindrome, γ is a convexed l-palindrome, and γ ≤t β.
From Definition 2 and Claim 1, it follows that an l-BFB palindrome α is a palindromic concatenation of l-blocks. In addition, for the total count 2nl of σl and 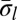 in α, α contains exactly nl
l-blocks, where each block contains one occurrence of σl and one occurrence of
in α, α contains exactly nl
l-blocks, where each block contains one occurrence of σl and one occurrence of 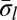 . When nl is even, α is of the form
. When nl is even, α is of the form 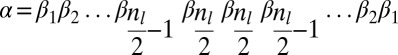 , each βi is an l-block. When nl is odd, α is of the form
, each βi is an l-block. When nl is odd, α is of the form 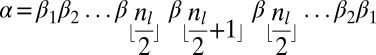 . In the latter case, say that
. In the latter case, say that 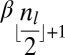 is the center of α, where in the former case say that the center of α is ɛ. Note that every l-block β appearing in α and different from its center occurs an even number of times in α. If the center of α is an l-block, this particular block is the only block which appears an odd number of times in α, whereas if it is an empty string then no block appears an odd number of times in α.
is the center of α, where in the former case say that the center of α is ɛ. Note that every l-block β appearing in α and different from its center occurs an even number of times in α. If the center of α is an l-block, this particular block is the only block which appears an odd number of times in α, whereas if it is an empty string then no block appears an odd number of times in α.
Now, let β be a 1-BFB palindrome with a count vector 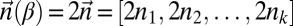 . It is helpful to depict β so that each character σl is at its own layer l, increasing with increasing l, as shown in Fig. 2A. As β is a concatenation of 1-blocks, we can consider the collection B1 = {m1β1, m2β2,…,mqβq} of these blocks, where each count mi is the number of distinct repeats of βi in β. For example, for the string in Fig. 2A, B1 = {2β1, β2, 2β3, 4β4}, where |B1| = n1 = 9, and β2 is the center of β. Masking from strings in B1 all occurrences of A and
. It is helpful to depict β so that each character σl is at its own layer l, increasing with increasing l, as shown in Fig. 2A. As β is a concatenation of 1-blocks, we can consider the collection B1 = {m1β1, m2β2,…,mqβq} of these blocks, where each count mi is the number of distinct repeats of βi in β. For example, for the string in Fig. 2A, B1 = {2β1, β2, 2β3, 4β4}, where |B1| = n1 = 9, and β2 is the center of β. Masking from strings in B1 all occurrences of A and 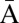 , each 1-block
, each 1-block 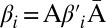 in B1 becomes a 2-BFB palindrome
in B1 becomes a 2-BFB palindrome 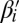 . Such 2-BFB palindromes may be further decomposed into 2-blocks, yielding a 2-block collection B2 (in Fig. 2B, B2 = {2β5, β6, 2β7}, where |B2| = n2 = 5). In general, for each 1 ≤ l ≤ k, masking in β all letters σr and
. Such 2-BFB palindromes may be further decomposed into 2-blocks, yielding a 2-block collection B2 (in Fig. 2B, B2 = {2β5, β6, 2β7}, where |B2| = n2 = 5). In general, for each 1 ≤ l ≤ k, masking in β all letters σr and 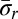 such that r < l defines a corresponding collection of l-block substrings of β. Each collection Bl contains exactly nl elements, as each l-block in the collection contains exactly two out of the 2nl occurrences of σl in the string (where one occurrence is reversed). The collection Bl+1 is obtained from Bl by masking occurrences of σl and
such that r < l defines a corresponding collection of l-block substrings of β. Each collection Bl contains exactly nl elements, as each l-block in the collection contains exactly two out of the 2nl occurrences of σl in the string (where one occurrence is reversed). The collection Bl+1 is obtained from Bl by masking occurrences of σl and 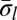 from the elements in Bl, and decomposing the obtained (l + 1)-BFB palindromes into (l + 1)-blocks. We may define Bk+1 =
from the elements in Bl, and decomposing the obtained (l + 1)-BFB palindromes into (l + 1)-blocks. We may define Bk+1 = 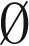 (where
(where 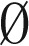 denotes an empty collection), because after masking in β all segments σ1,…,σk we are left with an empty collection of (k + 1)-blocks.
denotes an empty collection), because after masking in β all segments σ1,…,σk we are left with an empty collection of (k + 1)-blocks.
Fig. 2.

Layer visualization of a BFB palindrome 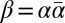 , where
, where 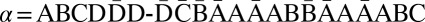 . A possible BFB sequence that produces α is
. A possible BFB sequence that produces α is 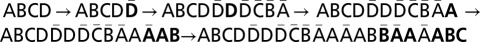 .
. 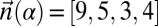 , and
, and 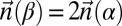 . (A–D) Layers 1–4 of β, respectively. In each layer l, the l-blocks composing the collection Bl are annotated as substrings of the form βi.
. (A–D) Layers 1–4 of β, respectively. In each layer l, the l-blocks composing the collection Bl are annotated as substrings of the form βi.
The algorithm we describe for the search variant of the BFB count-vector problem exploits the above-described property of BFB palindromes. Given a count vector 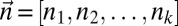 , the algorithm processes iteratively the counts in the vector one by one, from nk down to n1, producing a series of collections Bk, Bk−1,…, B1. Starting with Bk+1 =
, the algorithm processes iteratively the counts in the vector one by one, from nk down to n1, producing a series of collections Bk, Bk−1,…, B1. Starting with Bk+1 = 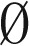 , each collection Bl in the series is obtained from the preceding collection Bl+1 in a two-step procedure: First, (l + 1)-blocks from Bl+1 are concatenated in a manner that produces an (l + 1)-BFB palindrome collection B′ of size nl (B′ may contain empty strings, which can be thought of as concatenations of zero elements from Bl+1). Then, Bl is obtained by “wrapping” each element β′ ∈ B′ with a pair of σl characters to become an l-block
, each collection Bl in the series is obtained from the preceding collection Bl+1 in a two-step procedure: First, (l + 1)-blocks from Bl+1 are concatenated in a manner that produces an (l + 1)-BFB palindrome collection B′ of size nl (B′ may contain empty strings, which can be thought of as concatenations of zero elements from Bl+1). Then, Bl is obtained by “wrapping” each element β′ ∈ B′ with a pair of σl characters to become an l-block 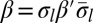 . We will refer to the first step in this procedure as collection folding, and to the second step as collection wrapping. For example, in Fig. 2D, the elements in B4 = {4β10} are folded to form a 4-palindrome collection B′ = {2β10β10,ɛ} of size n3 = 3. After wrapping each element of B′ by C to the left and
. We will refer to the first step in this procedure as collection folding, and to the second step as collection wrapping. For example, in Fig. 2D, the elements in B4 = {4β10} are folded to form a 4-palindrome collection B′ = {2β10β10,ɛ} of size n3 = 3. After wrapping each element of B′ by C to the left and 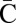 to the right, we get the 3-block collection
to the right, we get the 3-block collection 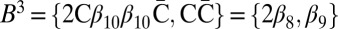 . Algorithm SEARCH-BFB
. Algorithm SEARCH-BFB in Fig. 3 gives the pseudocode for the described procedure, excluding the implementation of the folding phase, which is kept abstract here. We next discuss some restrictions over the folding procedure, and point out that greedy folding is nontrivial. Nevertheless, in SI Text we show an explicit implementation of a folding procedure, which guarantees that the search algorithm finds a BFB string provided that the input is a valid BFB count vector.
in Fig. 3 gives the pseudocode for the described procedure, excluding the implementation of the folding phase, which is kept abstract here. We next discuss some restrictions over the folding procedure, and point out that greedy folding is nontrivial. Nevertheless, in SI Text we show an explicit implementation of a folding procedure, which guarantees that the search algorithm finds a BFB string provided that the input is a valid BFB count vector.
Fig. 3.

Algorithm for the BFB count-vector problem.
Required Conditions for Folding.
Recall that the input of the folding procedure is an l-block collection B and an integer n, and the procedure should concatenate all strings in B in some manner to produce an l-BFB palindrome collection B′ of size n. Because both l-blocks and empty strings are special cases of l-BFB palindromes, when n ≥ |B| it is always possible to obtain B′ by simply adding n − |B| empty strings to B. Nevertheless, when n < |B|, there are instances for which no valid folding exists, as shown next.
For a pair of collections B and B′, B + B′ is the collection containing all elements in B and B′. When B″ = B + B′, we say that B = B″ − B′ (note that B″ − B′ is well-defined only when B″ contains B′). For some (possibly rational) number x ≥ 0, denote by xB the collection {⌊xm1⌋β1, ⌊xm2⌋β2,…, ⌊xmq⌋βq}. The operation mod2(B) yields the subcollection of B containing a single copy of each distinct element β with an odd count in B. For example, for B = {2β1, β2, 5β3, 6β4}, mod2(B) = {β2, β3}. Observe that 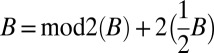 .
.
Claim 2.
Let B be an l-BFB palindrome collection such that mod2(B) = 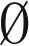 . Then, it is possible to concatenate all elements in B to obtain a single l-BFB palindrome.
. Then, it is possible to concatenate all elements in B to obtain a single l-BFB palindrome.
Proof:
By induction on the size of B. By definition, mod2(B) = 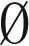 implies that the counts of all distinct elements in B are even. When B =
implies that the counts of all distinct elements in B are even. When B = 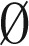 , the concatenation of all elements in B yields an empty string ɛ, which is an l-BFB palindrome as required. Otherwise, assume the claim holds for all collections B′ smaller than B. Let β ∈ B be an element such that for every β′ ∈ B, top(β′) ≤ top(β), and let B′ = B − {2β}. Note that mod2(B′) =
, the concatenation of all elements in B yields an empty string ɛ, which is an l-BFB palindrome as required. Otherwise, assume the claim holds for all collections B′ smaller than B. Let β ∈ B be an element such that for every β′ ∈ B, top(β′) ≤ top(β), and let B′ = B − {2β}. Note that mod2(B′) = 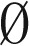 (the count parity is identical for every element in both B and B′), and from the inductive assumption it is possible to concatenate all elements in B′ into a single l-BFB palindrome α′. From Claim 1, the string α = βα′β is an l-BFB palindrome, obtained by concatenating all elements in B.□
(the count parity is identical for every element in both B and B′), and from the inductive assumption it is possible to concatenate all elements in B′ into a single l-BFB palindrome α′. From Claim 1, the string α = βα′β is an l-BFB palindrome, obtained by concatenating all elements in B.□
Claim 3.
Let B be an l-block collection. There is a folding B′ of B such that |B′| = |mod2(B)| + 1.
Proof:
Recall that 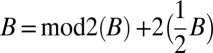 . All element counts in the collection
. All element counts in the collection 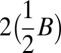 are even, hence
are even, hence 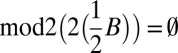 , and from Claim 2 it is possible to concatenate all elements in
, and from Claim 2 it is possible to concatenate all elements in 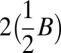 into a single l-BFB palindrome α. Thus, the collection B′ = mod2(B) + α is a folding of B of size |mod2(B)| + 1.□
into a single l-BFB palindrome α. Thus, the collection B′ = mod2(B) + α is a folding of B of size |mod2(B)| + 1.□
Claim 4.
For every folding B′ of an l-block collection B, |mod2(B′)| ≥ |mod2(B)|.
Proof:
Let β ∈ mod2(B) be an l-block repeating an odd number of times m in B. Therefore, β appears as a center of at least one element β′ that occurs an odd number of times in B′ (otherwise, β has an even number of distinct repeats as a substring of elements in B′, in contradiction to the fact that m is odd). Hence, for each β ∈ mod2(B) there is a corresponding unique element β′ ∈ mod2(B′), and so |mod2(B′)| ≥ |mod2(B)|.□
The SEARCH-BFB algorithm described in Fig. 3 tries in each iteration l to fold the block collection Bl+1 obtained in the previous iteration into an (l + 1)-BFB palindrome collection of size nl. When nl ≥ |mod2(Bl+1)| + 1, there always exists a folding as required: Bl+1 may be folded into a collection of size |mod2(Bl+1)| + 1 due to Claim 3, and additional nl − |mod2(Bl+1)| − 1 empty strings may be added to get a folding of size nl. On the other hand, when nl < |mod2(Bl+1)|, no folding as required exists, due to Claim 4. In the remaining case of nl = |mod2(Bl+1)|, the existence of an nl-size folding of Bl+1 depends on the element composition of Bl+1, as exemplified next.
algorithm described in Fig. 3 tries in each iteration l to fold the block collection Bl+1 obtained in the previous iteration into an (l + 1)-BFB palindrome collection of size nl. When nl ≥ |mod2(Bl+1)| + 1, there always exists a folding as required: Bl+1 may be folded into a collection of size |mod2(Bl+1)| + 1 due to Claim 3, and additional nl − |mod2(Bl+1)| − 1 empty strings may be added to get a folding of size nl. On the other hand, when nl < |mod2(Bl+1)|, no folding as required exists, due to Claim 4. In the remaining case of nl = |mod2(Bl+1)|, the existence of an nl-size folding of Bl+1 depends on the element composition of Bl+1, as exemplified next.
Consider the run of Algorithm SEARCH-BFB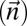 over the input count vector
over the input count vector 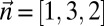 . Here, k = 3, and the algorithm starts by initializing the collection B4 =
. Here, k = 3, and the algorithm starts by initializing the collection B4 = 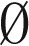 . In the first loop iteration l = 3, and the algorithm first tries to fold the empty collection B4 into a 4-BFB palindrome collection containing n3 = 2 elements. Because there are no elements in B4 to concatenate, the only way to perform this folding is by adding to B4 two empty strings, yielding the collection B′ = {2ɛ}, which after wrapping becomes
. In the first loop iteration l = 3, and the algorithm first tries to fold the empty collection B4 into a 4-BFB palindrome collection containing n3 = 2 elements. Because there are no elements in B4 to concatenate, the only way to perform this folding is by adding to B4 two empty strings, yielding the collection B′ = {2ɛ}, which after wrapping becomes 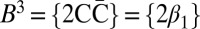 . In the next iteration l = 2, and B3 should be folded into a collection B′ of size n2 = 3. Among the possibilities to perform this folding are the following: B′a = {2β1, ɛ}, and B′b = {β1β1, 2ɛ}, which after wrapping become
. In the next iteration l = 2, and B3 should be folded into a collection B′ of size n2 = 3. Among the possibilities to perform this folding are the following: B′a = {2β1, ɛ}, and B′b = {β1β1, 2ɛ}, which after wrapping become 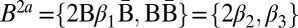 , and
, and 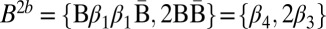 , respectively. Note that |mod2(B2a)| = |mod2(B2b)| = 1. Nevertheless, it is possible to fold B2a in the next iteration into the collection {β2β3β2} of size n1 = 1, whereas B2b cannot be folded into such a collection. The reason is that the only concatenation of all elements in B2b into a single palindrome is the concatenation β3β4β3, yet
, respectively. Note that |mod2(B2a)| = |mod2(B2b)| = 1. Nevertheless, it is possible to fold B2a in the next iteration into the collection {β2β3β2} of size n1 = 1, whereas B2b cannot be folded into such a collection. The reason is that the only concatenation of all elements in B2b into a single palindrome is the concatenation β3β4β3, yet 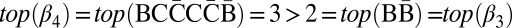 , and Claim 1 implies that this concatenation is not a valid BFB palindrome.
, and Claim 1 implies that this concatenation is not a valid BFB palindrome.
In SI Text, we define a property called the signature of a collection, and show how the exact minimum folding size depends on this signature (Table S1). We also show how to fold a collection in a manner that optimizes this signature, and guarantees for valid BFB count-vector inputs that the search algorithm finds an admitting BFB string.
Running Time
For a count vector 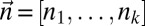 , let
, let 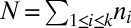 be the number of segments in a string corresponding to
be the number of segments in a string corresponding to 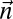 . Let
. Let 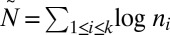 denote a number proportional to the number of bits in the representation of
denote a number proportional to the number of bits in the representation of 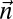 , assuming each count ni is represented by O(log ni) bits. In SI Text, we complete the implementation details of algorithms for the decision, search, and distance variants of the BFB count-vector problem, and show that these algorithms have the asymptotic running times of
, assuming each count ni is represented by O(log ni) bits. In SI Text, we complete the implementation details of algorithms for the decision, search, and distance variants of the BFB count-vector problem, and show that these algorithms have the asymptotic running times of 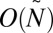 (bit operations), O(N), and O(Nlog
N) (under some realistic assumptions), respectively. For the decision and search variants, these running times are optimal, being linear in the input (for the decision variant) or output (for the search variant) lengths.
(bit operations), O(N), and O(Nlog
N) (under some realistic assumptions), respectively. For the decision and search variants, these running times are optimal, being linear in the input (for the decision variant) or output (for the search variant) lengths.
In practical terms, this has a significant effect on our ability to evaluate copy number signatures of BFB compared with the previous exponential time algorithm (12). To determine if a count vector consistent with BFB is in fact strong evidence for BFB, we have to check many count vectors. Analyzing the simulations we explain below required testing tens of millions of different count vectors, so even a small improvement in running time can have a large impact of the scope of analysis we can perform. However, the running time improvement with this algorithm is not small. For example, a count vector that took 9 s with the previous algorithm can be processed by the present algorithm in 1.2 × 10−5 s. A count vector that was abandoned after 30 h with the old algorithm now takes only 8.1 × 10−6 s. Thus, the improvement in running time is not of merely theoretical interest, and was required for making the current study possible.
Detecting Signatures of BFB
We can now describe the two features we will use to determine if a chromosome has undergone BFB. The first feature is based on the fold-back inversions that BFB produces. For a given region, we can find all of the breakpoints identified by sequencing and determine what proportion are fold-back inversions. We call this the fold-back fraction. The second feature relies on our algorithm that solves the BFB count-vector problems we have posed. For a given count vector, we can find the distance to the nearest count vector that could be produced by BFB using the distance metric we defined above. We call this the count-vector distance. For a particular count vector, we define a score s that combines these two features:
Here, f refers to the fold-back fraction, δ refers to the count-vector distance, and λ refers to the weight we give to the count-vector distance versus the fold-back fraction when calculating the score. When λ = 1, we are only looking at count-vector distance, whereas when λ = 0, we are only using fold-back fraction and ignoring the count vectors.
Results
To determine whether our two proposed features could identify BFB against the complex backdrop of a cancer genome, we simulated rearranged chromosomes. Our overall goal was to simulate cancer chromosomes that were highly rearranged yet had not undergone BFB to see if evidence for BFB appeared in them, suggesting that using such evidence would lead to false positives. Conversely, we also wanted to simulate chromosomes whose rearrangements included BFB to determine if a proposed BFB signature was sensitive enough to identify BFB when it occurred. Because it is not clear how to faithfully simulate cancer genome rearrangements, we used a wide range of simulation parameters so we could understand how different assumptions affect the features’ ability to identify BFB.
We began with a pair of unrearranged chromosomes and then introduced 50 rearrangements to each. Each rearrangement was an inversion, a deletion, or a duplication. Duplications were either direct or inverted and could be tandem or interspersed. The type of each rearrangement was chosen from a distribution. In some chromosome pairs, we imitated BFB by successively duplicating and inverting segments of one end of one chromosome for each round of BFB. The number of BFB rounds varied from 2 to 10. Then, we calculated the copy counts and breakpoints for the chromosome pair and introduced error to the copy counts according to a random model and also randomly deleted or inserted breakpoint observations. For each combination of rearrangement type distribution and number of BFB rounds, we simulated 5,000 chromosome pairs with BFB and 15,000 without BFB. Complete details are in SI Text.
We first examined the usefulness of count-vector distance alone in identifying BFB by setting λ = 1 in our score function (Eq. 1). For each chromosome pair, we found all contiguous count vectors of a given length and calculated their scores, as described above and in SI Text. We used the minimum score s over all of these subvectors in the chromosome as a score for the whole chromosome. Then, for varying thresholds, we classified all chromosomes with a score lower than the threshold as having been rearranged by BFB. The performance of this classification varied with the parameters used to simulate the chromosomes, but typical results can be seen in Fig. 4A. The solid lines show receiver operating characteristic (ROC) curves for different count-vector lengths for the simulation with eight rounds of BFB and a distribution that yields roughly equal probabilities of the other rearrangement types. Consistent with previous observations, short count vectors that are perfectly consistent with BFB can be found in many chromosomes, even if BFB did not occur. So, even with a score threshold of zero, they would still be classified as consistent with BFB. For example, 63% of chromosomes without any true BFB rearrangements in Fig. 4A had a count vector of length 6, perfectly consistent with BFB.
Fig. 4.

Simulation and pancreatic cancer results. (A) ROC curves for different count-vector lengths with and without fold-back fractions for the simulation of eight BFB rounds and equally likely other rearrangements. (B) Observed copy counts and copy counts compatible with BFB on the short arm of chromosome 12 in pancreatic cancer sample PD3641. The presence of fold-back inversions and the count vector’s consistency with BFB suggests that this portion of chromosome 12 underwent BFB cycles.
In contrast, examining longer count vectors produced a better classification. For instance, setting the score threshold to 0.10, count vectors of length 12 could achieve a true positive rate (TPR) of 67% and a false positive rate (FPR) of only 10%. However, this performance must be considered in the context of an experiment seeking evidence for BFB. Chromosomes that have undergone BFB are probably rare. If only 1 in 100 chromosomes tested underwent BFB, then a test with an FPR of even 1% will produce mostly false discoveries. Achieving this FPR with count vectors of length 12 with the chromosomes in Fig. 4A would result in a TPR of only 16%. A more appropriate target FPR for screening many samples, say 0.1%, could not be achieved with count vectors alone.
Next, we incorporated fold-back inversions into the scoring function by setting λ = 0.5. ROC curves using this approach are shown by dashed lines in Fig. 4A. Incorporating fold-back fractions into the scoring leads to better discrimination of chromosomes with and without BFB rearrangements; the test in Fig. 4A that combines count vectors of length 12 and fold-back inversions can achieve a TPR of 48% with an FPR of 0.1% by setting the score threshold to 0.27. This suggests that it could detect BFB in a large dataset without being overwhelmed by false discoveries.
Of course, these conclusions depend on our simulation resembling actual cancer rearrangements and BFB cycles. A true specification of cancer genome evolution is unknown and in any case varies from cancer to cancer. Recognizing this complication, we repeated the analysis in Fig. 4A for the different rearrangement distributions, number of BFB rounds, and count-vector lengths. For each combination, we recorded the score threshold needed to achieve FPRs of 0.1, 1, and 5%, and the respective expected TPRs. The full results are shown in Dataset S1 and ROC curves are shown in Figs. S1–S5. Generally, different simulations showed the same trends. Fold-back inversions alone were better at identifying BFB than count vectors alone, but the combination of both features provided the best classification. By examining a wide range of simulation parameters, we illustrate how changes in assumptions about cancer genome evolution and BFB influence the appropriateness and expected outcomes of tests for BFB.
We applied our method to a publicly available dataset of copy number profiles from 746 cancer cell lines (13). We found three chromosomes with count vectors of length 12 nearly consistent with BFB: chromosome 8 from cell line AU565, chromosome 10 from cell line PC-3, and chromosome 8 from cell line MG-63 (SI Text). Although the patterns of copy counts on these chromosomes do bear the hallmarks of BFB, our simulations suggest that labeling chromosomes as having undergone BFB based on these count vectors would lead to an FPR between 1 and 10%. Given that thousands of chromosomes were examined, many of which were highly rearranged, the consistency of these copy counts with BFB may be spurious.
We also applied our method to paired-end sequencing data from seven previously published pancreatic cancer samples (14). We estimated copy numbers from the reads and used breakpoints as reported by the original investigators (Fig. S6). We examined count vectors of length 8 and chose a threshold score of 0.18, which would give an FPR of 0.1% based on simulations where the non-BFB rearrangement types are roughly equally likely. We identified two chromosomes that showed evidence for BFB, both from the same sample, PD3641. The first was the long arm of chromosome 8. This chromosome was identified by the original investigators as likely being rearranged by BFB. Our analysis suggests that, barring rearrangements that differ significantly from any of our simulations, this chromosome did indeed undergo BFB cycles. We also found evidence for BFB rearrangements from a count vector spanning 10 megabases on the short arm of chromosome 12 (Fig. 4B and Fig. S7). Thus, we were able to recover evidence for BFB previously identified by hand curation. And, by combining count vector and fold-back analysis, we found an additional strong BFB candidate that would not be apparent without modeling and simulation.
Discussion
Some 80 years after Barbara McClintock’s discovery of the BFB mechanism, it is seeing renewed interest in the context of tumor genome evolution. Recent publications have claimed, based on empirical observations of segmentation counts and other features, that their data counts are consistent with BFB. The main technical contribution of the paper is an efficient algorithm for detecting if given segmentation counts can indeed be created by BFB cycles. That algorithm turns out to be nontrivial, requiring a deep foray into the combinatorics of BFB count vectors, even though its final implementation is straightforward and fast. Experimenting with the implementation reveals that in fact (i) there is a big diversity of count vectors created by true BFB cycles, not all of which are easily recognizable as BFB; and (ii) at least for short count vectors, it is often possible to create BFB-like vectors by non-BFB operations. Thus, being “consistent with BFB” and “caused by BFB” are not equivalent. Fortunately, our results also suggest that using longer count vectors, and additional information of fold-backs, gives stronger prediction of BFB, even in the presence of noise and diploidy. Although assembly of these highly rearranged genomes continues to be difficult, recent advances in long single-molecule sequencing will provide additional spatial information that will improve the resolving power of our algorithm. As more cancer genomes are sequenced, including single-cell sequencing, the method presented here will be helpful in determining the extent and scope of BFB cycles in the evolution of the tumor genome.
Materials and Methods
Details on the algorithm, and on the simulation methods are available in SI Text.
Code Availability.
Java and Python code used to analyze chromosomes is available at www.bitbucket.org/mckinsel/bfb.
Estimating Pancreas Tumor Copy Number.
The pancreas tumor data were downloaded from the European Genome-Phenome Archive, accession no. EGAS00000000064. The data were paired-end reads; each end was 37 bases long. We aligned the reads with Bowtie (17). Then we used readDepth (18) for segmentation and integer copy number estimation.
Supplementary Material
Acknowledgments
This research was supported by grants from the National Institute of Health (5R01-HG004962 and U54 HL108460) and the National Science Foundation (NSF-CCF-1115206).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
†We assume that genomic segments σ satisfy 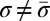 , therefore strings of the form
, therefore strings of the form 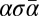 will not be considered palindromes.
will not be considered palindromes.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1220977110/-/DCSupplemental.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10(8):551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carr AM, Paek AL, Weinert T. DNA replication: Failures and inverted fusions. Semin Cell Dev Biol. 2011;22(8):866–874. doi: 10.1016/j.semcdb.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 4.McClintock B. The Stability of broken ends of chromosomes in Zea Mays. Genetics. 1941;26(2):234–282. doi: 10.1093/genetics/26.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DePinho RA, Polyak K. Cancer chromosomes in crisis. Nat Genet. 2004;36(9):932–934. doi: 10.1038/ng0904-932. [DOI] [PubMed] [Google Scholar]
- 6.Bignell GR, et al. Architectures of somatic genomic rearrangement in human cancer amplicons at sequence-level resolution. Genome Res. 2007;17(9):1296–1303. doi: 10.1101/gr.6522707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitada K, Yamasaki T. The complicated copy number alterations in chromosome 7 of a lung cancer cell line is explained by a model based on repeated breakage-fusion-bridge cycles. Cancer Genet Cytogenet. 2008;185(1):11–19. doi: 10.1016/j.cancergencyto.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Hillmer AM, et al. Comprehensive long-span paired-end-tag mapping reveals characteristic patterns of structural variations in epithelial cancer genomes. Genome Res. 2011;21(5):665–675. doi: 10.1101/gr.113555.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim G, et al. An integrated mBAND and submegabase resolution tiling set (SMRT) CGH array analysis of focal amplification, microdeletions, and ladder structures consistent with breakage-fusion-bridge cycle events in osteosarcoma. Genes Chromosomes Cancer. 2005;42(4):392–403. doi: 10.1002/gcc.20157. [DOI] [PubMed] [Google Scholar]
- 10.Hicks J, et al. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res. 2006;16(12):1465–1479. doi: 10.1101/gr.5460106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selvarajah S, et al. Genomic signatures of chromosomal instability and osteosarcoma progression detected by high resolution array CGH and interphase FISH. Cytogenet Genome Res. 2008;122(1):5–15. doi: 10.1159/000151310. [DOI] [PubMed] [Google Scholar]
- 12.Kinsella M, Bafna V. Combinatorics of the breakage-fusion-bridge mechanism. J Comput Biol. 2012;19(6):662–678. doi: 10.1089/cmb.2012.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bignell GR, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463(7283):893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campbell PJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467(7319):1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter NP. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat Genet. 2007;39(7, Suppl):S16–S21. doi: 10.1038/ng2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiang DY, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009;6(1):99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller CA, Hampton O, Coarfa C, Milosavljevic A. ReadDepth: a parallel R package for detecting copy number alterations from short sequencing reads. PLoS ONE. 2011;6(1):e16327. doi: 10.1371/journal.pone.0016327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.