

In this report, we introduce a simple method to pattern multiple cell types in a spatially controlled manner within predefined microenvironments. Spatial distribution of cells and interactions between neighboring cells in native microenvironments are of fundamental importance in determining cell fate decisions, such as differentiation, migration, and growth. [1–7] These interactions also serve an important role in developmental biology, organ development, tissue homeostasis, and cancer. [8–10] All these processes occur in various complex architectural organizations. Controlling the spatial distribution of different cell types in defined geometries can replicate these intricate native organizations, which can lead to useful models for several fields.
Native tissues are made of complex architectural organizations of multiple cell types. Parenchymal and non-parenchymal cells generate homotypic and heterotypic cell interactions which all regulate tissue and organ functions. [5,6,10] Tissue geometry also takes a role in cell fate decision and overall functions of an organ. [1,4,7,10] Spatial and geometrical distribution of different cells is also crucial in embryonic development. [9,11,12] The interplay between different spatially organized germ layers [9] and interactions of stem cells and surrounding stromal cells in defined microenvironments regulate stem cell differentiation into particular phenotypes; all of these associations eventually lead to organogenesis and tissue specification. [11,12] In addition, the intricate interactions between epithelial and mesenchymal layers are of importance in creating tissue architectures and organ development. [10] Tissue complexity can also be found in the tumor microenvironment in which heterotypic interactions between various spatially arranged cell types regulate tumor growth, invasion, and metastasis. [8] Mimicking these complex cell-cell interactions in vitro can thus be used to understand the processes involved in developmental biology, tissue homeostasis and pathologies such as cancer or be applied to tissue engineering and regenerative medicine.
Microfluidic devices have been previously utilized to replicate such complex cell-cell interactions in defined microenvironments, though it is challenging to use these devices to pattern multiple cell types with different spatial arrangements, and the associated device fabrication and cell seeding process are complex and tedious, inhibiting the high-throughput production. [13–15] Stencils [12,16] and micromechanical substrates [2] have also been utilized to pattern multiple cell types, but these methods require subsequent manipulations and changes of the substrates at each step which are cumbersome. It is also challenging to generate different patterned shapes by using these methods. In another approach, cell membranes were modified to promote their attachment to different cells to form cell aggregates, [3] though it is challenging to control the geometry of the tissues and spatial distribution of multiple cell types by using this method. Microstructures with different geometries were used to create microtissues of one cell type. [17–19] The static nature of the microstructures inhibits sequential patterning of multiple cell types within a single geometry. Given previous efforts, a simple method to control spatial distributions of multiple cell types within defined microenvironments would be highly useful.
Herein, we introduce a simple patterning method to control spatial arrangements of multiple cell types in defined geometries by utilizing the shape changing properties of poly(N-isopropylacrylamide) (PNIPAAm)-based dynamic microstructures. PNIPAAm is a thermoresponsive polymer possessing a lower critical solution temperature (LCST) of ~32 °C which goes to swollen state below LCST and turns to shrunken state above LCST. [20,21] PNIPAAm has been previously used to coat cell culture platforms and to fabricate hydrogel-based templates, largely with the purpose of generating cell based tissue constructs. [21] In one of our earlier studies, we fabricated PNIPAAm-based microwells in order to develop harvestable microtissues. [22] In a later study, we showed the use of thermoresponsive micromolds for fabricating multicompartment hydrogels from non-photocrosslinkable materials, which was challenging to do by using conventional static micromolding techniques and photolithographic methods. [23] These hydrogels were also shown to encapsulate either different particles or multiple cell types within different compartments. [23] However, it is challenging to control the proximity between different cells and the resulting cell-cell interactions within hydrogel systems. Controlling cell-cell interactions in defined microenvironments can better recapitulate multicellular organizations. In this study, we utilized PNIPAAm-based dynamic microwells to pattern multiple cell types in a spatially controlled manner to obtain multi-layered aggregates with controlled cell-cell interactions.
Dynamic microwells with circular and square shapes were fabricated using a soft lithographic technique (see Supporting Information). To test their temperature dependent shape change properties, these dynamic microwells were subjected to different incubation times at both 24 °C and 37 °C. After 30 min of incubation at 24 °C, circular microwells swelled and retained their circularity (Figure 1a). After increasing the temperature from 24 °C to 37 °C, circular microwells significantly increased their surface areas within 2 h and conserved their circularity (Figure 1a,b). The overall time response of circular microwells to the temperature rise from 24 °C to 37 °C showed a gradual increase in surface areas (Figure 1b). The thermoresponsiveness of square microwells were also analyzed in the same manner. After 30 min of incubation at 24 °C, square microwells swelled and exhibited square-like shapes (Figure 1c). After the temperature was raised from 24 °C to 37 °C, square microwells returned to their original square shapes with significantly increased surface areas within 2 h (Figure 1c,d). Similar to circular microwells, surface areas of square microwells gradually increased in response to incubation at 37 °C with different time intervals (Figure 1d). In light of these results, dynamic microwells demonstrate significant changes in surface areas at two different temperatures by conserving their geometries, facilitating the spatial patterning of multiple cell types at different temperatures.
Figure 1.

Temperature-dependent shape change properties of dynamic microwells. (a) Time lapse images and (b) corresponding responsiveness plot for circular microwells (n = 3). (c) Time lapse images and (d) responsiveness plot for square microwells (n = 3). Both microwell types were first incubated at 24 °C and then moved to 37 °C. During the incubation period at 37 °C, images of both microwell types were taken every 5 min for the first 30 min incubation period, every 10 min for the subsequent 30 min incubation period, and the last images were taken at the 2 h time point. The responsiveness is represented with change in surface areas of microwells. Error bars: ± sd and *** shows statistically significant difference (p < 0.001).
We showed the capability of these dynamic microwells to control spatial organization of multiple cell types in defined microenvironments by patterning two different cell types for potential use in various applications, such as developmental biology, cancer biology, and tissue engineering. Figure 2 illustrates the experimental procedure to pattern two different cell types with dynamic microwells (see also Supporting Information). After fabrication, microwells were placed at 24 °C for 30 min to allow them to reach their ambient temperature areas. A cell suspension of the first cell type was then seeded onto the microwells at 24 °C and immediately washed with phosphate buffered saline (PBS) to rinse off undocked cells. Cell-seeded microwells were then incubated at 37 °C for 2 h to allow microwells to shrink, resulting in more free space. A cell solution of the second cell type was then seeded onto the microwells at 37 °C and undocked cells were immediately rinsed off by washing with PBS. Microwells containing two different spatially patterned cell types were then placed at 37 °C for further incubation. By using this patterning process, two different cell types were spatially organized in a single geometry, either circular or square.
Figure 2.
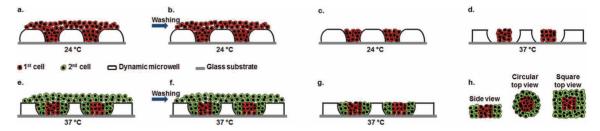
Schematic diagram of spatially controlled patterning of two different cell types with dynamic microwells. (a) Seeding the first cell type (red) at 24 °C when microwell structures were at swollen state. (b) Washing microwells to rinse off undocked cells on microwell surfaces. (c) Undocked cells were washed off the microwell surfaces. (d) Incubation at 37 °C to allow microwell structures to shrink, resulting in more free space for the second cell type. (e) Seeding the second cell type (green) within microwells. (f) Subsequently washing microwells to rinse off undocked cells on the surface. (g) Two cell types were spatially distributed within microwells and further incubated at 37 °C. (h) Side and top views of the resulting microtissues containing two spatially organized cell types.
Human hepatoblastoma, HepG2, cells having parenchymal characteristics, were chosen as the first cell type due to their relevance in tumor models [24] and tissue engineering. [22,25] Human umbilical vein endothelial cells (HUVECs) are non-parenchymal and were selected as the second cell type due to their relevance in the cancer microenvironment [8] and tissue vascularization. [26] HepG2 cells (red) were first seeded within both circular and square microwells. HepG2 aggregates formed circular-like shapes in circular microwells and square-like shapes in square microwells by filling the microwell surfaces that resulted from PNIPAAm swelling at ambient temperature (Figure 3a). After the incubation at 37 °C, green fluorescent protein (GFP)-labeled HUVECs were then seeded on both circular and square microwells. GFP-HUVECs were patterned around HepG2 aggregates in both microwell types by filling the free space that resulted from PNIPAAm expansion at 37 °C (Figure 3a). The resulting aggregates in circular microwells had two different spatially organized cell types in circular geometries (Figure 3a). HepG2 cells possessed surface areas similar to GFP-HUVECs in circular microwells (Figure 3b). Small amounts of GFP-HUVECs overlapped around HepG2 cells. Surface areas of the resulting circular aggregates were significantly larger than that of either HepG2 cells or GFP-HUVECs (Figure 3b). The resulting microtissues in square microwells had two spatially distributed cell types in square geometries (Figure 3a). GFP-HUVECs occupied significantly more space than HepG2 cells in square microwells (Figure 3c). Surface area of the resulting square microtissues was significantly larger than that of either HepG2 cells or GFP-HUVECs. Similar to circular microwells, few GFP-HUVECs overlapped around HepG2 cells in square microwells. To showcase that other non-parenchymal cells can be patterned, 3T3 fibroblasts were substituted for GFP-HUVECs as the second cell type (Supporting Information). Fibroblasts (3T3 cells) are non-parenchymal and relevant to both tumor microenvironment [8] and tissue engineering. [2,5] By using the same patterning method, HepG2 cells (red) were first patterned within both circular and square microwells. The resulting HepG2 aggregates formed circular shapes in circular microwells and square-like shapes in square microwells (Supporting Information (SI), Figure 1a). After incubation at 37 °C, 3T3 fibroblasts (green) were patterned around HepG2 aggregates within both microwell types (SI, Figure 1a). Surface areas of HepG2 cells and 3T3 fibroblasts were similar for circular microwells. The resulting aggregates in circular microwells exhibited significantly larger surface areas than that of either HepG2 cells or 3T3 fibroblasts (SI, Figure 1b). For square microwells, 3T3 fibroblasts occupied remarkably more space than HepG2 cells and the resulting microtissues kept significantly more space than either HepG2 cells or 3T3 fibroblasts (SI, Figure 1c). Similar to experiments with GFP-HUVECs, few 3T3 fibroblasts overlapped around HepG2 cells in both circular and square microwells. These results demonstrate that dynamic microwells are potentially useful for controlling the spatial organization of multiple cell types in defined microenvironments.
Figure 3.

Spatially controlled patterning of HepG2 cells (red) and GFP-HUVECs (green) within dynamic circular and square microwells. (a) HepG2 cells were the first patterned cell type in both circular and square microwells. GFP-HUVECs were patterned as the second cell type and spatially organized around HepG2 aggregates by filling the free space that resulted from microwell expansion at 37 °C. Merged images show that spatial distribution of two cell types was controlled in a single geometry, either circular or square. Areas of both cell types and overall aggregates were measured and plotted for (b) circular microwells (n = 3) (c) square microwells (n = 3). (Error bars: ± sd and statistical differences: ** p < 0.01; *** p < 0.001).
Microwells containing HepG2 cells and GFP-HUVECs were incubated for 3 days to observe the morphological changes of patterned cells. Within 1 day, GFP-HUVECs were located more towards the sides of circular and square microwells without overlapping HepG2 aggregates. GFP-HUVECs also started spreading and forming endothelial networks around HepG2 aggregates in both microwell types after 1 day of incubation (Figure 4a). At day 3, GFP-HUVECs had formed endothelial networks around HepG2 aggregates within both circular and square microwells (Figure 4a). For circular microwells, the shapes of HepG2 aggregates, endothelial networks, and composite aggregates conserved their circularity at day 3. Within square microwells, both endothelial network of GFP-HUVECs and HepG2 aggregates started to become circular at day 3 (Figure 4a). On the other hand, 3T3 fibroblasts started overlapping over HepG2 aggregates in circular and square microwells after 1 day of incubation (SI, Figure 2). It was observed that fibroblasts (3T3 cells) overlapped more over HepG2 cells at day 3 (data not shown). These results could be attributed to interactions between different cell types within different geometries, suggesting that dynamic microwells could potentially control the interplay between multiple spatially arranged cell types.
Figure 4.
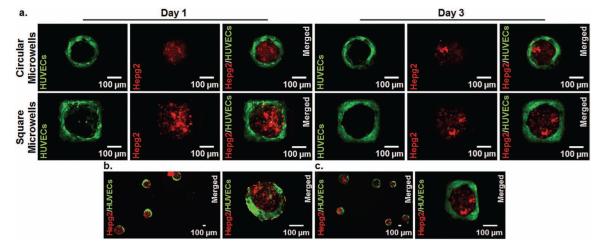
a) Microwells containing HepG2 cells (red) and GFP-HUVECs (green) were cultured for 3 days to observe the morphology of cells. Images of both HepG2 and GFP-HUVECs layers were taken at day 1 and day 3 for both circular and square microwells. HepG2 and GFP-HUVECs layers were shown in different images and their merged images were given for each microwell. Day 1 images show that GFP-HUVECs started spreading and generating an endothelial network around HepG2 aggregates. Within 3 days, GFP-HUVECs formed dense endothelial networks around HepG2 layers. (Day 1 and day 3 images are independent from each other for both microwell types). The resulting microtissues containing HepG2 cells and GFP-HUVECs were recovered from (b) circular microwells and (c) square microwells at day 3.
The resulting microtissues can be recovered from microwells by gently flowing PBS over the surface of the microwells. Microwells may also detach from the substrates at day 3, which may ease the retrieval of the resulting microtissues. Aggregates containing HepG2 cells and GFP-HUVECs were recovered from both circular and square microwells (Figure 4b,c). After retrieval from microwells, aggregates conserved their multi-layered structures containing HepG2 cells and GFP-HUVECs at different layers (Figure 4b,c).
Cell concentrations in this study were optimized for the microwell sizes and shapes used. For different sizes and shapes of microwells and for different applications, cell concentrations and seeding process may need further refinements. Furthermore, microwells with different thermoresponsive behaviors or with different dimensions could be fabricated by changing PNIPAAm prepolymer solutions or by using micromolds with different dimensions. Using higher concentrations of the second cell type and insufficient washing steps may cause the second cell type to wrap over the first cell type. It was observed that after seeding the first cell type, small agitations and improper washing may cause the detachment of aggregates of the first cell type from the microwells. This may lead to the formation of aggregates of the second cell type within the corresponding microwells. Stability of the cells could be improved by using adhesion proteins within microwells in future applications.
Different cell types could be substituted for the cells used in this study. One could pattern multiple cell types within different geometries in a spatially controlled manner to study early stages of development and tissue morphogenesis. It could also be possible to study cancer dynamics, such as metastasis and angiogenesis, by patterning their associated cell types with native spatial organizations. Another potential application could be to fabricate microtissues with native biological complexities for use in regenerative medicine and drug discovery. Furthermore, by using modular tissue engineering methods, [27] the resulting microtissues retrieved from microwells could be assembled into complex architectures with spatially controlled cell placement.
In summary, we described a simple method to control the spatial distributions of multiple cell types in predefined geometries. Dynamic microwells possess shape changing properties at different temperatures due to the thermoresponsiveness of PNIPAAm. This feature was exploited to pattern multiple cell types at different temperatures. Biologically relevant cell types were spatially patterned within dynamic circular and square microwells. The second cell type was spatially arranged around the first cell type in a defined microenvironment. The ability to control spatial organization of various cell types in defined microenvironments can be used to replicate different native biological complexities containing intricate cell-cell interactions, such as those found in development, cancer, and tissues. Thus, the presented method could potentially be useful in a number of fields, including tissue engineering, developmental biology, cancer biology, and drug discovery.
Supplementary Material
Acknowledgements
This work was financially supported by the U.S. Army Research Office through the Institute for Soldier Nanotechnologies at MIT under the project DAAD-19-02-D-002, the MIT-Portugal Program (MPP-09Call-Langer-47), the NIH (DE013023 and DE016516 (R.L.), HL092836, DE019024, EB012597, AR057837, DE021468, and HL099073 (A.K.)), and the Office of Naval Research.
Footnotes
Experimental Section Experimental Details can be found in the Supporting Information.
Supporting Information Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Halil Tekin, Department of Electrical Engineering and Computer Science Massachusetts Institute of Technology Cambridge, MA, 02139, USA; Center for Biomedical Engineering Department of Medicine Brigham and Women’s Hospital Harvard Medical School Boston, MA 02139, USA; David H. Koch Institute for Integrative Cancer Research Massachusetts Institute of Technology Cambridge, MA, 02139, USA.
Jefferson G. Sanchez, Center for Biomedical Engineering Department of Medicine Brigham and Women’s Hospital Harvard Medical School Boston, MA 02139, USA; Department of Chemical Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA
Christian Landeros, Center for Biomedical Engineering Department of Medicine Brigham and Women’s Hospital Harvard Medical School Boston, MA 02139, USA; Department of Biological Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA.
Karen Dubbin, Center for Biomedical Engineering Department of Medicine Brigham and Women’s Hospital Harvard Medical School Boston, MA 02139, USA; Department of Materials Science and Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA.
Prof. Ali Khademhosseini, Center for Biomedical Engineering Department of Medicine Brigham and Women’s Hospital Harvard Medical School Boston, MA 02139, USA; David H. Koch Institute for Integrative Cancer Research Massachusetts Institute of Technology Cambridge, MA, 02139, USA; Department of Chemical Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA; Department of Biological Engineering Massachusetts Institute of Technology Cambridge, MA, 02139, USA; Wyss Institute for Biologically Inspired Engineering Harvard University Boston, MA, 02115, USA.
References
- [1].Nelson CM, VanDuijn MM, Inman JL, Fletcher DA, Bissell MJ. Science. 2006;314:298. doi: 10.1126/science.1131000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hui EE, Bhatia SN. Proc. Natl. Acad. Sci. USA. 2007;104:5722. doi: 10.1073/pnas.0608660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gartner ZJ, Bertozzi CR. Proc. Natl. Acad. Sci. USA. 2009;106:4606. doi: 10.1073/pnas.0900717106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Science. 1997;276:1425. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- [5].Bhatia SN, Yarmush ML, Toner M, Biomed J. Mater. Res. 1997;34:189. doi: 10.1002/(sici)1097-4636(199702)34:2<189::aid-jbm8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- [6].Bhatia SN, Balis UJ, Yarmush ML, Toner M. FASEB J. 1999;13:1883. doi: 10.1096/fasebj.13.14.1883. [DOI] [PubMed] [Google Scholar]
- [7].Nelson CM, Jean RP, Tan JL, Liu WF, Sniadecki NJ, Spector AA, Chen CS. Proc. Natl. Acad. Sci. USA. 2005;102:11594. doi: 10.1073/pnas.0502575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hanahan D, Weinberg RA. Cell. 2011;144:646. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [9].Li LH, Xie T. Annu. Rev. Cell Dev. Biol. 2005;21:605. doi: 10.1146/annurev.cellbio.21.012704.131525. [DOI] [PubMed] [Google Scholar]
- [10].Nelson CM, Bissell MJ. Annu. Rev. Cell Dev. Biol. 2006;22:287. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rossant J, Tam PPL. Development. 2009;136:701. doi: 10.1242/dev.017178. [DOI] [PubMed] [Google Scholar]
- [12].Toh Y-C, Blagovic K, Yu H, Voldman J. Integr. Biol. 2011;3:1179. doi: 10.1039/c1ib00113b. [DOI] [PubMed] [Google Scholar]
- [13].Chiu DT, Jeon NL, Huang S, Kane RS, Wargo CJ, Choi IS, Ingber DE, Whitesides GM. Proc. Natl. Acad. Sci. USA. 2000;97:2408. doi: 10.1073/pnas.040562297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tien J, Nelson CM, Chen CS. Proc. Natl. Acad. Sci. USA. 2002;99:1758. doi: 10.1073/pnas.042493399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Torisawa Y.-s., Mosadegh B, Luker GD, Morell M, O’Shea KS, Takayama S. Integr. Biol. 2009;1:649. doi: 10.1039/b915965g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jinno S, Moeller HC, Chen CL, Rajalingam B, Chung BG, Dokmeci MR, Khademhosseini A. J. Biomed. Mater. Res., Part A. 2008;86A:278. doi: 10.1002/jbm.a.32030. [DOI] [PubMed] [Google Scholar]
- [17].Karp JM, Yeh J, Eng G, Fukuda J, Blumling J, Suh K-Y, Cheng J, Mahdavi A, Borenstein J, Langer R, Khademhosseini A. Lab Chip. 2007;7:786. doi: 10.1039/b705085m. [DOI] [PubMed] [Google Scholar]
- [18].Khademhosseini A, Langer R, Borenstein J, Vacanti JP. Proc. Natl. Acad. Sci. USA. 2006;103:2480. doi: 10.1073/pnas.0507681102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yuan B, Li Y, Wang D, Xie Y, Liu Y, Cui L, Tu F, Li H, Ji H, Zhang W, Jiang X. Adv. Funct. Mater. 2010;20:3715. [Google Scholar]
- [20].Tekin H, Ozaydin-Ince G, Tsinman T, Gleason KK, Langer R, Khademhosseini A, Demirel MC. Langmuir. 2011;27:5671. doi: 10.1021/la200183x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tekin H, Sanchez JG, Tsinman T, Langer R, Khademhosseini A. AIChE J. 2011;57:3249. doi: 10.1002/aic.12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tekin H, Anaya M, Brigham MD, Nauman C, Langer R, Khademhosseini A. Lab Chip. 2010;10:2411. doi: 10.1039/c004732e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tekin H, Tsinman T, Sanchez JG, Jones BJ, Camci-Unal G, Nichol JW, Langer R, Khademhosseini A. J. Am. Chem. Soc. 2011;133:12944. doi: 10.1021/ja204266a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Smrekar B, Wightman L, Wolschek MF, Lichtenberger C, Ruzicka R, Ogris M, Rodl W, Kursa M, Wagner E, Kircheis R. Gene Ther. 2003;10:1079. doi: 10.1038/sj.gt.3301965. [DOI] [PubMed] [Google Scholar]
- [25].Liu VA, Bhatia SN. Biomed. Microdevices. 2002;4:257. [Google Scholar]
- [26].Rouwkema J, Rivron NC, van Blitterswijk CA. Trends Biotechnol. 2008;26:434. doi: 10.1016/j.tibtech.2008.04.009. [DOI] [PubMed] [Google Scholar]
- [27].Nichol JW, Khademhosseini A. Soft Matter. 2009;5:1312. doi: 10.1039/b814285h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.