

Abstract
Mammalian homologues of the Drosophila canonical Transient Receptor Potential (TRPC) protein have been proposed to encode the store-operated Ca2+ influx (SOC) channel(s). This study examines the role of TRPC1 in the SOC mechanism of retinal cells. htrpc1 transcript was detected in bovine retinal and in human adult retinal pigment epithelial (ARPE) cells. Western blot analysis also confirmed the expression of TRPC1 protein in neuronal cells including retina and ARPE cells. To determine the role of TRPC1 protein in retinal cells, TRPC1 was recombinantly expressed in ARPE cells and changes in intracellular Ca2+ were analyzed. ARPE cells stably transfected with htrp1 cDNA displayed 2-fold higher Ca2+ influx with no significant increase in the basal influx. Consistent with this the overexpressed TRPC1 protein was localized in the plasma membrane region of ARPE cells. Interestingly, both bovine retinal tissues and ARPE cells showed that TRPC1 protein co-localizes and could be co-immunoprecipitated with β-tubulin. Disruption of tubulin by colchicine significantly decreased both plasma membrane staining of the TRPC1 protein and Ca2+ influx in ARPE cells. These results suggest that TRPC1 channel protein is expressed in retinal cells, further, targeting/retention of the TRPC1 protein to the plasma membrane in retinal cells is mediated via its interaction with β-tubulin.
Keywords: Transient receptor potential protein, Store-operated calcium entry, β-tubulin, Trafficking, Retina
Introduction
Ca2+ is one of the most universal signal-transduction element, which plays an important role in regulating a great variety of cellular processes (Berridge, 1998). Retinal cells displaying coordinated Ca2+ activity are necessary for the development of the nervous system (Ohmasa & Saito, 2004). It has been proposed that Ca2+ signaling within clusters in the retinal pigment epithelium may serve to coordinate cell cycle events (Owens & Kriegstein, 1998). In neuronal cells entry of Ca2+ is mediated via two pathways, either via the voltage-gated ion channels (activated in response to voltage change) or store operated Ca2+ channels (SOCC). These SOC channels are present in the plasma membranes and are activated upon depletion of internal Ca2+ store, termed as store-operated Ca2+ entry (SOCE) (Putney, 1991). The molecular events involved in the regulation of SOCE have not yet been established; part of this is due to the unknown identity of the SOCE channel. Further, it was originally thought that SOCE was only present in nonexcitable cells; however, recent studies have shown that SOCE is a not only present, but is essential for many neuronal functions (Berridge, 1998; Micci & Christensen, 1998). Identification of Transient Receptor Potential (TRP) mutants in Drosophila suggests that TRP proteins function as SOCE channel. Till date, 28 mammalian members of the TRP super family have been identified, which are involved in numerous cellular functions (Berridge, 1998; Montell, 2001; Clapham, 2003). TRP proteins have been identified in chicken retina and amacrine cells, suggesting that these could be important for normal physiology of these retinal cells (Sosa et al., 2002; Crousillac et al., 2003). However, not much is known about the expression of TRPs in retinal pigment epithelium (RPE) cells. Further, in view of the relationship between Ca2+ signaling and TRP proteins, understanding the expression of TRPs would be important to understand the function of these ion channels.
In Drosophila, mutations in the trp gene cause retinal degeneration and neuronal cell death (Minke, 2002), indicating that mammalian TRP proteins could have a significant role in retinal function. Several observations support the view that the RPE cells regulate the development of the neural retina. Neural retinal cells when dissociated can reaggregate and can functionally differentiate by the addition of RPE cells (Vollmer & Layer, 1986; Rothermel et al., 1997). Further, Raymond and Jackson (1995) have shown that genetically ablating RPE cells in transgenic mice result in severe disruption of the underlying neural retinal structure. Study of albino animals also shows that when RPE is devoid of melanin, the neural retina shows developmental defects, including increased cell proliferation and cell death (Ilia & Jeffery, 2000). The signaling mechanisms by which RPE exerts its effects on the developing neural retina are largely unknown. Ca2+ waves have been shown to play an important role in controlling retinal cell division (Catsicas & Mobbs, 2001). However, the channels which could mediate this effect are largely unknown. Here, as a first step we have shown that indeed TRP proteins are expressed in RPE cells and contribute towards Ca2+ signaling in these cells.
TRPC protein have been identified in both excitable and non-excitable cells, and have been demonstrated as candidates for store operated calcium channels (SOCC). Overexpression of TRPC in various cell types leads to an increase in Ca2+ influx upon agonist stimulation (Minke & Cook, 2002). Whereas, expression of TRPC in antisense orientation resulted in the decrease of Ca2+ influx, which suggests that TRPC channels are critical for SOCE. (Liu et al., 2000; Minke & Cook, 2002). Although it is well established that some members of TRPC proteins can be activated upon store depletion and others are subjected to a gating mechanism that operates through phospholipase C, the molecular mechanism involved in the activation of TRPC is still an unresolved issue.(Liu et al., 2000; Putney, 2003). Our previous research demonstrates that TRPC1 functions as a store-operated Ca2+ influx channel, a process critical for normal function of most cells (Singh et al., 2001; Bollimuntha et al., 2004). Here we have shown that TRPC1 is expressed in retinal tissues including RPE cells. Further, we have shown that in retinal and in brain tissues, TRPC1 interacts with neuronal specific cytoskeletal protein β-tubulin. This interaction is critical for the plasma membrane localization and in the function of TRPC1 protein in RPE cells. Thus, overall our results indicate that TRPC1 association with cytoskeletal is critical for its function as well as its localization/retention to the plasma membrane.
Materials and methods
ARPE cell culture transfection and crude membrane preparation
ARPE cells were cultured and stably transfected, by G418 selection, using lipofectAMINE and pcDNA3TRPC1 vector as described earlier (Liu et al., 2000). Cells were harvested by scraping in ice-cold phosphate-buffered saline (PBS) containing 1% (v/v) aprotinin (Sigma, St. Louis, MO) and centrifugation for 5 min at 2000g. The cell pellet was resuspended in a lysis buffer containing (in mM): 10 Tris-HCl (pH 8.0), 1 MgCl2, 0.5 4-(2-aminoethyl)-bezenesulfonyl fluoride hydrochloride (AEBSF) (ICN Biomedicals, Inc., Aurora, OH), 0.1 phenylmethysulfonyl fluoride (PMSF) (Calbiochem, La Jolla, CA) and frozen at −80°C for at least 2 h before use. Crude membranes were prepared from cell lysates by homogenization and centrifugation of as described previously (Liu et al., 2000). Protein concentration was determined by using the Biorad protein assay (microassay procedure).
RNA isolations, synthesis of the first strand cDNA, and reverse transcription-polymerase chain reaction (RT-PCR) analysis
Fresh bovine eye balls were obtained from the local slaughter house and retina was isolated and frozen immediately in liquid nitrogen. Total RNA was extracted from the frozen tissues and ARPE cells using TRIzol reagent (Life Technologies, Inc., Gaitherburg, MD) and was treated with deoxyribonuclease I (Life Technologies, Inc.) at a concentration of 1 unit of DNase I /1 μg of RNA in a buffer containing 20 mM Tris-HCl (pH 8.4), 2 mM MgCl2, 50 mM KCl, for 15 min at room temperature. The reaction was terminated by adding ethylene diamine tetracetic acid (EDTA) at a final concentration of 2.5 mM and heated at 65°C for 10 min. Sequences specific for the trpc-genes were used as described previously (Bollimuntha et al., 2004). First strand cDNA synthesis and RT-PCR was performed as described previously (Liu et al., 2000; Bollimuntha et al., 2004). After PCR, 10 μl of the RT-PCR product was analyzed on a 1% agarose gel.
Immunoprecipitation, western blotting, and mass spectroscopy analysis
Crude membranes were treated with 0.5% Nonidet P-40 or with 1.5 mM Octylglucoside + 0.5 M KI and the solubilizate was collected by centrifugation at 45,000 g for 60 min and precleared by incubation with an excess of the protein A beads and centrifugation. 200 μg of the cleared sample was incubated with either 10 μg of monoclonal anti-HA (Roche Research Center, Nutley, NJ) or polyclonal anti-Tubulin (Sigma) antibody for overnight at 4°C. Immunocomplexes were pulled down with 30 μl of 50% protein A beads for 2 h at 4°C. Beads were washed four times with wash buffer as described in Lockwich et al. (2001) and treated with sodium dodecyl sulphate (SDS) solubilization buffer. SDS-PAGE and western blotting detected TRPC1 protein as described before (Liu et al., 2000, Bollimuntha et al., 2004). Anti-HA and anti-TRPC1 were used at 1:1000 dilutions. The secondary antibodies and conditions for the enhanced chemiluminescence (ECL) reaction were as described previously (Singh et al., 2001). For mass spectroscopy analysis, immunocomplexes pulled down by TRPC1 antibody or IgG control antibody were trypsined (10 units) overnight at room temperature. 10 μl of samples containing peptides were injected by autosampler onto a 0.3 × 1-mm trapping column (PepMap C18; LC Packings) using a CapLC system (Micromass, Beverly, MA). Peptides were eluted at 250 nl/min and chromato-graphed on a 50-μm × 5-cm Biobasic C18 column (New Objective, Cambridge, MA) with a gradient of 5–40% acetonitrile over 20 min followed by 80% acetonitrile for 5 min. The eluants were directed into a quadruple time-of-flight mass spectrometer (QTOF2; Micromass, Beverly, MA) and ionized immediately using an electrosprayer. The mass spectrometer was operated in standard MS/MS switching mode with the three most intense ions in each survey scan subjected to MS/MS analysis. In addition, the “include function” of the instrument operating software was used to program MS/MS analysis of precursor ions of all possible tryptic peptides based on the structure of the proteins pre identified by MALDITOF MS (calculated as doubly and triply charged ions). Peptide identification and MS/MS data analyses utilized Micromass software ProteinLynx™ Global Server, MassLynx™ Version 3.5, and the Swiss-Protein and NCBI protein sequence databases.
Yeast two-hybrid assay
N-TRPC1 or C-TRPC1 was subcloned into pGBKT7 (GAL4 DNA binding domain; Clontech, Mountain View, CA) and used to transform the yeast reporter strain AH 109 (Clontech). Cells were selected by growth on SD-trp− plates and retransformed using human brain library (Clontech) and then plated on SD-trp−, leu− and SD trp−, leu−, his−, ade2− plates. The latter was used to select positive clones and interaction was confirmed by β-Galactosidase assay (Clontech Matchmaker system). Plasmid DNA was isolated from positive clones, E. coli were transformed, DNA was isolated, sequenced, and analyzed using BLAST as described in Singh et al., (2004).
Confocal microscopy
RPE cells expressing HA-TRPC1 were cultured on coverslips encoded with poly L lysine for 24 h. Cells were fixed, permeabilized, and treated with either monoclonal anti-HA, polyclonal anti-TRPC1, or polyclonal anti-tubulin antibody at 1:100 dilution for 1 h. Cells were then washed and probed with either rhodamine-linked or florescein isothiocyanate (FITC)-linked secondary antibodies as described before (Singh et al., 2004). Confocal images were collected using an MRC 1024-krypton/argon Zeiss laser scanning confocal equipped with an inverted photomicroscope.
[Ca2+]i measurements
Fura2 fluorescence in single cells was measured as described earlier (Singh et al., 2002, 2004) by using an Photonics monochrometer attached to an inverted Olympus ×70 microscope with a Fluor 40× oil-immersion objective. Images were acquired using an enhanced CCD camera (OCRA) and the PCI software (Universal Imaging Corporation, PA). Analog plots of the fluorescence ratio (340/380) in single cells are shown.
Results
TRPC1 protein is expressed in retinal cells including RPE
We used multiple approach using molecular, biochemical, and physiological techniques to identify the presence of TRP proteins in bovine retina. RT-PCR was performed using TRPC specific primers (described in Bollimuntha et al., 2004). As indicated in Fig. 1A, only trpc1, trpc5, and trpc6 gene products were observed (500 bp as expected) in whole bovine retina, suggesting that these TRP genes are expressed in bovine retinal tissues. The lack of expression of trpc2, trpc3, and trpc5 was not due to their primer design or other experimental limitations as desired polymerase chain reaction (PCR) products were obtained when plasmid DNA was used as template (data not shown). Semiquantitative measurement of RT-PCR product showed that trpc1 was expressed the most, followed by trpc5 and trpc6 gene. To demonstrate if these trp genes are also expressed in retinal pigment epithelium (RPE) cells, mRNA was isolated from cultured ARPE-19 cells and RT-PCR was performed. Using TRPC specific primers we have demonstrated that trpc1 was expressed in ARPE cells, whereas, other trp transcripts were not amplified under these conditions. Overall, these results suggest that TRPC1 should be the primary storedependent Ca2+ channel present in ARPE cells (Fig. 1B). RT-PCR was also performed using 5 μg of retinal or ARPE mRNA (for 40 PCR cycles), which also displayed similar results and no expression of other trp genes was observed (data not shown). Gene products obtained from bovine retina and ARPE cells were cloned and sequenced, which confirmed that the PCR products obtained were trpc genes (data not shown).
Fig. 1.

Expression of TRPC1 in retinal cells. RT-PCR products (∼ 500-base pair region) amplified from mRNA isolated from fresh bovine retina (A) or cultured ARPE cells (B). TRPC specific forward and reverse primers were used along with 0.5 μg of mRNA for 25 cycles. C, D: Detection of TRPC1 protein (from bovine heart, liver, kidney, brain, retina, and ARPE cells) using anti-TRPC1 antibodies. Conditions for SDS-polyacrylamide gel electrophoresis and western blot analysis were the same as described in Singh et al. (2001). The TRPC1 protein is indicated by an arrow. E, F,G: Localization of TRPC1 in ARPE cells detected using anti-TRPC1 antibody followed by incubation with rhodamine-linked anti-rabbit IgG (F). No fluorescence was detected when the primary antibody was not added (E). Localization in ARPE cells over expressing TRPC1 detected using anti-HA antibodies (G)
Endogenous TRPC1 protein expression was determined via western blot analysis using TRPC1 specific polyclonal antibodies (Liu et al., 2000; Singh et al., 2001). TRPC1 protein was detected in the plasma membranes isolated from bovine tissues (heart, brain, liver, kidney, and retina cells, Fig. 1C) and also from crude membranes isolated from ARPE cells (Fig. 1D). To confirm the specificity of TRPC1 antibody human submandibular gland cells overexpressing HA-tagged TRPC1 protein was used as a control (Fig. 1D). This blot was stripped and re-probed with HA antibodies, which showed the same band in control samples (data not shown). Based on these results, we can confirm that a significant amount of TRPC1 is expressed in the plasma membrane of neuronal and nonneuronal cells including retina. TRPC6 antibody (obtained from Sigma) failed to show endogenous TRPC6 protein in retinal cells (data not shown); lack of signal could be either due to very low expression of the TRPC6 protein or due to the specificity of the TRPC6 antibody.
To define the expression and localization of TRPC1 proteins immunoflourescence technique was adopted. ARPE cells were cultured on glass coverslips for 24 h and detected using polyclonal TRPC1 antibody. As indicated in Figs. 1F and 1G, punctate staining of the fluorescently labeled TRPC1 protein was observed along the plasma membrane confirming the expression of the protein in the ARPE cells. No immunoreactivity was observed in the absence of the primary antibody (Fig. 1E). Similar results were also obtained when HA-tagged TRPC1 was overexpressed and immunoflourescence was performed using monoclonal HA antibodies (Fig. 1G). These results are consistent with our previous results, where significant expression of TRPC1 was observed in the plasma membrane (Liu et al., 2000, Bollimuntha et al., 2004).
Physical interaction between TRPC1 and β-tubulin
A rat brain cDNA library was screened using a yeast two-hybrid (Y2H) system to identify proteins interacting with the N-terminus (aa 1-349) or C-terminus of TRPC1 (aa 643-793, Fig. 2A). Interacting proteins were screened followed by the isolation of plasmid and were sequenced to identify the gene. In our multiple screening, only the N-terminus of TRPC1 showed significant interaction with β-tubulin protein and was identified as a possible interacting partner for NTRPC1 (Fig. 2A shows the Y2H interactions obtained with these clones). C-TRPC1 did not showed any interaction with this protein (the results were negative in three trials). Further, tubulin plasmid was isolated from the positive clone and tested against CTRPC1 and no interaction was detected (data not shown). Only the N-TRPC1/tubulin clone showed a significant increase in β-galactosidase units (18 units) which was comparable to that obtained in the positive controls (25 units).
Fig. 2.
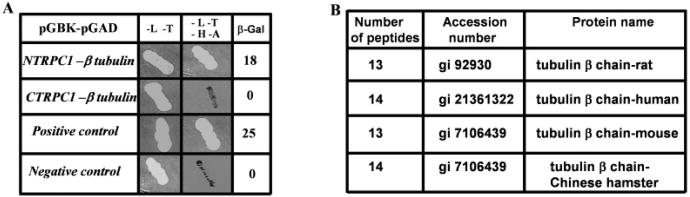
Interaction of TRPC1 with β-tubulin. A: Yeast two-hybrid interactions between the N (aa 1-349) or C-terminus (aa 643-793) of TRPC1 (NTRPC1 and CTRPC1) and β-tubulin protein. Quantitative β-gal assay were 18 and 25 for NTRPC1/tubulin and for the positive controls, 0 indicates no β-gal activity. B: Proteins identified by mass spectrometer, TRPC1-specific immunoprecipitate was trypsined and individual peptides were sequenced and matched against the protein data bank.
Although yeast two-hybrid is accepted as a reliable method for examining protein interactions, there are few limitations associated with this technique. Further, yeast two-hybrid was performed using rat brain cDNA library. Thus, to study proteins interacting in retinal cells, we performed additional experiments using mass spectrophotometer. Crude membranes were isolated from bovine retinal tissues and solubilized using OG and KI as described in Singh et al. (2004). Detergent solubilized membranes were incubated with polyclonal anti-TRPC1 antibody. Immunocomplexes were pulled down using protein A agarose beads, washed thoroughly (4 times as described in Singh et al., 2004), trypsinized, and peptides were detected using LC-MS-MS system. As indicated in Fig. 2B, peptides obtained from MS-MS indicated that in bovine retinal cells, TRPC1 protein specifically interacts with β-tubulin. This interaction was specific since immunocomplex pull down using anti rabbit IgG showed no pull down of the β-tubulin protein (data not shown). In aggregate, these data suggest that endogenous TRPC1 in neuronal cells associates with β-tubulin. These results are also consistent with other observations, where it was reported that both TRPC3 and TRPC4 interacts with cytoskeletal proteins (Lockwich et al., 2001; Merry et al., 2002).
Disruption of tubulin mislocalizes TRPC1 protein in ARPE cells
To investigate the interaction in RPE cells, we overexpressed HA-tagged TRPC1 protein in ARPE cells. The interaction between TRPC1/tubulin was further confirmed by immunoprecipitation and immunolocalization studies. Anti-HA antibody, anti-tubulin, or anti-TRPC1 antibodies were used to immunoprecipitate TRPC1 protein from detergent extracts of crude membranes isolated from ARPE cells transiently expressing HA-TRPC1. As indicated in Fig. 3A, TRPC1 was detected in both immunoprecipitates where either HA antibodies or tubulin antibodies were used to pull down the complex (Fig. 3A). In reverse immunoprecipitation experiments, the tubulin protein was also detected in immunoprecipitates using HA and tubulin antibodies (data not shown). Antibody controls were also performed where immunocomplex was pulled down using HA antibodies in ARPE cells, which do not overexpress HA-TRPC1 protein to rule out the possibility of nonspecific interactions.
Fig. 3.

Immunoprecipitation and co-localization of TRPC1 with tubulin. A: Co-immunoprecipitation of TRPC1 with the cytoskeletal protein tubulin. Crude membranes prepared from ARPE cells transiently expressing HA-TRPC1 were solubilized and immunoprecipitation was performed as described in experimental procedures. Proteins in the IP were detected using SDS PAGE and immunoblotting. Antibodies used for IP and IB are indicated in the figure. Control IPs is shown by crude membranes of HA-TRPC1 expressing ARPE cells. B: Co-localization of HA-TRPC1 with endogenous tubulin proteins in ARPE cells. Anti-HA antibody, anti tubulin antibody, and FITC or rhodamine-conjugated secondary antibodies were used to detect transiently expressed HA-TRPC1 and endogenous tubulin in ARPE cells. Overlay of the images is shown in the right panel (yellow signal). Confocal pictures of the transiently expressing HA-TRPC1 and tubulin proteins in ARPE cells treated with colchicine (lower panels). Arrows in the images show the protein localization.
To assess the reliability and confirmation of this interaction, immunolocalization studies were performed. Localization of overexpressed HA-TRPC1 (using monoclonal antibodies) and endogenous tubulin protein (using polyclonal antibodies) in ARPE cells was examined by immunoflourescence using confocal microscopy. Fig. 3B (upper panel) demonstrates that HA-TRPC1 signal is primarily in the plasma membrane and subplasma membrane regions. Tubulin being a cytoskeletal protein was also localized in the subplasma membrane region and some at the plasma membrane region. Overlay of both the images demonstrated significant overlap of the two fluorescence signals (Fig. 3B, yellow signal). Single labeling was also performed using single antibodies, which also showed similar results and no stress fibers were observed with tubulin staining in these cells (data not shown). To further confirm this interaction and to determine the functional consequences of this interaction, ARPE cells were treated with colchine (100 μM) for 3 h. Similar results were also obtained with lower colchine doses (1 μM) as shown in Fig. 3B. Staining of ARPE cell treated with colchicine showed an altered tubulin staining, where a diffused cytoplasmic staining of the tubulin protein was observed. Interestingly, the plasma membrane staining of the TRPC1 protein was also severely disrupted, with most of the HA-TRPC1 protein present in the cytoplasm (Fig. 3B). Both tubulin and HA-TRPC1 proteins showed a significant co-localization, however, the punctate plasma membrane staining of HA-TRPC1 was absent upon colchicine treatment. In aggregate, our data suggest that TRPC1 interacts with tubulin and disruption of tubulin leads to mislocalization of the TRPC1 protein in RPE cells.
Thapsigargin stimulated Ca2+ influx in decreased upon disruption with tubulin
Ca2+ measurement was performed to determine if SOCE mechanism is present in RPE cells. RPE cells were loaded with FURA-2, and fluorescent measurement (340/380) was performed to detect changes in cytosolic Ca2+. Thapsigargin (Tg, 2 μM), a SERCA pump blocker induced a rapid release of Ca2+ from intracellular stores as demonstrated by the first peak in Fig. 4A, since no external Ca2+ is present the increase in cytosolic Ca2+ ([Ca2+]i) will be due to release of internal Ca2+ stores. When Ca2+ was readded to the cells, a robust second increase in [Ca2+]i was obtained, which represents the Ca2+ influx component (Figs. 4A & 4B), through the SOCE channel. Since TRPC1 was only expressed in RPE cells (as evident with the RT-PCR data, Fig. 1B), it could be stipulated that this increase in cytosolic Ca2+ would be via the TRPC1 channel activity. Interestingly, similar results were obtained by other investigators who had demonstrated that RPE cells do exhibit SOCE properties; however, no TRP proteins were identified in these experiments (Ryan et al., 1999; Himpens & Vereecke, 2000). Ca2+ influx was also measured in cells treated with colchicine for 6 h. As indicated in Figs. 4C and 4D addition of Tg in a Ca2+ free media showed an increase in the cytosolic Ca2+. This increase in cytosolic Ca2+ was similar as that of untreated cells (Figs. 4A & 4B); however, the Ca2+ influx measured upon readdition of 1 mM external Ca2+ was drastically reduced in the ARPE cells which were subjected to colchicine (Figs. 4C & 4D). The mean value showed a 40–50% decrease (as compared with control) in the amount of Ca2+ influx entering these cells. Whereas, no change in the basal Ca2+ level measured by the addition of 1 mM Ca2+ (without Tg stimulation) was observed. These results are in accordance with our immunoflourescence studies where colchicine treatment disrupts tubulin as well as the plasma membrane localization of the TRPC1 protein. Decrease in Ca2+ influx, as well as TRPC1 protein, reiterates the assumption that indeed tubulin has got a substantial role in the localization and function of TRPC1 protein and disruption of tubulin will lead to an decreased plasma membrane staining as well as reduced Ca2+ influx in ARPE cells.
Fig. 4.

Colchicine treatment decrease thapsigargin stimulated TRPC1 activity. Ca2+ influx were measured in Tg-stimulated cells in a Ca2+-free buffer, followed by addition of 1 mM Ca2+ to the medium. A, C: Fluorescence traces in either control ARPE cells (A) or colchine-treated ARPE cells (B) stimulated with Tg. B, D: Bar graphs showing the relative Ca2+ influx in the absence and in the presence of extracellular Ca2+. “*” indicate values that are significantly different from that of the respective control condition (P < 0.02, number of cells is indicated in each case). Internal Ca2+ release and basal Ca2+ influx were not altered by colchine treatment.
Discussion
The data presented above demonstrate that TRPC1 is expressed in bovine retinal as well as in RPE cells. RPE cells are highly specialized epithelium cells and serves as multifunctional and indispensable component of the vertebrate eye. To understand the signaling mechanisms present in RPE, we investigated storedependent Ca2+ channels present in RPE and in neural retina. We used ARPE-19 cells, which are derived from nontransformed, human RPE cell line, which display a normal kariology (Dunn et al., 1996). This cell line has been previously used as an alternative to primary cultures because of its availability and the stability of its features in prolonged cultivation. To establish the expression of various TRPC proteins, we performed RT-PCR on mRNA isolated from both bovine retina and RPE cells. Our results indicate that TRPC1 is expressed predominately in both bovine retina and in RPE cells. These results were further confirmed using TRPC1 specific antibody, which detected ∼80 kDa protein band. These results are consistent with our previous results where a similar band was detected in salivary gland cells (Liu et al., 2000; Singh et al., 2001). The specificity of the TRPC1 antibody was further tested, where HA-tagged TRPC1 was transiently overexpressed in RPE cells and western blots were performed using anti-TRPC1 antibody. RPE cells overexpressing TRPC1 showed a significant increase in the TRPC1 protein levels. Presence of TRPC1 in retinal cells is critical, since TRPC1 exhibits many features, which are common to Drosophila TRP proteins, and Drosophila TRP proteins are known to have a significant role in phototransduction (Montell, 2001). Although, the function of TRPC1 is largely unknown in human retina, it could be postulated that mammalian TRPC1 could have a similar function as that of its counterpart in Drosophila and more research is needed to understand the function of TRPC1 protein in retinal function.
A great deal of attention has been paid on the role of RPE in neural retina development, whereas, little is known about the molecular mechanisms and the signaling processes present in RPE cells itself. Processes such as development of the neural retina are controlled in part by the adjacent RPE cells, which require Ca2+ signaling. In retinal cells, Ca2+ entry occurs via many plasma membrane ion channels mainly the store-operated Ca2+ channels or via the voltage-operated Ca2+ channels (Osborne et al., 2001). Investigations using patch-clamp techniques on cultured or freshly isolated RPE cells have demonstrated that voltage-operated Ca2+ channels (similar to L type channels) are expressed in RPE cells and contribute towards signal transduction pathways by communicating between RPE and photoreceptors (Rosenthal & Strauss, 2002). Whereas, the store-operated Ca2+ channels (activated by store depletion per se) are largely unknown. Our results indicate that in bovine retinal tissues TRPC1 is expressed predominantly. This expression of TRPC1 was further confirmed in RPE cells. Our results are also consistent with other published reports, where it was shown that both TRPC1 and TRPC4 were expressed in the adult chicken retina (Crousillac et al., 2003). Since both TRPC1 and TRPC4 are store-operated Ca2+ channels, it is possible that TRPC4 could also contribute towards store-operated calcium influx. Interestingly, this group also demonstrated that TRPC1 co-localizes with a Ca2+-sensitive bNOS, which could be of greater relevance during neural degeneration.
Our results also indicate a functionally significant interaction between TRPC1 and cytoskeletal protein β-tubulin. This association was observed in both retinal and brain tissues and was observed for both endogenous and exogenously expressed TRPC1 in retinal cells. In RPE cells, tubulin is localized in three distinct patterns: (1) in a single apical cilium, (2) in a network under the apical plasma membrane, and (3) in longitudinally arranged bundles near the lateral membranes and the nucleus (Topp et al., 1996). This is consistent with our studies where a significant staining of tubulin was observed at the apical end, whereas, on the basolateral end the staining was significantly decreased. To further confirm the interaction between TRPC1 and tubulin, RPE cells were treated with colchine, which inhibits tubulin dimer formation resulting in a loss of tubulin function (Downing & Nogales, 1999). Interestingly, addition of colchicine disrupted the plasma membrane staining of the TRPC1 protein. However, both tubulin and TRPC1 proteins showed a significant co-localization, suggesting that functional tubulin is critical for its plasma membrane localization/retention. To demonstrate this more physiologically, we performed Ca2+ imaging on RPE cells. Addition of thapsigargin showed an initial increase in cytosolic Ca2+ ([Ca2+]i), and since no external Ca2+ was present this increase must be due to the release of Ca2+ from internal stores, which will activate TRPC1 channel in the plasma membrane. Addition of external Ca2+ increased [Ca2+]i, which was significantly decreased upon colchicine treatment. These results are consistent with our confocal data, where a decrease in the plasma membrane staining of TRPC1 was observed.
We have also demonstrated that the interaction between TRPC1 and β-tubulin is mediated via the N-terminal domain. Interestingly, all TRPC proteins have a high homology region at the N-terminus region, which consists of 3–4 ankyrin repeats and a coiled coil domain. These are the speculated regions through which TRP proteins could interact with other candidate proteins, responsible for their function and regulation. Also recently using microarray analysis, 30 genes were identified which were altered upon retinal degeneration. Interestingly, one such gene was the endoplasmic reticulum Ca2+-ATPase gene, which was upregulated in degenerative retina (Sato et al., 2003). Although TRPC1 was not directly identified in this study, it could still be speculated that TRPC1 could be involved in retinal degeneration, since Ca2+-ATPase is directly involved in regulating TRPC1 channel activity. Further, TRPC1 homologue in Drosophila has been shown to have a role in retinal degeneration (Hong et al., 2002). Based on these experimental experiences, we can hypothesize that the regulation of intracellular Ca2+ levels may be a potentially effective therapy to prevent progressive retinal degeneration, and Ca2+ channels could be used as specific targets to prevent retinal degeneration. Alternatively, TRPC1 could very well contribute towards fluid regulation in RPE cells, since our previous studies had shown that overexpression of TRPC1 protein in rat salivary gland cells increases fluid secretion (Singh et al., 2001).
Presence of TRPC1 protein in retinal and RPE cells opens new possible target proteins, which could be involved in several retinal functions. Further, decrease in Ca2+ influx in RPE cells upon treatments with colchicine suggests that β-tubulin is critical for this event. Immunolocalization studies reveal that TRPC1 is mislocalized, when ARPE cells are treated with colchine. Taken together our results suggest that TRPC1 proteins are expressed in ARPE cells and contribute towards the functional role of retinal cells. Further, our results identified a novel interaction between TRPC1 protein and tubulin protein. Tubulin protein is critical for cellular function, thus, it interaction with TRPC1 indicates that TRPC1 must have a critical role in neuronal function and more research is needed to establish this relationship.
Acknowledgments
This work was supported by research grant (ND-BRIN) and ND-EPSCoR granted to Brij B. Singh. We thank Dr(s) Gene Homandberg, Masaru Miyagi, and Indu Ambudkar for constant encouragement, reagents, instruments, and support. We specially thank Ashley Bansal, Ryan Carruth, and Tami Casavan for technical assistance.
References
- Berridge MD. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bollimuntha S, Singh BB, Shavali S, Sharma SK, Ebadi M. TRPC1-mediated inhibition of MPP+ neurotoxicity in humanSH-SY5Y neuroblastoma cells. Journal of Biological Chemistry. 2005;280:2132–2140. doi: 10.1074/jbc.M407384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catsicas M, Mobbs P. The GABAB receptors regulate chick retinal calcium waves. Journal of Neuroscience. 2001;21:897–910. doi: 10.1523/JNEUROSCI.21-03-00897.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Crousillac S, LeRouge M, Rankin M, Gleason E. Immunolocalization of the TRPC channel subunits 1 and 4 in the chiken retina. Visual Neuroscience. 2003;20:453–463. doi: 10.1017/s0952523803204107. [DOI] [PubMed] [Google Scholar]
- Downing KH, Nogales E. Crystallographic structure of tubulin: Implications for dynamics and drug binding. Cell Structure and Function. 1999;24:269–275. doi: 10.1247/csf.24.269. [DOI] [PubMed] [Google Scholar]
- Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Experimental Eye Research. 1996;62:155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- Himpens B, Vereecke J. Intra- and intercellular Ca2+-signal transduction. ScienceSTKE. 2000;62:501–563. [PubMed] [Google Scholar]
- Hong YS, Park S, Geng C, Baek K, Bowman JD, Yoon J, Pak WL. Single amino acid change in the fifth transmembrane segment of the TRP Ca2+ channel causes massive degeneration of photoreceptors. Journal of Biological Chemistry. 2002;277:33884–33889. doi: 10.1074/jbc.M204075200. [DOI] [PubMed] [Google Scholar]
- Ilia M, Jeffery G. Retinal cell addition and rod production depend on early stages of ocular melanin synthesis. Journal of Comparative Neurology. 2000;420:437–444. [PubMed] [Google Scholar]
- Liu X, Wang W, Singh BB, Lockwich T, Jadlowiec J, O'Connell B, Wellner R, Zhu MX, Ambudkar IS. TRPC1 a candidate protein for the store-operated Ca2+ influx mechanism in salivary gland cells. Journal of Biological Chemistry. 2000;275:3403–3411. doi: 10.1074/jbc.275.5.3403. [DOI] [PubMed] [Google Scholar]
- Lockwich T, Singh BB, Liu X, Ambudkar IS. Stabilization of cortical actin induces internalization of transient receptor potential 3 (Trp3)-associated caveolar Ca2+ signaling complex and loss of Ca2+ influx without disruption of Trp3-inositol trisphosphate receptor association. Journal of Biological Chemistry. 2001;276:42401–42408. doi: 10.1074/jbc.M106956200. [DOI] [PubMed] [Google Scholar]
- Mery L, Strauss B, Dufour JF, Krause KH, Hoth M. The PDZ-interacting domain of TRPC4 controls its localization and surface expression in HEK293 cells. Journal of Cell Science. 2002;115:3497–3508. doi: 10.1242/jcs.115.17.3497. [DOI] [PubMed] [Google Scholar]
- Micci MA, Christensen BN. Exchange in catfish retina horizontal cells: Regulation of intracellular Ca2+ store function. American Journal of Physiology. 1998;274:1625–1633. doi: 10.1152/ajpcell.1998.274.6.C1625. [DOI] [PubMed] [Google Scholar]
- Minke B. The TRP calcium channel and retinal degradation. Advances in Experimental Medicine and Biology. 2002;514:601–622. doi: 10.1007/978-1-4615-0121-3_34. [DOI] [PubMed] [Google Scholar]
- Minke B, Cook B. TRP channel proteins and signal transduction. Physiological Review. 2002;82:429–452. doi: 10.1152/physrev.00001.2002. [DOI] [PubMed] [Google Scholar]
- Montell C. Physiology, phylogeny, and functions of the TRP superfamily of cation channels. Science STKE. 2001;90 doi: 10.1126/stke.2001.90.re1. REI. [DOI] [PubMed] [Google Scholar]
- Ohmasa M, Saito T. GABAA-receptor-mediated increase in intracellular Ca2+ concentration in the regenerating retina of adult newt. Neuroscience Research. 2004;49:219–227. doi: 10.1016/j.neures.2004.02.015. [DOI] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR. Patterns of intracellular calcium fluctuation in precursor cells of the neocortical ventricular zone. Journal of Neuroscience. 1998;18:5374–5388. doi: 10.1523/JNEUROSCI.18-14-05374.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW., Jr The capacitative model for receptor-activated calcium entry. Advances in Pharmacology. 1991;22:251–269. doi: 10.1016/s1054-3589(08)60037-x. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr Capacitative calcium entry in the nervous system. Cell Calcium. 2003;34:339–344. doi: 10.1016/s0143-4160(03)00143-x. [DOI] [PubMed] [Google Scholar]
- Raymond SM, Jackson IJ. The retinal pigmented epithelium is required for development and maintenance of the mouse neural retina. Current Biology. 1995;5:1286–1295. doi: 10.1016/s0960-9822(95)00255-7. [DOI] [PubMed] [Google Scholar]
- Rosenthal R, Strauss O. Ca2+-channels in the RPE. Advances in Experimental Medicine and Biology. 2002;514:225–235. [PubMed] [Google Scholar]
- Rothermel A, Willbold E, Degrip WJ, Layer PG. Pigmented epithelium induces complete retinal reconstitution from dispersed embryonic chick retinae in reaggregation culture. Proceedings of the Royal Society (London) 1997;264:1293–1302. doi: 10.1098/rspb.1997.0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan JS, Baldridge WH, Kell ME. Purinergic regulation of cation conductances and intracellular Ca2+ in cultured rat retinal pigment epithelial cells. Journal of Physiology. 1999;520:745–759. doi: 10.1111/j.1469-7793.1999.00745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Ohguro H, Ohguro I, Mamiya K, Takano Y, Yamazaki H, Metoki T, Miyagawa K, Ishikawa F, Nakazawa M. Study of pharmacological effects of nilvadipine on RCS rat retinal degradation by micro array analysis. Biochemistry and Biophysical Research Communication. 2003;306:826–831. doi: 10.1016/s0006-291x(03)01092-1. [DOI] [PubMed] [Google Scholar]
- Sosa R, Hoffpauir B, Rankin ML, Bruch RC, Gleason EL. Metabotropic glutamate receptor 5 and calcium signaling in retinal amacrine cells. Journal of Neurochemistry. 2002;81:973–983. doi: 10.1046/j.1471-4159.2002.00883.x. [DOI] [PubMed] [Google Scholar]
- Singh BB, Lockwich T, Bandyopadhyay B, Liu X, Bollimuntha S, Brazer SC, Combs C, Das S, Leenders M, Sheng Z, Knepper M, Ambudkar SV, Ambudkar IS. VAMP-2-dependent exocytosis is involved in plasma membrane insertion of TRPC3 channels and contributes to agonist-stimulated Ca2+ influx. Molecular Cell. 2004;15:635–646. doi: 10.1016/j.molcel.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Singh BB, Liu X, Tang J, Zhu MX, Ambudkar IS. Calmodulin regulates Ca2+-dependent feedback inhibition of store-operated Ca2+ influx by interaction with a site in the C-terminus of TrpC1. Molecular Cell. 2002;9:739–750. doi: 10.1016/s1097-2765(02)00506-3. [DOI] [PubMed] [Google Scholar]
- Singh BB, Zheng C, Liu X, Lockwich T, Liao D, Zhu M, Birnbaumer L, Ambudkar IS. TRPC1 dependent enhancement of salivary gland fluid secretion: Role of store-operated calcium entry. FASEB Journal. 2001;15:1652–1654. doi: 10.1096/fj.00-0749fje. [DOI] [PubMed] [Google Scholar]
- Topp KS, Bisla K, Saks ND, Lavail JH. Centripetal transport of herpes simplex virus in human retinal pigment epithelial cells in vitro. Neuroscience. 1996;71:1133–1144. doi: 10.1016/0306-4522(95)00497-1. [DOI] [PubMed] [Google Scholar]
- Vollmer G, Layer PG. An in vitro model of proliferation and differentiation of the chick retina: Coaggregates of retinal and pigment epithelial cells. Journal of Neuroscience. 1986;6:1885–1896. doi: 10.1523/JNEUROSCI.06-07-01885.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]