

Abstract
Mammalian homologues of the Drosophila canonical transient receptor potential (TRP) proteins have been implicated to function as plasma membrane Ca2+ channels. This study examined the role of TRPC1 in human neuroblastoma (SH-SY5Y) cells. SH-SY5Y cells treated with an exogenous neurotoxin, 1-methyl-4-phenylpyridinium ion (MPP+) significantly decreased TRPC1 protein levels. Confocal microscopy on SH-SY5Y cells treatment with MPP+ showed decreased plasma membrane staining of TRPC1. Importantly, overexpression of TRPC1 reduced neurotoxicity induced by MPP+. MPP+-induced α-synuclein expression was also suppressed by TRPC1 overexpression. Protection of SH-SY5Y cells against MPP+ was significantly decreased upon the overexpression of antisense TRPC1 cDNA construct or the addition of a nonspecific transient receptor potential channel blocker lanthanum. Activation of TRPC1 by thapsigargin or carbachol decreased MPP+ neurotoxicity, which was partially dependent on external Ca2+. Staining of SH-SY5Y cells with an apoptotic marker (YO-PRO-1) showed that TRPC1 protects SH-SY5Y neuronal cells against apoptosis. Further, TRPC1 overexpression inhibited cytochrome c release and decreased Bax and Apaf-1 protein levels. Interpretation of the above data suggests that reduction in the cell surface expression of TRPC1 following MPP+ treatment may be involved in dopaminergic neurodegeneration. Furthermore, TRPC1 may inhibit degenerative apoptotic signaling to provide neuroprotection against Parkinson’s disease-inducing agents.
Parkinson’s disease is a progressive neurodegenerative disorder associated with selective loss of the dopaminergic neurons in the substantia nigra pars compacta (1). Neurotoxins, such as 1-methyl-4-phenylpyridinium ion (MPP+),1 cause selective nigral dopaminergic lesions and cause Parkinsonian syndrome (2–5). Although the underlying cause of dopaminergic cell death or the molecular mechanism by which these cells degenerate is still not fully understood, several molecular mechanisms have been proposed to play a role that includes overproduction of reactive oxygen species, impairment of mitochondrial respiration, disturbances of Ca2+ homeostasis, and excitotoxicity (6–10). Of these, Ca2+ homeostasis is believed to play an important role, because Ca2+ has both stimulatory and inhibitory roles in the cell death process. Release of Ca2+ from the endoplasmic reticulum (ER) followed by Ca2+ influx from the extracellular environment induces oxidative stress, which could activate cell death cascades (11).
Ca2+ concentration is very tightly regulated in neuronal cells. Disturbances in neuronal Ca2+ homeostasis have been implicated in a variety of neuropathological conditions (12). Hypotheses about how such disturbances might cause neurodegeneration have largely focused on excessive concentration of cytosolic Ca2+ ([Ca2+]i), which could cause overactivation of a variety of potentially destructive processes (11). More recently, several lines of evidence suggests that neuronal toxicity is not simply a function of increased [Ca2+]i. For example, treatments with AMPA or KCl can increase [Ca2+]i up to 1–2 μm in cortical neurons, without causing toxicity (13), whereas equally high Ca2+ loads are toxic when entering via the N-methyl-d-aspartate channels but not when entering via the voltage-dependent Ca2+ channels (13), suggesting that the source of increased [Ca2+]i or the calcium channel itself is critical for neuronal cell death. Further, in some neurons decreasing [Ca2+]i is toxic, whereas in others a modest increase in [Ca2+]i can be neuroprotective, indicating a “set point” mechanism for the effect of [Ca2+]i (11, 13).
The action of Ca2+ ion is mediated by several mechanisms, which are highlighted by the central role of Ca2+ in apoptotic processes and neuronal excitotoxicity. Mitochondria not only functions as an ATP producer but also functions as a regulator of intracellular Ca2+ homeostasis. Increased mitochondrial Ca2+ overload as a result of excitotoxicity has been associated with the generation of superoxide and may induce the release of proapoptotic mitochondrial proteins, proceeding through DNA fragmentation/condensation and culminating in cell demise by apoptosis (14–16). Apoptosis is a controlled cellular process that is accompanied by the activation of certain caspases (17), up-regulation of proapoptotic protein Bax (18), release of cytochrome c, and increase in intracellular Ca2+ (19, 20). Ca2+ enters the cytoplasm from two sources: it is either released from the intracellular stores, or it enters through the plasma membrane (21, 22). The important initiating step in the rise of cytosolic Ca2+ is the intracellular release of Ca2+, which is initiated by the activation of the G protein-coupled receptor followed by generation of the second messenger inositol 1,4,5-trisphosphate. Inositol 1,4,5-trisphosphate binds to its receptor in the ER to release Ca2+ from internal stores. Depletion of the intracellular stores leads to the opening of the plasma membrane Ca2+ channels, which are known as receptor-operated Ca2+ channels (23). The addition of thapsigargin (a SERCA pump blocker) initiates a passive release of Ca2+ from the internal stores, which could activate plasma membrane Ca2+ channels known as store-operated Ca2+ channels.
Mammalian homologues of the transient receptor potential (trp) genes have been suggested as the plasma membrane Ca2+ influx channels (21–24). In recent years, 28 mammalian members of the TRP superfamily has been identified that are classified into TRPC, TRPV, TRPM, TRPML, TRPP, and ANKTM1 subfamilies (22, 25). TRP superfamily members are non-voltage-gated cation channels except for TRPM5, which displayed voltage modulation (26), and are known to be activated by diverse stimuli including heat, pressure, pH, Ca2+, and activation of G protein-coupled receptors. These TRP channels not only function as Ca2+ influx channels but are also involved in pain transduction, osmoregulation, mechanosensitivity, cell growth, differentiation, and other as yet uncharacterized functions (25). Both overexpression and down-regulation has shown that TRPC1, TRPC4, and TRPV6 exhibit properties associated with store-operated Ca2+ channels and could be activated by store depletion per se; however, the expression of these TRP proteins is very restricted. TRPC1 is the only exception; it is ubiquitously expressed and is localized in the plasma membrane (24–27). Targeting of TRP proteins to the plasma membrane is essential for its function, which is dependent on its ability to interact with other proteins. TRPC1 interaction with caveolin 1 and TRPV2 interaction with a chaperone protein RGA is critical for their trafficking to the plasma membrane (28, 29). Further, TRPC3, TRPC5, and TRPC6 have been shown to be present in vesicles and are translocated to the membrane upon stimulation (30, 31).
Our previous studies demonstrate that TRPC1 functions as Ca2+ influx channel (24, 32). The present study was undertaken to determine the effects of MPP+ on the expression/function of TRPC1 in human dopaminergic SH-SY5Y cells. Our data demonstrate that expression of TRPC1 is significantly reduced in cells treated with MPP+. Further, overexpression of the TRPC1 protected dopaminergic neurons from cellular toxicity elicited by MPP+, and down-regulation of TRPC1 reduced TRPC1 protein levels and an increased cell death. Finally, we show that critical proapoptotic factors are inhibited in TRPC1-overexpressing cells treated with MPP+. Together our data reveal an important role of TRPC1 in the protection of dopaminergic neurons against Parkinson’s disease-inducing agents.
EXPERIMENTAL PROCEDURES
SH-SY5Y Cell Culture, Transformation, and Reagents
SH-SY5Y neuroblastoma cells were obtained from the American Type Culture Collection (Manassas, VA). They were cultured in a medium containing minimum essential medium, F-12 medium, Hanks’ balanced salt solution (2:1:1) with 10% fetal bovine serum (Biofluids), 1 unit/ml penicillin, and 1 μg/ml streptomycin and maintained at 37 °C with 95% humidified air and 5% CO2. Culture medium was changed twice weekly. SH-SY5Y cells were maintained in complete medium until reaching 90% confluence and then were trypsinized, centrifuged, and resuspended in the same medium as described above. MPP+ was added to culture wells and was present during the duration of the experiment (12–24 h) unless otherwise stated. Agents being tested for protective/inhibitory effects were added 15 min prior to the introduction of the toxic drug. SH-SY5Y cells were either stably or transiently transfected using TRPC1 antisense cDNA or adenovirus encoding trpc1 gene as described earlier (24, 32). 1-Methyl-4-phenylpyridinium ion (MPP+) and LaCl3 were obtained from Sigma. Thapsigargin, carbachol, and BAPTA-AM were obtained from Calbiochem; 2-aminoethoxydiphenyl borate (2APB) was obtained from Tocris-Cookson.
mRNA Isolation, Synthesis of the First Strain cDNA, and RT-PCR Analysis
Total RNA was extracted from SH-SY5Y cells using TRIzol reagent (Invitrogen) and was treated with deoxyribonuclease I (Invitrogen) at a concentration of 1 unit of DNaseI/1 μg of RNA in a buffer containing 20 mm Tris-HCl (pH 8.4), 2 mm MgCl2, 50 mm KCl for 15 min at room temperature. The reaction was terminated by adding EDTA at a final concentration of 2.5 mm and heated at 65 °C for 10 min. The primer sequences used for TRPC, TRPV, and TRPM are given in Table I. First strand cDNA synthesis and RT-PCR were performed as described previously (24). After 30 PCR cycles, 10 μl of the RT-PCR product was analyzed on a 1% agarose gel, cloned into TA cloning vector (Invitrogen) and confirmed either by sequencing or restriction analysis.
Table I.
Oligonucleotide sequences of the PCR primers
| Proteins | Forward primer sequence (5′–3′) | Reverse primer sequence (5′–3′) |
|---|---|---|
| TRPC1 | gcaatgataccttccattcgttc | cgatgcactaggcagcagatc |
| TRPC2 | aggctctggtgcagcgctac | gcgggaaccagaggtctagag |
| TRPC3 | ccactgtagaagaaagtttcaag | cagcatgctgggattcagtttc |
| TRPC4 | ctgaagaaggcctgaccgaag | cctgtaaccccagtgtgtcc |
| TRPC5 | gggaaatagaaaacatccaag | gaggagcagatgctggatg |
| TRPC6 | ccaggaagatgcagagatg | ggaagtcttcgcattatctattg |
| TRPV1 | ggctctatgatcgcaggag | caggagttctgcagcaggaac |
| TRPV2 | gaccagcaagtacctcac | catgcaggactgtgttgccc |
| TRPV3 | ctgacttcctcatgcacaag | gaggatgtacttcaggatctc |
| TRPV4 | gtgcctgggcccaagaaagc | catccttgggctggaagaag |
| TRPV5 | cacatagcagccctctatg | tccgcttctgcatcaggtg |
| TRPV6 | caagttctgcagatggttcc | gcaaaggttttgttgggctg |
| TRPM1 | ctcatgagtttcggagtagc | ccacacctggttggatattg |
| TRPM2 | gtggactggctgttccgag | gctagttgtccttcaggtag |
| TRPM3 | cacctgatgaccaaggaatg | cttgtgtttatcttctggagtg |
| TRPM4 | gccggagaaggagcagagc | cacccatggccaccaccttg |
| TRPM5 | caagatcatcgtggtagagc | tccagaacatgtctgcgttg |
| TRPM6 | caggtgttactgtggccg | gagggattccaactgtccag |
| TRPM7 | agcactttgaccaagaggg | gcatcttctttgagggcatc |
| TRPM8 | agccaggctcagcatgagg | ctgaagatcttgcgcatgcg |
Cell Viability (MTT) Assay
SH-SY5Y cells were seeded on 96-well plates at a density of 0.5×106 cells/well. The cultures were grown for 24 h followed by addition of fresh medium containing MPP+. Cell viability was determined by MTT assay as described before (33). After incubation for 12 h with the desired drug, 30 μl of MTT reagent (0.5 mg/ml MTT in phosphate-buffered saline containing 10 μm HEPES) was added to each well and incubated in a CO2 incubator for 2 h. The medium was aspirated from each well, and the culture plate was dried at 37 °C for 1 h. The resulting formazan dye was extracted with 100 μl of 0.04 n HCl in isopropanol, and the absorbance was measured in a micro plate reader (Molecular Device, Sunnyvale, CA) at 570 and 630 nm.
Membrane Preparations and Western Blotting
SH-SY5Y cells were cultured and transfected as described earlier (24, 32). The cells were harvested, lysed, and stored at −80 °C. Crude membranes were prepared from cell lysates (34). Mitochondrial membranes were isolated as described by Muralikrishnan and Ebadi (35). Protein concentration was determined by using the Bio-Rad protein assay. 25 μg of the proteins were resolved on 4–20% SDS-PAGE gels, transferred to polyvinylidene difluoride membranes, and probed with respective antibodies. A 1:1000 dilution of the primary antibody was used to probe for TRPC1, SERCA2B, Apaf-1, Bax, and α-synuclein, respectively. For α-synuclein blot, 10 μg of the whole cell lysates were resolved on a 15% gel and probed with α-synuclein antibodies. Peroxidase-conjugated respective secondary antibodies were used to label the proteins. The proteins on the membrane were detected using ECL reagent (Pierce) and analyzed using Lumiimager (Roche Applied Science).
Confocal Microscopy
For immunofluorescence SH-SY5Y cells were grown overnight on coverslips, washed twice with phosphate-buffered saline, and fixed for 30 min using 3% paraformaldehyde. The cells were then permeabilized using cold methanol and blocked for 20 min using 5% donkey serum. For TRPC1 staining, the cells were treated with anti-TRPC1 antibody at 1:100 dilutions for 1 h. The cells were washed (three times with phosphate-buffered saline 0.5% bovine serum albumin) and labeled with rhodamine-linked anti rabbit secondary antibody (1:100 dilution) (24, 30). Confocal images were collected using a MRC 1024-krypton/argon laser scanning confocal equipped with a Zeiss LSM 510 Meta photomicroscope.
Vybrant Staining Assay
Vybrant apoptosis assay kit (Molecular Probes, Eugene, OR) was used to evaluate apoptosis as per the manufacturer’s instructions. This kit distinguishes apoptotic and necrotic cells by propidium iodide dye and lipid dye (YO-PRO-1) staining. The cells were visualized using a fluorescence microscope using a 10× objective. The dead and necrotic cells exhibit red fluorescence, whereas apoptotic cells have a green fluorescence. The total apoptotic and necrotic cells were counted, and the percentages of cells exhibiting apoptosis/necrosis were calculated.
RESULTS
TRPC1 Transcriptome in SH-SY5Y Cells
To investigate the expression of various TRP proteins, RT-PCR was performed. 10 ng of total mRNA isolated from SH-SY5Y cells was used for cDNA synthesis using primers specific for each trp genes. Analysis of the amplified fragments showed that TRPC1, 3, and 5 were expressed in SH-SY5Y cells, whereas expression of TRPC2, TRPC4, and TRPC6 was not observed under similar conditions (Fig. 1A, top panel). The lack of expression of TRPC2, TRPC4, and TRPC6 was not due to their primer design or other experimental limitations because the desired PCR products were obtained when plasmid DNA was used as template (data not shown). As indicated in Fig. 1A, a 455-base pair TRPC1, a 690-base pair TRPC3, and a 595-base pair of TRPC5 fragments were amplified. Although these PCR products matched their respective base pair sizes, we sequenced these products, which confirmed the presence of TRPC1, TRPC3, and TRPC5 transcripts (data not shown). To investigate whether other TRP members are also expressed, RT-PCR was performed using specific TRPV and TRPM primers. Analysis of TRPV transcripts indicate that TRPV2, TRPV3, and TRPV4 were expressed in SH-SY5Y cells (Fig. 1A, middle panel). Semi-quantitative analysis of the PCR products indicate that TRPV4 was expressed more, followed by TRPV3 and TRPV2. Expression of TRPM transcripts was also determined using RT-PCR method, using TRPM-specific primers. As indicated in Fig. 1A (bottom panel), only TRPM1, TRPM3, and TRPM5 were expressed in SH-SY5Y cells, whereas TRPM2, TRPM4, TRPM6, and TRPM8 were not detected. RT-PCR using TRPM5 primers resulted in two bands. The top band corresponded with the right size and TRPM5 product, and the bottom band is perhaps an alternative spliced product of TRPM5 gene. Expression of multiple TRP transcripts in SH-SY5Y cells indicates that these TRP channels could have an important role in the physiology/function of the dopaminergic neuronal cells.
Fig. 1. TRP channel transcriptome in SH-SY5Y cells and expression of TRPC1 in the presence of MPP+.

A, RT-PCR on SH-SY5Y cells using trpc-, trpv-, and trpm-specific primers. B, Western blot on crude membranes prepared from SH-SY5Y cells treated with MPP+ (250 μm) in a time-dependent manner (0, 1, 3, 6, 10, and 24 h). The upper blot was probed using anti-TRPC1 antibody, and the lower blot was probed with anti-SERCA2B antibodies.
Effect of MPP+ on TRPC1 Expression
To investigate the role of MPP+, SH-SY5Y cells were incubated with 250 μm of MPP+, and TRPC1 protein levels were investigated. As indicated in Fig. 1B, no significant changes in the TRPC1 protein levels were observed when incubated with MPP+ for an initial period of 6 h. However, prolonged incubation with MPP+ (10–24 h) showed a significant decrease in the TRPC1 levels. Densitometry on these bands revealed that 70–80% of the TRPC1 protein was reduced after 10 h of incubation with MPP+, which remained the same even after 24 h of incubation. In contrast, blots probed with anti-SERCA2B antibody showed no significant decrease in their respective protein levels (Fig. 1B, lower panel). Similar results were also obtained using actin or tubulin antibodies, where no significant decrease in their protein levels was observed after 24 h of incubation with MPP+ (data not shown). In aggregate these results suggest that the decrease in the TRPC1 protein level observed upon MPP+ treatment is specific and could be the direct effect of the drug.
MPP+ Treatment Alters TRPC1 Plasma Membrane Localization
To study the role of MPP+ on the expression/localization of the TRPC1 protein, confocal microscopy was performed. Localization of the endogenous TRPC1 protein displayed a punctate plasma membrane staining along with some subplasma membrane staining (Fig. 2A). Incubation of SH-SY5Y cells without the TRPC1 antibody showed no staining (data not shown). These results are consistent with our previous findings, where TRPC1 protein was expressed in the plasma membrane (24, 29). In contrast, MPP+ treatment for 10 h on control SH-SY5Y cells showed a dramatic decrease in the plasma membrane staining of TRPC1 (Fig. 2A). Similar results were also obtained upon 24 h of incubation with MPP+ where TRPC1 was localized primarily in the cytosol rather than in the plasma membrane (data not shown).
Fig. 2. MPP+ induces TRPC1 translocation.

A, localization of endogenous TRPC1 protein in SH-SY5Y (control) shows no treatment, (+ MPP+) shows cells treated with 250 μm MPP+ for 10 h. Anti-TRPC1 antibody and rhodamine-conjugated secondary antibody were used to detect endogenous TRPC1 protein. B, localization of the TRPC1 protein in cells either overexpressing TRPC1 or in TRPC1 down-regulated cells subjected to MPP+ treatment for 10 h. C, detection of TRPC1 protein in SH-SY5Y cell membrane. Lane 1, control; lane 2, cells transfected with antisense htrpc1; lane 3 cells overexpressing TRPC1.
Confocal studies performed with cells overexpressing TRPC1 and treated with MPP+ showed a punctate plasma membrane staining similar to that of the control cells. In contrast, a diffused pattern of staining was observed for cells where TRPC1 expression was down-regulated using trpc1 antisense construct and treated with MPP+ for 10 h (Fig. 2B). Further, consistent with our previous results, TRPC1 protein levels were significantly reduced in cells overexpressing antisense trpc1 construct (Fig. 2C) (24). These results suggest that MPP+ treatment not only decreases TRPC1 protein levels but also alters its localization, and overexpression of TRPC1 restores this effect by reverting to the control conditions.
Overexpression of TRPC1 Protects SH-SY5Y Cells from Cell Death
To evaluate the role of TRPC1 in SH-SY5Y cells, we transiently overexpressed TRPC1 using adenoviral method. A multiplicity of infection of 5 for Ad-TRPC1 was used to infect SH-SY5Y cells; we have previously used this virus to overexpress TRPC1 protein in human submandibular gland cells (32). As indicated in Fig. 3A, Western blots were performed on crude membranes isolated from control untreated cells, and TRPC1 protein was detected. Cells treated for 10 h with MPP+ had a significant decrease in TRPC1 protein levels. This decrease in TRPC1 protein levels was similar as observed in Fig. 1A. In contrast, MPP+ treatment on SH-SY5Y cells overexpressing TRPC1 (using Ad-TRPC1; multiplicity of infection of 5) showed a significant increase in the levels of the TRPC1 protein (Fig. 3A). SH-SY5Y cells infected with a multiplicity of infection of 5 of control virus (Ad-Luciferase) showed no increase in the TRPC1 protein level upon 10–24 h of MPP+ treatment (data not shown). These blots were reprobed with SERCA2B antibodies, which showed no decreases in their protein levels (Fig. 3A, lower panel).
Fig. 3. Overexpression of TRPC1 protects SH-SY5Y neuronal cells from MPP+.

A, Western blot on crude membranes prepared from control SH-SY5Y cells, or TRPC1-overexpressing cells treated with MPP+ (250 μm) for 10 h. The upper blot was probed using anti-TRPC1 antibody, and the lower portion of the same blot was probed with anti-SERCA2B antibodies. IB, immunoblot. B, MTT assays performed on control and TRPC1-overexpressing cells treated with 250 μm of MPP+ treated for 10 h. C and D, Western blot on whole cell lysates prepared from control SH-SY5Y cells or TRPC1-overexpressing cells treated with MPP+ (250 μm) for 10 h as well as with TRPC1 down-regulated cells treated with MPP+. The lower panel in D shows the same blot reprobed with actin antibodies. The blot was probed using anti-α synuclein antibodies overnight followed by the addition of peroxidase-conjugated anti-goat secondary antibody. The proteins were detected using ECL and exposed to an x-ray film or Lumiimager.
MPP+ is a toxic metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine produced by monoamine oxidase B and causes neurodegeneration. Thus, to investigate whether TRPC1 has a role in the protection of dopaminergic SH-SY5Y neuronal cells, MTT assays were performed. SH-SY5Y cells were grown on a 96-well plate and treated with the desired drugs both in control and in transiently overexpressing TRPC1 cells. SH-SY5Y cells treated with MPP+ showed a decrease in the cell survival (50–60% reduction in cells as compared with control untreated cells) (Fig. 3C). Interestingly, SH-SY5Y cells overexpressing TRPC1 showed a significant increase in cell survival (70–80% cell survived, p < 0.03; Fig. 3C).
The two consistent pathologic hallmarks of Parkinson’s disease are the loss of vulnerable neuronal populations and the presence of Lewy bodies and Lewy neurites. These latter pathologies are intracellular aggregates of several proteins including α-synuclein. Synucleins are a family of small, highly charged proteins and are known to be up-regulated in Parkinson’s disease patients and also in cells subjected to MPP+ treatment (2, 5, 33). Thus, Western blots were performed to investigate whether TRPC1 has a role in the regulation of α-synuclein. Whole cell lysates were prepared from control, TRPC1-overexpressing, and TRPC1 down-regulated cells both in the presence and in the absence of Parkinson’s disease-inducing drugs. As indicated in Fig. 3D, control cells treated with MPP+ showed up-regulation of α-synuclein. However, SH-SY5Y cells overexpressing TRPC1 had a significant decrease in α-synuclein protein levels upon MPP+ treatment (Fig. 3D). An increased level of α-synuclein was observed when TRPC1 was down-regulated using antisense construct (Fig. 3D). Further, these experimental conditions showed no effect on the expression of actin (Fig. 3D, lower panel). Collectively, these findings implicate a casual role for TRPC1 in modulating the expression of α-synuclein and perhaps its subsequent aggregation.
Activation of TRPC1 Increases Its Neuroprotection Ability in SH-SY5Y Cells
To further examine the role of TRPC1 in the protection of SH-SY5Y cells, we transiently transformed SH-SY5Y cells with antisense trpc1 cDNA, and MTT assays were performed. SH-SY5Y cells expressing antisense trpc1 cDNA showed a significant decrease in the protection of SH-SY5Y cells treated with MPP+. Importantly, this decrease was significantly lower than control cells treated with MPP+ (p < 0.05, a 39% decrease as compared with 50% in control cells) (Fig. 4). Activation of TRPC1 using muscarinic agonist carbachol significantly increased the protection of SH-SY5Y cells from MPP+. Interestingly, stimulation in the absence of external Ca2+ showed more protection against MPP+-mediated toxicity (Fig. 4). Activation of TRPC1 with SERCA pump blocker thapsigargin also showed increased cell survivability and was partially dependent on external Ca2+ (Fig. 4). Thus, increased cell survivability was dependent on the activation of TRPC1, however independent of Ca2+ influx, because in the absence of Ca2+ more protection was observed. Pretreatment of TRPC1 cells with intracellular Ca2+ chelators BAPTA-AM increased cell survivability. However, pretreatment of TRPC1-overexpressing cells with La3+ significantly decreased cell survivability (Fig. 4). Although the addition of an ER antagonist 2APB showed a slight decrease in the protection elicited by TRPC1 overexpression, this decrease was not significant. Thus, altogether these results confirm that activation of TRPC1 is critical for the protection of dopaminergic neurons, which is partially dependent on the Ca2+ influx ability of TRPC1.
Fig. 4. Activation of TRPC1 increases SH-SY5Y protection from MPP+.

Shown is a representative bar graph with the percentage of cell survival as detected via MTT assay. Control cells, TRPC1 expressing cells as Ad-TRPC1 (TRPC1), or as antisense (AS) were treated with MPP+. These data are the averages of three independent experiments performed in triplicate. *, values significantly different (p < 0.05) from control value without any treatment. **, values significantly different (p < 0.05) from all sets of experiments. The cells were transiently transfected (for 24 h) with Ad-TRPC1 encoding virus or trpc1 antisense cDNA. Other details of the experiment are provided under “Experimental Procedures.” Carbachol (CCh), thapsigargin (Tg), La3+, and 2APB were added 10 min prior to the addition of MPP+.
Expression of the TRPC1 Prevents SH-SY5Y Cell Death via Inhibition of the Apoptotic Pathway
In neuronal cells, death occurs primarily by apoptosis and partially via necrosis. Thus, to elucidate the role of TRPC1 in the protection of SH-SY5Y neurons, we examined its effect both on apoptosis and on necrosis. To identify necrosis-mediated cell death propidium iodide dye was used, whereas to differentiate the cell death from apoptosis an apoptotic marker YO-PRO-1 was used. As indicated in Fig. 5A, control SH-SY5Y cells without any treatment showed little cell death (2 cells/100 cells) (Fig. 5A; average data are shown in Fig. 5B). Whereas cells treated with MPP+ showed a significant increase in cell death via the apoptotic pathway as measured by YO-PRO-1(Fig. 5) (24 cells/100 cells). Magnified image is shown as an inset, which indicates cell death upon MPP+ treatment. In contrast, cell death via the necrotic pathway was similar to that of control SH-SY5Y cells without any MPP+ treatment (propidium iodide staining; Fig. 5). These results are similar to previous studies showing that MPP+ causes cell death via the apoptotic pathway (21–23). SH-SY5Y cells overexpressing TRPC1 showed a remarkable decrease in the cell death via apoptosis in MPP+-treated cells (~50% reduction, 11 of 100 cells), whereas no significant decrease was observed in cell death via necrosis (Fig. 5A; average data are shown in Fig. 5B). Strikingly, apoptotic cell death was reverted when TRPC1 expression was down-regulated using antisense construct in MPP+-treated cells (39 of 100 cells, as shown in Fig. 5). In aggregate, the results presented above strongly suggest that TRPC1 protects SH-SY5Y neuronal cells via inhibiting apoptotically mediated cell death.
Fig. 5. TRPC1 overexpression decreases YO-PRO-1 staining in MPP+-treated SH-SY5Y cells.

A, marker for necrosis (propidium iodide staining) and apoptosis (YO-PRO-1) were used to stain control cells and cells overexpressing or down-regulating TRPC1 protein treated with MPP+. Rhodamine-conjugated propidium iodide and fluorescein isothiocyanate-conjugated YO-PRO-1 was added to cells treated with MPP+ for 12 h. Fluorescence images were taken immediately using either a 10× or 40× objective, and the red and green cells were counted along with total number of cells. The red box shows an enlarged image of fluorescein isothiocyanate-conjugated YO-PRO-1 on MPP+-treated cells using 100× objective. B, represents mean bar graph from 600–1100 cells in each group. *, values significantly different from its counterpart (p < 0.05). **, value significantly different from all experimental conditions (p < 0.03). DIC, differential interference contrast.
Overexpression of TRPC1 Affects the Expression of Proteins Responsible for Apoptotic Cell Death in SH-SY5Y Cells
To understand the neuroprotective function of TRPC1, we investigated proteins necessary for the apoptotically mediated cell death process. Consistent with our above results TRPC1 affects proteins required for the regulation of the apoptotic pathway. It has been known that the expression and/or intracellular distribution of the proapoptotic protein Bax is critical for apoptotically mediated cell death. Interactions between Bax and BH3 death domain proteins result in Bax membrane integration and permeabilization of the mitochondrial outer membrane, which releases cytochrome c. Once released into the cytosol, cytochrome c together with other proteins activates the caspase cascade of proteases, which mediates the biochemical and morphological alterations that are characteristic of apoptosis. Thus, to investigate that indeed TRPC1 has a role in apoptotically mediated cell death; we isolated mitochondria from different sets of cells and studied the presence of cytochrome c and Bax proteins in their membranes. As indicated in Fig. 6, cytochrome c protein was present in the membrane fraction of control SH-SY5Y cells. Whereas treatment with MPP+ decreased cytochrome c protein level in the mitochondrial membrane of SH-SY5Y cells (Fig. 6, top blot). In contrast, SH-SY5Y cells overexpressing TRPC1 protein showed a significant increase in the cytochrome c levels, treated with MPP+ (Fig. 6, top blot), whereas down-regulation of TRPC1 using antisence had decreased cytochrome c levels (data not shown). These results suggest that the release of cytochrome c is inhibited in cells overexpressing TRPC1. Western blot using Bax antibody showed that Bax protein levels were substantially increased in SH-SY5Y cells treated with MPP+. This increase in Bax protein levels was significantly reduced in cells overexpressing TRPC1 protein. Further, no significant change in the protein levels was observed in SERCA2B protein probed with SERCA2B antibodies on crude membrane fractions.
Fig. 6. Overexpression of TRPC1 decreases proteins required for apoptotic pathway.
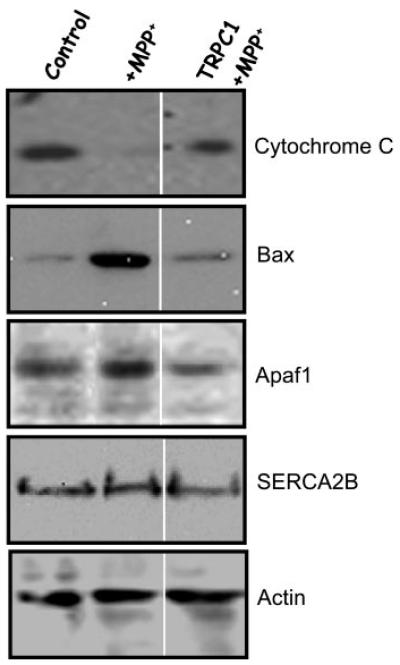
Shown is a Western blot performed on a mitochondrial membrane fraction. 25 μg of the membrane fraction was resolved on 4–20% SDS gel and probed with different antibodies. Bound cytochrome c was assayed using anti-cytochrome c antibody (top panel). Bax and SERCA2B proteins were identified using anti-Bax or anti-SERCA2B antibodies. Similarly, Apaf-1 and actin antibodies were used to detect proteins from the total cell lysates, respectively. Details about the antibodies and methods are described under “Experimental Procedures.”
During apoptotic neuronal death, cytochrome c is capable of binding to Apaf-1 (apoptotic protease-activating factor-1). This complex activates procaspase-9, resulting in caspase-mediated execution of apoptotic neuronal death. Thus, we investigated Apaf-1 proteins level in all sets of cells. TRPC1 overexpression significantly decreased the amount of Apaf-1 protein levels in MPP+-treated cells, suggesting that TRPC1 protects SH-SY5Y neurons by inhibiting the proapoptotic complex. Interestingly, it has been previously reported that cytosolic Ca2+ induced prominent degradation of Apaf-1 in human SH-SY5Y neuroblastoma cells (36). Taken together the data in Figs. 5 and 6 demonstrate that TRPC1 protects SH-SY5Y cells by inhibiting proteins important for apoptotic process.
DISCUSSION
In this study, we have demonstrated that MPP+, an exogenous neurotoxin involved in the degeneration of dopaminergic neurons, also inhibited TRPC1 expression. TRP proteins are a newly identified plasma membrane Ca2+ channels, which are present in most cell types. Our results indicate that MPP+ significantly decreases TRPC1 protein levels. This decrease in the TRPC1 protein levels was not a generalized affect, because the expression of the ER calcium pump (SERCA) protein was not affected. Since Ca2+ channels are critical for the normal physiology of neuronal cells, disruption of these channels might contribute toward the pathophysiology of several diseases including Parkinson’s disease. TRPC1 protein is known to be localized in the plasma membrane, where it functions as a Ca2+ channel (24). Confocal microscopy confirmed intense staining found in the plasma membrane of control cells that was disrupted in MPP+-treated cells. Thus, MPP+ not only decreases TRPC1 protein levels but also effects its localization. Further, the addition of MPP+ significantly reduced thapsigargin (a SERCA pump blocker) and carbachol-stimulated Ca2+ influx (data not shown). This decrease in Ca2+ influx could be due to the absence of functional TRPC1 in the plasma membrane. Interestingly, the internal stores were not affected upon MPP+ treatment (data not shown), which supports our Western blot data, where no decrease in the levels of the ER calcium pump SERCA2B was observed.
TRPC1 Protects SH-SY5Y Cells against MPP+-induced Cell Toxicity
MPP+ is a metabolic product of MPTP and is known to induce cell death in dopaminergic neurons (1, 3). To demonstrate the specific role of TRPC1, we transformed SH-SY5Y cells with either antisense TRPC1 constructs or overexpressed TRPC1 using adenoviral constructs. Consistent with our previous observation overexpression of TRPC1 showed a 2–3-fold increase in the TRPC1 protein levels, and down-regulation using antisense significantly decreased TRPC1 levels (24). Interestingly, TRPC1 overexpression not only increases TRPC1 protein in the plasma membrane but also decreases MPP+-mediated cell toxicity, whereas cells expressing antisense TRPC1 showed a decrease in the cell viability that was significantly less than control cells treated with MPP+. A decrease in the TRPC1 levels upon MPP+ treatment would attribute to a reduced store-mediated Ca2+ influx, which could lead to cell death. Several lines of evidence support this notion including the reported effect of Ca2+ antagonists, where a decrease in Ca2+ was responsible for various human diseases (37). Although our results indicate that TRPC1 is decreased upon MPP+ treatment, we cannot rule out other possibilities. SH-SY5Y neuroblastoma cell TRP transcriptome showed that apart from TRPC1 other TRP transcripts are also present in these cells. Expression of other TRP proteins could still contribute toward the physiological function of SH-SY5Y cells especially TRPM5 because this is the only TRP channel that is modulated by voltage change (26), and further research is needed to understand whether other TRP proteins are also altered upon MPP+ treatment.
To demonstrate whether activation of TRPC1 is necessary for its neuroprotective role, we stimulated TRPC1 both in the presence and in the absence of Ca2+. Our results indicate that activation of the TRPC1 protein by either thapsigargin or carbachol showed an increased protection of dopaminergic SH-SY5Y neurons. Ca2+ mobilization via the TRPC1 channel protein was not essential for the protection of MPP+-induced cell death. However, Ca2+ did play a role in neuroprotection. Inhibition of intracellular Ca2+ mobilization by the absence of external Ca2+ showed more protection than in the presence of Ca2+. Activation of TRPC1 could either translocate itself to the plasma membrane or facilitate its interaction with other regulatory proteins. A similar mechanism has been demonstrated in TRPC3, TRPC5, and TRPC6 channels, where an increased plasma membrane staining was observed upon stimulation (30, 31). Nonspecific blocking of TRPC1 channels using lanthanum significantly decreased TRPC1-mediated protection. Whereas the addition of 2APB, a nonspecific TRP channel blocker, showed no protection of SH-SY5Y cells against MPP+, also no significant decrease in the neuroprotective role of TRPC1 was observed in the presence of 2APB. These results are inconsistent with other published studies where the addition of 2-APB prevented Ca2+-induced permeability transition pores in non-synaptosomal brain mitochondria (38). Because these studies were performed on isolated mitochondria, 2APB could function differently. Further, 2APB not only affects store-operated Ca2+ entry channels (39) but also affects other proteins, such as the sarco-endoplasmic reticulum Ca2+ ATPase pump (40), the voltage-dependent K+ channels (41), gap junctions (42), and TRPV channels (43). Interestingly, in Jurkat T cells 2-APB inhibits Ca2+ efflux from mitochondria, thereby accumulating Ca2+ that could activate proapoptotic proteins leading to cell death (44).
Neuroprotective Role of TRPC1 Is Mediated by Inhibition of the Apoptotic Complex
A number of studies have suggested that alteration in cytoplasmic free Ca2+ is important for programmed cell death (45–47). Further, in dopaminergic neurons the addition of MPP+ along with dopamine stimulates Ca2+ release from mitochondria, which resulted in a disturbed Ca2+ homeostasis (9, 48). Thus, to understand the mechanism via which TRPC1 protects dopaminergic neurons, we investigated the proteins required for apoptosis. We observed in the present study that exposure to MPP+ led to an increased Bax expression, whereas overexpression of TRPC1 significantly decreased the level of the Bax protein. Bax is a pro-cell death driving force within the central decision point at the onset of apoptosis, and the ratio of Bax to cell death repressors including Bcl-xL modulates the activation of downstream effectors of cell death (18). It was also stated that up-regulation of Bax, rather than down-regulation of Bcl-xL, was more important in this cell death paradigm (49). Thus, collectively our results indicate that both TRPC1 and Bax function reciprocally, and alterations of TRPC1 alter Bax levels in SH-SY5Y cells. In fact, a critical role of Bax in dopaminergic cell death has been demonstrated in Parkinson models (50) and Parkinson’s patients (51). It has been shown that overexpression of Bax in cultured cells causes a loss of ER Ca2+ content that enhances the participation of Bax into the mitochondrial membrane leading to cell death (52). Overexpression of the TRPC1 protein can significantly contribute in the refilling of the stores and thus could inhibit Bax-mediated toxicity and activation of the apoptotic pathway.
MPP+-induced cell death also involves changes in intracellular Ca2+ concentration. Our results indicate that SH-SY5Y cells treated with MPP+ had a mode of cell death similar to that with dopaminergic neurons (53). Although whether TRPC1 overexpression prevents mitochondrial dysfunction as reported for Parkinson’s disease (54) was not directly addressed in our study, the decreased Bax level, decrease in cytochrome c release(s), and protection of apoptotic cell death point to the possibility that mitochondrial dysfunction may be involved. Bax is believed to exert its action primarily at the outer mitochondrial membrane by decreasing the mitochondrial transmembrane potential and promoting leakage of cytochrome c into the cytosol, leading to caspase activation and cell death (55). Loss of mitochondrial Ca2+ is shown to be accompanied by release of cytochrome c, indicating an increase in permeability of both the inner and outer mitochondrial membranes (43). Recent research reveals that there is a direct interaction taking place between cytochrome c released from mitochondria and the inositol 1,4,5-trisphosphate receptor present on the ER during apoptosis (56). Collectively our results suggest that TRPC1 may be an important Ca2+-regulating channel in these cells, and any decrease in this protein may severely impair cell viability.
In conclusion, the present finding demonstrates that MPP+ treatment decreases plasma membrane staining of TRPC1. Further, overexpression of TRPC1 showed an increased protection of dopaminergic SH-SY5Y cells, regardless of its Ca2+ mobilization properties. We hypothesized that TRPC1 protects SH-SY5Y neurons by inactivating/inhibiting the translocation of the key proteins necessary for apoptotically mediated cell death. However, it remains to be seen whether TRPC1 activation further inhibits the translocation of the key proteins required for apoptotic cell death. We have used the SH-SY5Y cell line, which is a subclone derived from the human neuroblastoma SK-N-SH cell line, which expresses tyrosine hydroxylase, dopamine-β-carboxylase, and dopamine transporter (57). Because SH-SY5Y exhibits unique properties similar to dopaminergic neurons in brain, it could be good source of preliminary investigation. However, cell lines cannot truly represent in vivo conditions. Thus, future studies will 1) establish the role of TRPC1 in the neuroprotection of cultured primary dopaminergic neurons as well as in brain, 2) elucidate the intracellular signaling mechanisms that are responsible for these effects, and 3) identify the relationship between the loss of TRPC1 protein and mitochondrial dysfunction.
Acknowledgments
We gratefully acknowledge Drs. Indu Ambudkar, Gene Homandberg, and Min Wu for valuable suggestions, support, reagents, and editing of the manuscript. We also thank Tammy Casavan for assistance with confocal microscopy.
Footnotes
This work was supported by North Dakota Biomedical Research Infrastructure Network, National Institutes of Health, by North Dakota Experimental Program to Stimulate Competitive Research (National Science Foundation), and by University of North Dakota School of Medicine and Health Sciences (to B. B. S.) and NINDS, National Institutes of Health Grant 2R01NS 34566-09 (to M. E.).
The abbreviations used are: MPP+, 1-methyl-4-phenylpyridinium ion; ER, endoplasmic reticulum; 2APB, 2-aminoethoxydiphenyl borate; RT, reverse transcription; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; TRP, transient receptor potential; SERCA, sarco-endoplasmic reticulum Ca2+ ATPase pump; AMPA, α-amino-5-methyl-3-hydroxy-4-isoxazolepropionic acid; BAPTA, 2-bis(O-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid tetra (acetoxymethyl ester).
REFERENCES
- 1.Hirsch EC, Hoglinger G, Rousselet E, Breidert T, Parain K, Feger J, Ruberg M, Prigent A, Cohen-Salmon C, Launay JM. J. Neural Transm. 2003;3:89–100. doi: 10.1007/978-3-7091-0643-3_6. [DOI] [PubMed] [Google Scholar]
- 2.Sandler M, Carter SB, Hunter KR, Stern GM. Nature. 1973;241:439–443. doi: 10.1038/241439a0. [DOI] [PubMed] [Google Scholar]
- 3.Ballard PA, Tetrud JW, Langston JW. Neurology. 1985;35:949–956. doi: 10.1212/wnl.35.7.949. [DOI] [PubMed] [Google Scholar]
- 4.Moser A, Kompf D. Life Sci. 1992;50:1885–1891. doi: 10.1016/0024-3205(92)90549-5. [DOI] [PubMed] [Google Scholar]
- 5.Meissner W, Prunier C, Guilloteau D, Chalon S, Gross CE, Bezard E. Mol. Neurobiol. 2003;28:209–218. doi: 10.1385/MN:28:3:209. [DOI] [PubMed] [Google Scholar]
- 6.Beal MF. Ann. Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 7.Fahn S, Cohen G. Ann. Neurol. 1992;32:804–812. doi: 10.1002/ana.410320616. [DOI] [PubMed] [Google Scholar]
- 8.Tatton WG, Olanow CW. Biochim. Biophys. Acta. 1999;1410:195–213. doi: 10.1016/s0005-2728(98)00167-4. [DOI] [PubMed] [Google Scholar]
- 9.Sheehan JP, Swerdlow RH, Parker WD, Miller SW, Davis RE, Tuttle JB. J. Neurochem. 1997;68:1221–1233. doi: 10.1046/j.1471-4159.1997.68031221.x. [DOI] [PubMed] [Google Scholar]
- 10.Doble A. Pharmacol. Ther. 1999;81:163–221. doi: 10.1016/s0163-7258(98)00042-4. [DOI] [PubMed] [Google Scholar]
- 11.Ermak G, Davies KJ. Mol. Immunol. 2002;38:713–721. doi: 10.1016/s0161-5890(01)00108-0. [DOI] [PubMed] [Google Scholar]
- 12.Lehotsky J, Kaplan P, Babusikova E, Strapkova A, Murin R. Physiol. Res. 2003;52:269–274. [PubMed] [Google Scholar]
- 13.Chen Q, Surmeier DJ, Reiner A. Exp. Neurol. 1999;159:283–296. doi: 10.1006/exnr.1999.7135. [DOI] [PubMed] [Google Scholar]
- 14.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Histol. Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 15.Mochizuki H, Goto K, Mori H, Mizuno Y. J. Neurol. Sci. 1996;137:120–123. doi: 10.1016/0022-510x(95)00336-z. [DOI] [PubMed] [Google Scholar]
- 16.Tompkins MM, Basgall EJ, Zamrini E, Hill WD. Am. J. Pathol. 1997;150:119–131. [PMC free article] [PubMed] [Google Scholar]
- 17.Martin SJ, Green DR. Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 18.Pettmann B, Henderson CE. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- 19.Yu SP, Canzoniero LM, Choi DW. Curr. Opin. Cell Biol. 2001;13:405–411. doi: 10.1016/s0955-0674(00)00228-3. [DOI] [PubMed] [Google Scholar]
- 20.Paschen W, Frandsen A. J. Neurochem. 2001;79:719–725. doi: 10.1046/j.1471-4159.2001.00623.x. [DOI] [PubMed] [Google Scholar]
- 21.Minke B, Cook B. Physiol. Rev. 2002;82:429–472. doi: 10.1152/physrev.00001.2002. [DOI] [PubMed] [Google Scholar]
- 22.Montell C. Sci. STKE. 2001;90:re1. doi: 10.1126/stke.2001.90.re1. [DOI] [PubMed] [Google Scholar]
- 23.Putney JW., Jr. Trends Cell Biol. 2004;14:282–286. doi: 10.1016/j.tcb.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Wang W, Singh BB, Lockwich T, Jadlowiec J, O’Connell B, Wellner R, Zhu MX, Ambudkar IS. J. Biol. Chem. 2000;275:3403–3411. doi: 10.1074/jbc.275.5.3403. [DOI] [PubMed] [Google Scholar]
- 25.Moran MM, Xu H, Clapham DE. Curr. Opin. Neurobiol. 2004;14:362–369. doi: 10.1016/j.conb.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Hofmann T, Chubanov V, Gudermann T, Montell C. Curr. Biol. 2003;13:1153–1158. doi: 10.1016/s0960-9822(03)00431-7. [DOI] [PubMed] [Google Scholar]
- 27.Venkatachalam K, van Rossum DB, Patterson RL, Ma HT, Gill DL. Nat. Cell Biol. 2002;4:263–272. doi: 10.1038/ncb1102-e263. [DOI] [PubMed] [Google Scholar]
- 28.Barnhill JC, Stokes AJ, Koblan-Huberson M, Shimoda LM, Muraguchi A, Adra CN, Turner H. J. Cell. Biochem. 2004;91:808–820. doi: 10.1002/jcb.10775. [DOI] [PubMed] [Google Scholar]
- 29.Brazer SC, Singh BB, Liu X, Swaim W, Ambudkar IS. J. Biol. Chem. 2003;278:27208–27215. doi: 10.1074/jbc.M301118200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh BB, Lockwich TP, Bandyopadhyay BC, Liu X, Bollimuntha S, Brazer SC, Combs C, Das S, Leenders AG, Sheng ZH, Knepper MA, Ambudkar SV, Ambudkar IS. Mol. Cell. 2004;15:635–646. doi: 10.1016/j.molcel.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 31.Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE. Nat. Cell Biol. 2004;6:709–720. doi: 10.1038/ncb1150. [DOI] [PubMed] [Google Scholar]
- 32.Singh BB, Zheng C, Liu X, Lockwich T, Liao D, Zhu MX, Birnbaumer L, Ambudkar IS. FASEB J. 2001;15:652–654. doi: 10.1096/fj.00-0749fje. [DOI] [PubMed] [Google Scholar]
- 33.Shavali S, Carlson EC, Swinscoe JC, Ebadi M. J. Neurosci. Res. 2004;76:563–571. doi: 10.1002/jnr.20082. [DOI] [PubMed] [Google Scholar]
- 34.Lockwich TP, Liu X, Singh BB, Jadlowiec J, Weiland S, Ambudkar IS. J. Biol. Chem. 2000;275:11934–11942. doi: 10.1074/jbc.275.16.11934. [DOI] [PubMed] [Google Scholar]
- 35.Muralikrishnan D, Ebadi M. Brain Res. 2001;892:241–247. doi: 10.1016/s0006-8993(00)02994-2. [DOI] [PubMed] [Google Scholar]
- 36.Reimertz C, Kogel D, Lankiewicz S, Poppe M, Prehn JH. J. Neurochem. 2001;78:1256–1266. doi: 10.1046/j.1471-4159.2001.00503.x. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi T, Mori Y. Eur. J. Pharmacol. 1998;363:1–15. doi: 10.1016/s0014-2999(98)00774-2. [DOI] [PubMed] [Google Scholar]
- 38.Chinopoulos C, Starkov AA, Fiskum G. J. Biol. Chem. 2003;278:27382–27389. doi: 10.1074/jbc.M303808200. [DOI] [PubMed] [Google Scholar]
- 39.Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. J. Biochem. (Tokyo) 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- 40.Bilmen JG, Wootton LL, Godfrey RE, Smart OS, Michelangeli F. Eur. J. Biochem. 2002;269:3678–3687. doi: 10.1046/j.1432-1033.2002.03060.x. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Deshpande M, Payne R. Cell Calcium. 2002;32:209–216. doi: 10.1016/s0143416002001562. [DOI] [PubMed] [Google Scholar]
- 42.Harks EG, Camina JP, Peters PH, Ypey DL, Scheenen WJ, Van Zoelen EJ, Theuvenet AP. FASEB J. 2003;17:941–943. doi: 10.1096/fj.02-0786fje. [DOI] [PubMed] [Google Scholar]
- 43.Hu HZ, Gu Q, Wang C, Colton CK, Tang J, Kinoshita-Kawada M, Lee LY, Wood JD, Zhu MX. J. Biol. Chem. 2004;279:35741–35748. doi: 10.1074/jbc.M404164200. [DOI] [PubMed] [Google Scholar]
- 44.Prakriya M, Lewis RS. J. Physiol. 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bellomo G, Perotti M, Taddei F, Mirabelli F, Finardi G, Nicotera P, Orrenius S. Cancer Res. 1992;52:1342–1346. [PubMed] [Google Scholar]
- 46.Baffy G, Miyashita T, Williamson JR, Reed JC. J. Biol. Chem. 1993;268:6511–6519. [PubMed] [Google Scholar]
- 47.Magnelli L, Cinelli M, Turchetti A, Chiarugi VP. Biochem. Biophys. Res. Commun. 1994;204:84–90. doi: 10.1006/bbrc.1994.2429. [DOI] [PubMed] [Google Scholar]
- 48.Frei B, Richter C. FEBS Lett. 1986;198:99–102. doi: 10.1016/0014-5793(86)81192-9. [DOI] [PubMed] [Google Scholar]
- 49.Choi HJ, Kim SW, Lee SY, Moon YW, Hwang O. Exp. Neurol. 2003;181:281–290. doi: 10.1016/s0014-4886(03)00054-2. [DOI] [PubMed] [Google Scholar]
- 50.Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D, Korsmeyer SJ, Przedborski S. Proc. Natl. Acad. Sci. U. S. A. 2001;98:2837–2842. doi: 10.1073/pnas.051633998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tatton NA, Kish SJ. Neuroscience. 1997;77:1037–1048. doi: 10.1016/s0306-4522(96)00545-3. [DOI] [PubMed] [Google Scholar]
- 52.Pan Z, Bhat MB, Nieminen AL, Ma J. J. Biol. Chem. 2001;276:32257–32263. doi: 10.1074/jbc.M100178200. [DOI] [PubMed] [Google Scholar]
- 53.Anglade P, Vyas S, Hirsch EC, Agid Y. Histol. Histopathol. 1997;12:603–610. [PubMed] [Google Scholar]
- 54.Schapira AH, Mann VM, Cooper JM, Dexter D, Daniel SE, Jenner P, Clark JB, Marsden CD. J. Neurochem. 1990;55:2142–2145. doi: 10.1111/j.1471-4159.1990.tb05809.x. [DOI] [PubMed] [Google Scholar]
- 55.Vander Heiden MG, Thompson CB. Nat. Cell Biol. 1999;1:E209–E216. doi: 10.1038/70237. [DOI] [PubMed] [Google Scholar]
- 56.Darren B, Randen LP, Leela S, Natalia OG, Tomohiro K, Solomon HS. Nat. Cell Biol. 2003;5:1051–1061. [Google Scholar]
- 57.Lee HS, Park CW, Kim YS. Exp. Neurol. 2000;165:164–171. doi: 10.1006/exnr.2000.7460. [DOI] [PubMed] [Google Scholar]