

Abstract
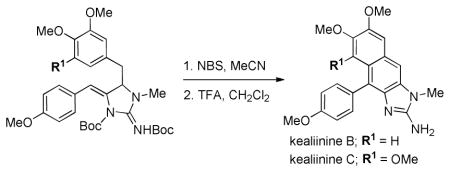
Synthesis of the reported structures of kealiinines B and C have been executed. An intermolecular electrophile-induced cyclization of a pendant arene on an ene-guanidine affords the tetracyclic, oxidized naphthimidazole cores.
Marine sponges of the class Calcarea provide a large number of small molecule alkaloids containing a 2-aminoimidazole core with interesting biological activities (Figure 1).1,2 For example, naamine D has been shown to be a competetive inhibitor of iNOS.3 Naamidine A has shown antimitogenic effects by inhibiting EGF-stimulated DNA synthesis in the growth of squamous tumor cells as well as cell cycle arrest in the G1 phase.4,5 As a result, interest has emerged to incorporate this heterocyclic scaffold as a pharmacophore in biomedical research.
Figure 1.
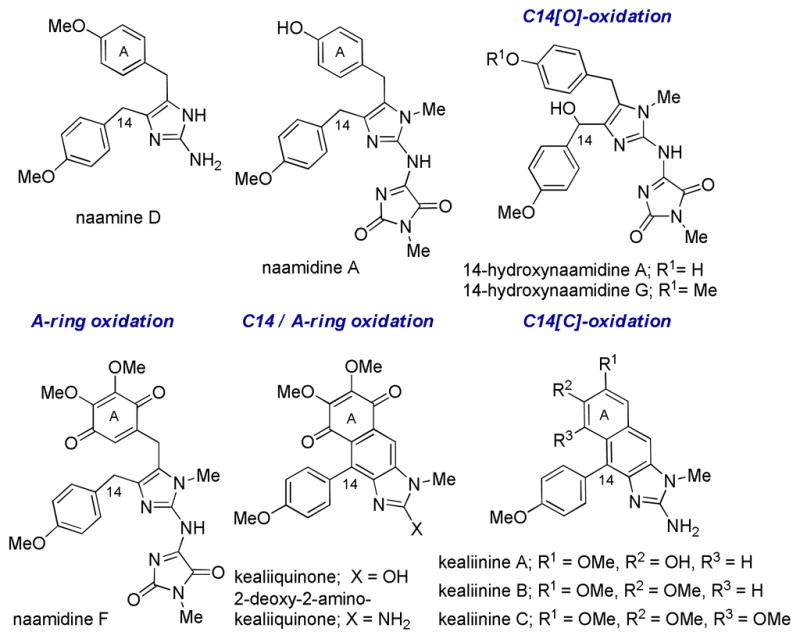
Representative Calcarea alkaloids
Within this class of natural products are several oxidized variants, including 14-hydroxynaamidines A and G6 which are hydroxylated at C14 and naamidine F7 which is isolated as the A-ring quinone. 2-Deoxy-2 aminokealiiquinone8 is oxidized both in the A-ring and at C14.
In 2004, a chemical investigation of the marine sponge Leucetta chagosensis by Proksch and co-workers afforded the new imidazole alkaloids kealiinines A-C.9 Related to 2-deoxy-2-aminokealiiquinone, these natural products are oxidized at C14 by virtue of a new C-C bond, but remain at the hydroquinone oxidation state in the A-ring. Although their biosynthesis has not been studied, it is reasonable to suggest that the kealiinines are intermediates between kealiiquinones and naamine derivatives by ring closure and aromatization.6,10 For example, naamines may be hydroxylated at C14 (Scheme 1, A). These intermediates might be subject to ionization to promote a Friedel-Crafts reaction (pathway 1). Oxidation would then provide the kealiinine skeleton. Alternatively, the hydroxylated intermediate may be subject to elimination (pathway 2) to give an intermediate which can undergo 6-π electrocyclization and oxidation to give the kealiinines. This biosynthetic strategy has also been explored in the laboratory by both Ohta and Lovely to access these naphthimidazole cores.11
Scheme 1.

Plausible biomimetic strategy.
Our strategy for synthesizing kealiinines B and C was also inspired by these hypothetical reactive intermediates that can be generated from the oxidized naamine (Scheme 1, B). If a cyclic ene-guanidine were reactive toward an electrophile, the intermediate formed would be reactive with a pendant nucleophile leading to Friedel-Crafts type alkylation or elimination / 6-π electrocyclization. Access to these ene-guanidines was envisaged to be straightforward with the development of 5-exo-dig selective propargylguanidine hydroaminations recently reported by both our group and Van der Eycken’s.12,13
Synthesis of the propargylguanidine precursors was initiated with a Cu(I)-catalyzed 3-component iminiumacetylide addition (A3-coupling)14 of 4-ethynylanisole, N-methylallylamine, and the aryl acetaldehydes 1 or 2 (Scheme 2).15 Although this reaction required elevated heating for extended reaction times due to the formation of stable enamine intermediates, 3 and 4 could be obtained in good yields on multi-gram scale. Deallylation of these intermediates with Pd(0) gave the secondary propargylamines 5 and 6.16 Guanylation of these intermediates with S-Me-N,N′-di-Boc-pseudothiourea and Hg(II) provided the propargylguanidines 7 and 8 in good yield.17
Scheme 2.

Substrate preparation.
We initiated our cyclization studies with the trimethoxy substituted arene for kealiinine C as it is symmetric (Scheme 2). To our surprise, cyclization of the propargylguanidine 7 gave 9 as a single regioisomer in good yield. Previous studies had shown that electron rich alkynes (e.g. pMeOPh-) typically gave poor 5-exo / 6-endo selectivity. Angle compression provided by substitution at the propargylic position presumably overrides this electronic selectivity through a Thorpe-Ingold effect. We next anticipated that activation of the ene-guanidine with NBS should generate the bromonium ion A. A key to this synthetic plan was that this bromonium ion can be opened via pathway a and not cyclization through pathway b to give intermediate B. While at first one might expect the formal products of a 5-exo cyclization (pathway b) to outcompete a 6-endo cyclization, there are several examples where the related electrophile-induced cyclizations are successful.18 Further, donation from the p-methoxy substituent might make this a formal 6-exo cyclization.19 In our experience the ene-guanidines are not very nucleophilic, suggesting that donation of the neighboring nitrogen lone pair is hampered by steric compression. This gave us hope that pathway a might be competitive.
To our delight treatment of 9 with NBS in acetonitrile provided the cyclized, fully aromatic and mono-Boc protected intermediate 10 in 56% isolated yield. The only other compound isolated from this reaction was brominated at C8 (circa 5%). While it does appear that the p-methoxyphenyl substituent on the ene-guanidine directs cyclization to give the 6-membered ring, we cannot rule out a 5-exo pathway followed by a pinacol rearrangement or that an elimination 6-π elctrocyclization is operative. Either way, any potential intermediates must funnel to intermediate C, which is capable of losing a Boc group and undergoing a 2 electron oxidation to give 10. Deprotection of this intermediate gave kealiinine C in good yield, and the structure was ultimately confirmed by X-ray crystallography.20 In the isolation paper only 1H-NMR data in CD3OD and DMSO-d6 were reported for both kealiinines B and C. Comparison of our spectra to that reported in DMSO-d6 shared very little agreement. However the TFA salt of synthetic kealiinine C did match the natural product well in CD3OD when referenced to the observed proton resonances for the natural product and not the solvent.
We next targeted kealiinine B to examine the regioselectivity of the oxidative cyclization relative to the A ring. Treatment of the propargyl guanidine 8 with AgOAc gave the ene-guanidine 11, also as a single regioisomer (Scheme 4). Exposure to NBS again delivered a single cyclized, fully aromatic and mono-Boc protected intermediate 12. This was presumed to be the correct isomer for kealiinine B as H8 was a singlet. Presumably attack of the bromonium ion is preferred by rotamer A as rotamer B would encounter more demanding steric interactions with the neighboring aromatic ring. Deprotection of 12 gave kealiinine B in 82% yield.
Scheme 4.

Synthesis of Kealiinine B
Again, the 1H NMR data for synthetic kealiinine B did not match that reported for natural compound in DMSO-d6. The free base did not match either, but it was a crystalline solid which permitted us to unambiguously assign the structure of our synthetic material via X-ray crystallography.21 Having now realized that kealiinine C only matched the natural material in CD3OD, we examined the TFA salt of kealiinine B in CD3OD and when aligned to the reported peaks there was excellent agreement for all proton resonances except for that at H6 which differed by 0.27 ppm.
Still concerned about the spectroscopic data, the Proksch laboratory was generous enough to compare our synthetic sample with a small amount of natural material by HPLC. This revealed two things: 1) that the natural sample was actually an inseperable mixture of kealiinines B and C and 2) the compounds did co-elute, suggesting they were indeed identical. We were also able to examine the original spectrum in CD3OD for the natural products (a mixture of B and C) which revealed that H6 in kealiinine B is actually at 7.74 ppm not 7.90 ppm as originally reported.22 This is in good agreement with our synthetic material, and gave us confidence that the structures of the natural products are correct and agree nicely with our synthetic materials.
The fact that the spectra were still not in agreement in DMSO-d6 was puzzling (most protons differing by 0.1 – 0.2 ppm). We hypothesized that this might be due to 1) a concentration effect, however dilution experiments did not reveal a change in the chemical shifts of the protons or 2) that the presence of both natural products in the sample might give rise to unique intermolecular interactions which change the chemical shifts, however titration of kealiinine C into a solution of B also showed no obvious change in chemical shifts. While we are confident, from the LC and 1H NMR data in CD3OD, that these are indeed the structures corresponding to the natural products, we must concede the possibility that alternate structures might exist that fit the DMSO-d6 data with better agreement.
In the original isolation of kealiinines A-C only biological activity for kealiinine A was reported. This was reported in a brine shrimp lethality assay with an IC50 = 20 μg / mL.9 The activity of kealiinines B and C were not reported, likely due to the small amounts of isolated material and the fact that they were isolated as an inseparable mixture. We evaluated our synthetic kealiinines B and C against several breast cancer cell lines, including a normal breast cell line (MCF-10A), a luminal ER+ cell line (MCF-7), an aggressive luminal ER+ line (T47D) and a “basal-like” triple negative cell line (MDA-MB-231). Kealiinine B showed antiproliferative activity against all of these cell lines with IC50’s ranging from ~ 10–13 μM (Figure 2). Kealiinine C, however, did not appreciably inhibit proliferation at concentrations below 100 μM.
Figure 2.
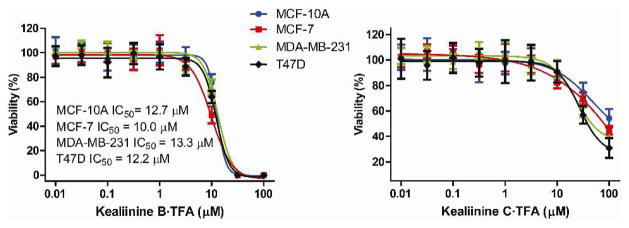
Antiproliferative activities.
The antiproliferative effects of members of this family have most extensively been studied for naamidine A, which is known to cause apoptosis through a caspase-3 dependent cascade after activation of the MAPK pathway.23 We examined the similarity of kealiinines B and C’s activity in this regard, and it appears that the antiproliferative effects arise from a caspase independent pathway (Figure 3). Treatment of MCF-10A or T47D cells, which are sensitive to caspase activation,24 with 30 μM concentrations of kealiinine B or C for 48 hrs showed no evidence of caspase-3 cleavage or PARP cleavage, known hallmarks of caspase activation (Lanes 2 and 3). Due do limited amounts of natural naamidine A, staurosporine was used as a positive control,25 and shows that indeed both cell lines are capable of undergoing caspase-3 dependent apoptosis (Lane 4).
Figure 3.
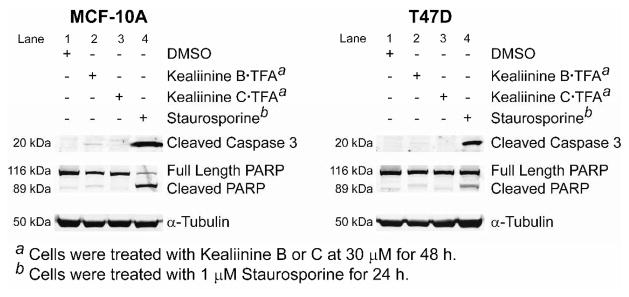
Interrogating Caspase 3 activity.
In conclusion, we have demonstrated that ene-guanidines, derived from the hydroamination of propargylguanidines, are useful intermediates for the synthesis of the reported structures for kealiinines B and C via an electrophilic activation / cyclization sequence. The ability to prepare these compounds individually has allowed us to provide independent and comprehensive characterization for these purported natural products, as well as an independent evaluation of their antiproliferative activity. Current efforts are directed to understand the mechanism by which kealiinine B inhibits cell proliferation.
Supplementary Material
Scheme 3.

Synthesis of Kealiinine C
Acknowledgments
This research was supported by grants from the National Institutes of Health ((R01-GM 090082 and R01-CA140296). KMG thanks the DOD Breast Cancer Research Program for a postdoctoral fellowship (W81XWH-09-1-0431). We are very grateful to Prof. Carl Lovely (UT-Arlington) for helpful discussions and for sharing spectral and x-ray data to confirm the identity of these products. We also thank Prof. Peter Proksch (UNI-Düsseldorf) for assistance with comparisons of synthetic and natural samples.
Footnotes
Supporting Information Available Experimental and characterization details, including 1H and 13C NMR spectra for all compounds. Biological protocols as well as Western Blots. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Sullivan JD, Giles RL, Looper RE. Curr Bioact Compd. 2009;5:39. [Google Scholar]
- 2.Koswatta PB, Lovely CJ. Nat Prod Rep. 2011;28:511. doi: 10.1039/c0np00001a. [DOI] [PubMed] [Google Scholar]
- 3.Dunbar DC, Rimoldi JM, Clark AM, Kelly M, Hamann MT. Tetrahedron. 2000;56:8795. [Google Scholar]
- 4.Copp BR, Fairchild CR, Cornell L, Casazza AM, Robinson S, Ireland CM. J Med Chem. 1998;41:3909. doi: 10.1021/jm980294n. [DOI] [PubMed] [Google Scholar]
- 5.James RD, Jones DA, Aalbersberg W, Ireland CM. Mol Can Ther. 2003;2:747. [PubMed] [Google Scholar]
- 6.Mancini I, Guella G, Debitus C, Pietra F. Helv Chim Acta. 1995;78:1178. [Google Scholar]
- 7.Carroll AR, Bowden BF, Coll JC. Aust J Chem. 1993;46:1229. [Google Scholar]
- 8.Fu X, Schmitz FJ, Tanner RS, Kelly-Borges M. J Nat Prod. 1998;61:384. doi: 10.1021/np970453q. [DOI] [PubMed] [Google Scholar]
- 9.Hassan W, Edrada R, Ebel R, Wray V, Berg A, van Soest R, Wiryowidagdo S, Proksch P. J Nat Prod. 2004;67:817. doi: 10.1021/np0305223. [DOI] [PubMed] [Google Scholar]
- 10.Akee R, Carroll T, Yoshida W, Scheuer P, Stout T, Clardy J. J Org Chem. 1990;55:1944. [Google Scholar]
- 11.(a) Kawasaki I, Taguchi N, Yamashita M, Ohta S. Chem Pharm Bull. 1997;45:1393. [Google Scholar]; b) Koswatta PB, Lovely CJ. Chem Commun. 2010;46:2148. doi: 10.1039/b926285g. [DOI] [PubMed] [Google Scholar]; (c) Lima HM, Sivappa R, Yousufuddin M, Lovely CJ. Org Lett. 2012;14:2274. doi: 10.1021/ol300704w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gainer MJ, Newbold NR, Takahashi Y, Looper RE. Angew Chem Int Ed. 2011;50:684. doi: 10.1002/anie.201006087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ermolat’ev DS, Bariwal JB, Steenackers HPL, De Keersmaecher SCJ, Van der Eycken EV. Angew Chem Int Ed. 2010;49:9465. doi: 10.1002/anie.201004256. [DOI] [PubMed] [Google Scholar]
- 14.For leading references see: Wei C, Li Z, Li CJ. Synlett. 2004:1472.Zani L, Bolm C. Chem Commun. 2006:4263. doi: 10.1039/b607986p.Li CJ. Acc Chem Res. 2010;43:581. doi: 10.1021/ar9002587.Koradin C, Polborn K, Knochel P. Angew Chem Int Ed. 2002;41:2535. doi: 10.1002/1521-3773(20020715)41:14<2535::AID-ANIE2535>3.0.CO;2-M.Gommermann N, Koradin C, Polborn K, Knochel P. Angew Chem Int Ed. 2003;42:5763. doi: 10.1002/anie.200352578.
- 15.Both arylacetaldehydes are commercially available or obtainable by the LiAlH4 reduction of the more readily available phenylacetic acid to the alcohol, followed by IBX oxidation to the aldehyde. See Pelphrey PM, Popov VM, Joska TM, Beierlein JM, Bolstad ES, Fillingham YA, Wright DL, Anderson AC. J Med Chem. 2007;50:940. doi: 10.1021/jm061027h.
- 16.Guibe F, Garro-Helion F, Merzouk A. J Org Chem. 1993;58:6109. [Google Scholar]
- 17.Horwell D, Guo Z, Cammidge A. Synth Commun. 2000;40:2933. [Google Scholar]
- 18.(a) Padwa A, Lee H, Rashatasakhon P, Rose M. J Org Chem. 2004;69:8209. doi: 10.1021/jo048647f. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Prasad CVC, Somers PK, Hwang CK. J Am Chem Soc. 1989;111:5330–5334. [Google Scholar]; (c) Bedford SB, Bell KE, Bennett F, Hayes CJ, Knight DW, Shaw DE. J Chem Soc Perkin Trans I. 1999;15:2143. [Google Scholar]
- 19.A similar donation strategy has been used to favor 5-endo-dig over 5-exo-dig cyclizations. David WK, Redfern AL, Gilmore J. Tetrahedron Lett. 1998;39:8909.
- 20.This structure has been deposited at the Cambridge Crysallographic Database (CCDC No. 887954).
- 21.This structure has been deposited at the Cambridge Crysallographic Database (CCDC No. 887953).
- 22.See “Spectral comparisons” in the Supporting Information for more details.
- 23.LaBarbera DV, Modzelewska K, Glazar AI, Gray PD, Kaur M, Liu T, Grossman D, Harper MK, Kuwada SK, Moghal N, Ireland CM. Anticancer Drugs. 2009;20:425. doi: 10.1097/CAD.0b013e32832ae55f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Annu Rev Cell Dev Biol. 1999;15:269. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 25.Chae HJ, Kang JS, Byun JO, Han KS, Kim DU, Oh SM, Kim HM, Chae SW, Kim HR. Pharmacol Res. 2000;42:373. doi: 10.1006/phrs.2000.0700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.