

Abstract
The sole Food and Drug Administration-approved treatment for acute stroke is tissue-type plasminogen activator (tPA), but tPA aggravates impairment of cerebrovasodilation during hypotension in a newborn pig photothrombotic model of stroke. Coupling to carrier red blood cells (RBC) enhances thrombolytic effects of tPA, while reducing its side effects. ATP- and Ca-sensitive K channels (Katp and Kca) are important regulators of cerebrovascular tone and mediate cerebrovasodilation during hypotension. Mitogen-activated protein kinase, a family of at least three kinases, ERK, p38, and c-Jun-N-terminal kinase (JNK), is upregulated after photothrombosis. This study examined the effect of photothrombosis on Katp- and Kca-induced cerebrovasodilation and the roles of tPA and JNK during/after injury. Photothrombosis blunted vasodilation induced by the Katp agonists cromakalim, calcitonin gene-related peptide, and the Kca agonist NS 1619, which was aggravated by injection of tPA. In contrast, both pre- or post-injury thrombosis injection of RBC-tPA and JNK antagonist SP 600125 prevented impairment of Katp- and Kca-induced vasodilation. Therefore, JNK activation in thrombosis impairs K channel-mediated cerebrovasodilation. Standard thrombolytic therapy of central nervous system ischemic disorders using free tPA poses the danger of further dysregulation of cerebrohemodynamics by impairing cation-mediated control of cerebrovascular tone, whereas RBC-coupled tPA both restores reperfusion and normalizes cerebral hemodynamics.
Keywords: Cerebral circulation, Newborn, Plasminogen activators, Signal transduction, Ischemia
Introduction
Neonatal stroke may occur in as many as 1 in 4,000 births [1]. Maternal and perinatal coagulopathy predispose to perinatal stroke [2, 3], with 30% of neonatal strokes being due to thrombosis [4]. A better understanding of the pathophysiologic responses that occur in children after ischemic stroke is needed to develop mechanism-based approaches to therapy. One contributor to neurological damage after ischemic stroke may be cerebrovascular dysfunction. Hypotension leads to loss of cerebrovascular regulation promoting tissue ischemia, while cerebrovaso-constriction associated with hypocapnia contributes to periventricular leukomalacia in the perinate [5]. We have shown that pial artery dilation in response to hypotension and hypercapnia is blunted in a piglet photothrombotic model of ischemic stroke [6, 7].
Relaxation of blood vessels can be mediated by several mechanisms, including cGMP, cAMP, and K+ channels [8]. Membrane potential of vascular muscle is a major determinant of vascular tone, and activity of K+ channels is a major regulator of membrane potential [8]. Activation or opening of these channels increases K+ efflux, producing hyperpolarization of vascular muscle. Membrane hyperpolarization closes voltage-dependent calcium channels and causes relaxation of vascular muscle. Direct measurements of membrane potential and K+ current in vitro indicate that several types of K+ channels are present in cerebral blood vessels. In addition, a number of pharmacological studies using activators and inhibitors have provided functional evidence that K+ channels, especially ATP-sensitive (Katp) and calcium-sensitive (Kca) channels, regulate cerebrovascular tone [8]. Cromakalim and calcitonin gene-related peptide (CGRP) are prototypical Katp agonists; NS 1619 a Kca agonist. Vasodilation induced by these drugs is as an index of the intactness of K channel function after traumatic brain injury and cerebral ischemia [9, 10]. Pial artery dilation in response to hypotension is due to activation of Katp and Kca channels [11], giving functional significance to intactness of K channel function.
Vascular injection of recombinant tissue-type plasminogen activator (tPA) is the only Food and Drug Administration-approved treatment for stroke [12]. However, tPA exhibits deleterious as well as beneficial effects that profoundly constrain its clinical utility. In addition to its salutary effect of reperfusion mediated by dissolution of intravascular thrombi, tPA contributes to bleeding, excito-toxic neuronal cell death [13], and increases stroke infarct volume in mice [14]. Systemic administration of tPA within a clinically relevant timeframe after phototothrombotic vascular injury in piglets aggravates impairment of cerebrovsodilation [6, 7], which is blocked by co-administration of a PA inhibitor-1-derived peptide [6].
Contemporaneous studies demonstrate that anchoring tPA on carrier red blood cells (RBC) endows the resultant complex, RBC-tPA, with dramatically prolonged circulation time enabling thromboprophylaxis to be attained at relatively low doses compared with its soluble counterpart [15–17]. In rodent models of thrombosis and traumatic brain injury and a piglet model of photothrombosis, RBC-tPA provided effective thromboprophylaxis, rapid reperfusion, neuroprotection, and reduction in mortality, all without associated hemorrhage [7, 18, 19].
Mitogen-activated protein kinase (MAPK), a family of at least three kinases (extracellular signal-related kinase—ERK-, p38, and c-Jun-N-terminal kinase—JNK), is upregulated after cerebral ischemia [20–22]. tPA impairs dilation during hypotension following photothrombosis through upregulation of JNK MAPK [7]. This study more fully characterized this observation by examining of the effects of tPA and RBC-tPA after photothrombosis on Katp- and Kca-induced pial artery dilation and JNK and p38 MAPK activation in K channel-mediated cerebrovasodilation.
Materials and Methods
Closed Cranial Window Technique and Cerebral Hypoxia/ Ischemia
Newborn pigs (1–5 days, 1.2–1.6 Kg) of either sex were studied. All protocols were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Animals were anesthetized with isoflurane (1–2 MAC), maintained with a-chloralose (30–50 mg/kg supplemented with 5 mg/kg/h iv). A catheter was inserted into a femoral artery to monitor blood pressure. The trachea was cannulated and the animals were ventilated with room air. A heating pad was used to maintain the animals at 37–39°C, monitored rectally.
A cranial window was placed in the parietal skull of these anesthetized animals. This window consisted of three parts: a stainless steel ring, a circular glass coverslip, and three ports consisting of 17-gauge hypodermic needles attached to three precut holes in the stainless steel ring. For placement, the dura was cut and retracted over the cut bone edge. The cranial window was placed in the opening and cemented in place with dental acrylic. The volume under the window was filled with a solution, similar to cerebrospinal fluid (CSF), of the following composition (in mM): 3.0 KCl, 1.5 MgCl2, 1.5 CaCl2, 132 NaCl, 6.6 urea, 3.7 dextrose, and 24.6 NaHCO3. This artificial CSF was warmed to 37°C and had the following chemistry: pH 7.33, pCO2 46 mm Hg, and pO2 43 mm Hg, which was similar to that of endogenous CSF. Pial arterial vessel diameter was measured with a microscope, a camera, a video output screen, and a video microscaler.
Induction of photothrombosis was based on that described for the newborn pig [23] but in our studies, we used the area of the closed cranial window to expose two to three main and one to three smaller arteries supplying the middle cerebral artery territory. Arterial occlusion was achieved by photothrombosis in which a stable thrombus consisting of aggregating platelets, fibrin, and other blood components is formed in response to endothelial peroxidative damage. The photochemical reaction occurs due to interaction of iv photosensitizing dye erythrosine B (20 mg/kg iv) and the focused beam of a solid state laser operated at 532 nm, power of 200 mW, average intensity of 60–75 W/cm2, and durations of up to 3–5 min using a Snake Creek minilaser (Hallstead, PA, USA).
Protocol
Two types of pial vessels, small arteries (resting diameter, 120–160 μm) and arterioles (resting diameter, 50–70 μm) were examined to determine whether segmental differences in the effects of H/I could be identified. Typically, 2–3 ml of artificial CSF were flushed through the window over a 30-s period and excess CSF was allowed to run off through one of the needle ports.
Twelve experimental groups of animals were studied (all n=5): (1) sham control, vehicle pre-treated; (2) photothrombosis, vehicle pre-treated; (3) photothrombosis, pre-treated with free tPA (2 mg/kg iv); (4) photothrombosis, pre-treated with RBC-tPA conjugate (0.1 mg/kg iv); (5) photothrombosis, pre-treated with the JNK MAPK antagonist, SP 600125 (1 mg/kg iv); (6) photothrombosis, pre-treated with the p38 MAPK antagonist SB 203580 (1 mg/kg iv); (7) sham control, vehicle post-treated; (8) photothrombosis, post-treated with vehicle; (9) photothrombosis, post-treated with tPA; (10) photothrombosis, post-treated with RBC-tPA; (11) photothrombosis, post-treated with SP 600125; and (12) photothrombosis post-treated with SB 203580. The vehicle for all agents was 0.9% saline, except for the MAPK antagonists, which was dimethyl sulfoxide (stock) diluted with saline, with a maximal ratio of 1:1,000. These two types of vehicle–CSF controls had no significant effect on pial artery diameter. In sham control animals, responses to cromakalim, CGRP, and NS 1619 (10−8, 10−6 M) were obtained initially and again 2.5 h later in the presence of agent vehicle. In photothrombosis vehicle animals, responses to vasoactive stimuli were obtained initially and then again 2.5 h post-insult in the presence of vehicle. In drug-treated animals, drugs were administered either 30 min before photothrombosis (pre-treatment) or 2 h after photothrombosis (post-treatment).
Preparation of RBC-tPA
RBCs were isolated by centrifugation from fresh anti-coagulated (heparin, 1,000 U/Kg) animal blood. Biotinylated tPA was coupled to biotinylated RBC via streptavidin, producing RBC-tPA complexes possessing 5×104 tPA molecules per RBC, as described previously [15, 16]. A vehicle control with RBCs in heparin plus uncoupled tPA has previously been observed to have no effect on pial artery diameter [24].
ELISA
Commercially available enzyme-linked immunosorbent assay (ELISA) kits were used to quantity CSF p38, and JNK MAPK (Assay Designs) concentration. Phosphorylated MAPK isoform enzyme values were normalized to total form and then expressed as percent of the control condition.
Statistical Analysis
Pial artery diameter values were analyzed using analysis of variance for repeated measures. If the value was significant, the data were then analyzed by Fisher’s protected least significant difference test. The sample populations studied were normally distributed and the power was 0.89. An α level of p<0.05 was considered significant in all statistical tests. Values are represented as mean±SEM of the absolute value or as percentage changes from control value.
Results
tPA Aggravates, but RBC-tPA Prevents, Impairment of Pial Artery Dilation in Response to Katp and Kca Channel Agonists After Photothrombosis
Cromakalim, CGRP, and NS 1619 (10−8, 10−6) elicited reproducible dilation of pial small arteries. Vasodilation in response to all 3 K channel agonists was blunted after photothrombosis, and the impairment was aggravated by pre- and post-injury treatment with tPA (2 mg/kg iv) (Figs. 1, 2, and 3). Pre-treatment was 30 min prior to whereas post-treatment was 2 h after injury. In contrast, pre- and post-treatment with RBC-tPA (0.1 mg/kg iv) prevented impairment of pial artery dilation in response to cromakalim, CGRP, and NS 1619 (Figs. 1, 2, and 3). Similar observations were made in pial arterioles. For example, cromakalim (10−8, 10−6) dilated pial arterioles by 17±1% and 32±2%, 6±1% and 8±1%, 1±1% and 2±1%, 1±1% and 2±1%, 14±1% and 24±2%, and 9±1% and 14± 1%, n=5, for sham control, photothrombosis, photothrombosis+ tPA pre-injury treatment, photothrombosis+ tPA post-injury treatment, photothrombosis+RBC-tPA pre-injury treatment, and photothrombosis+RBC-tPA post-injury treatment, respectively.
Fig. 1.

Influence of cromakalim (10−8, 10− 6 M) on pial artery diameter before injury (control), 2.5 h after photothrombotic injury (PTI), 2.5 h after PTI treated with tPA (2 mg/kg iv), 2.5 h after PTI treated with RBC-tPA (0.1 mg/kg iv), 2.5 h after PTI treated with SP 600125 (1 mg/kg iv), and 2.5 h after PTI treated with SB 203580 (1 mg/kg iv), n=5. a Pretreatment 30 min prior to PTI, b post-treatment 2 h after PTI. *p<0.05 compared with corresponding control value +p<0.05 compared with corresponding PTI nontreated value
Fig. 2.
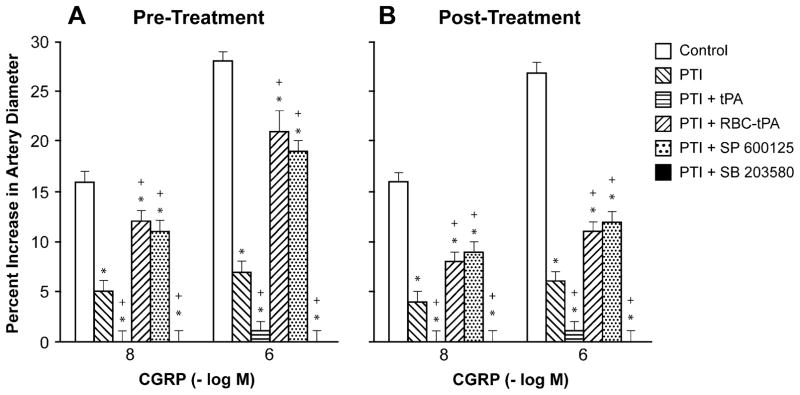
Influence of calcitonin gene-related peptide (CGRP; 10−8, 10−6 M) on pial artery diameter before injury (control), 2.5 h after photothrombotic injury (PTI), 2.5 h after PTI treated with tPA (2 mg/kg iv), 2.5 h after PTI treated with RBC-tPA (0.1 mg/kg iv), 2.5 h after PTI treated with SP 600125 (1 mg/kg iv), and 2.5 h after PTI treated with SB 203580 (1 mg/kg iv), n=5. a Pretreatment 30 min prior to PTI, b post-treatment 2 h after PTI. *p<0.05 compared with corresponding control value +p<0.05 compared with corresponding PTI nontreated value
Fig. 3.
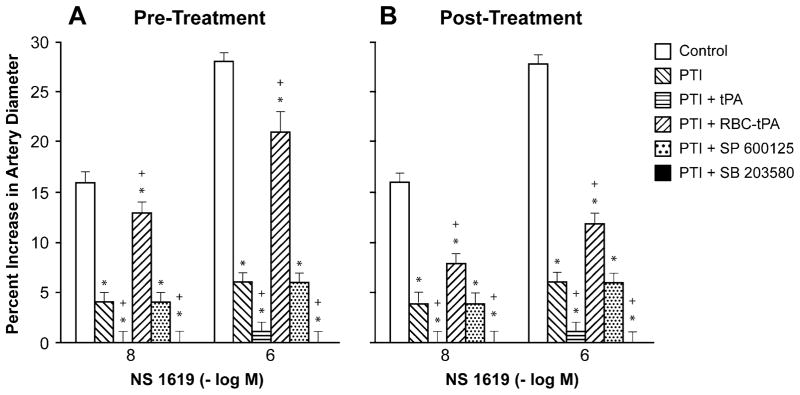
Influence of NS 1619 (10−8, 10−6 M) on pial artery diameter before injury (control), 2.5 h after photothrombotic injury (PTI), 2.5 h after PTI treated with tPA (2 mg/kg iv), 2.5 h after PTI treated with RBC-tPA (0.1 mg/kg iv), 2.5 h after PTI treated with SP 600125 (1 mg/kg iv), and 2.5 h after PTI treated with SB 203580 (1 mg/kg iv), n=5. a Pretreatment 30 min prior to PTI, b post-treatment 2 h after PTI. *p<0.05 compared with corresponding control value +p<0.05 compared with corresponding PTI nontreated value
The JNK MAPK Antagonist SP 600125 Prevents but the p38 MAPK Antagonist SB 203580 Aggravates Impairment of Pial Artery Dilation in Response to Katp and Kca Channel Agonists after Photothrombosis
Pre- and post-injury treatment with SP 600125 (1 mg/kg iv) prevented impairment of dilation of small pial arteries in response to cromakalim and CGRP, but not NS 1619 after photothrombosis (Figs. 1, 2, and 3). This dose of SP 600125 inhibited JNK MAPK upregulation after photothrombosis [7]. In contrast, SB 203580 aggravated impairment of all 3 K channel agonists after photothrombosis (Figs. 1, 2, and 3) at a dose that inhibited p38 MAPK upregulation [7]. Similar observations were made in pial arterioles. For example, cromakalim (10−8, 10−6) dilated pial arterioles by 18±1% and 32±2%, 4±1% and 7±1%, 1± 1% and 2±1%, 1±1% and 2±1%, 14±1% and 24±2%, and 11±1% and 14±1%, n=5, for sham control, photothrombosis, photothrombosis+tPA pre-injury treatment, photothrombosis+ tPA post-injury treatment, photothrombosis+SP 600125 pre-injury treatment, and photothrombosis+SP 600125 post-injury treatment, respectively.
RBC-tPA Blocks JNK but Potentiates p38 MAPK Upregulation After Photothrombosis
Photothrombosis increased CSF JNK MAPK, which was aggravated by tPA (105±3%, 236±12%, and 347±20%, n=5 for sham control, photothrombosis and photothrombosis+ tPA, respectively). Pre-and post-injury treatment with RBC-tPA blocked increases in CSF JNK MAPK (103±3%, 108±5%, and 116±5%, n=5, for sham control, photothrombosis+RBC-tPA pre-injury, and photothrombosis+ RBC-tPA post-injury treatment, respectively). On the other hand, p38 MAPK was elevated by photothrombosis, modestly elevated further by tPA, but potentiated by RBC-tPA pre-injury treatment (102±2%, 180±17%, 204± 19%, and 302±13%, n=5). Similar observations were made for tPA and RBC-tPA post-injury treatment.
Blood Chemistry
Blood chemistry values were collected before and after all experiments. There were no statistical differences between sham control, photothrombosis, and photothrombosis antagonist-treated animals. There were no differences in mean arterial blood pressure among groups.
Discussion
Several new findings emerged from this study. First, pial artery dilation in response to Katp and Kca channel agonists was impaired after photothrombosis which was aggravated by tPA administered either pre- or post-injury. In contrast, pre- or post-injury administration of RBC-tPA prevented impairment of vasodilation in response to both K channel subtype agonists. Since cerebral autoregulation during hypotension is dependent on intact Katp and Kca function [11], these data suggest that current thrombolytic therapy with tPA to treat central nervous system (CNS) ischemic disorders appears to disrupt autoregulation by impairing cation-mediated control of cerebrovascular tone, thereby aggravating cerebral damage. CGRP is an endogenous activator of the Katp channel in the piglet cerebral circulation [8, 9] and its use therefore provides physiological perspective to observations in this study.
Second, administration of the JNK MAPK antagonist SP 600125 either pre- or post-injury prevented impairment of Katp but not Kca-mediated pial artery dilation. The p38 MAPK antagonist SB 203580 aggravated impairment of both Katp and Kca channel agonist-mediated dilation post-injury. Photothrombosis upregulated CSF JNK MAPK, which was aggravated by tPA but blocked by RBC-tPA administered pre- or post-injury, similar to recent studies [7]. The p38 isoform of MAPK was similarly upregulated by photothrombosis, only modestly potentiated by tPA, but robustly increased by RBC-tPA, similar to recent studies [7]. Taken together, these data suggest that upregulation of JNK MAPK appears to be deleterious, whereas p38 MAPK may protect Katp-mediated cerebrovasodilation in the setting of focal thrombotic cerebral ischemia. For Kca-mediated cerebrovasodilation, although tPA similarly aggravates dilation, the mechanism is independent of JNK and is uncertain at present. Nonetheless, p38 protected Kca-mediated cerebrovasodilation, similar to that observed for the Katp channel. Therefore, the mechanism by which RBC-tPA protects Kca-mediated cerebrovasodilation differs from its effect on Katp in that while p38 upregulation appears beneficial in both settings, but there was little evidence of JNK being deleterious. No role for the third MAPK isoform, ERK MAPK, in impaired cerebrovasodilation to hypotension or hypercapnia after photothrombosis has been observed [6, 7]. However, in unrelated studies using a distinct model of injury, global cerebral hypoxia/ ischemia, tPA-impaired Katp and Kca-mediated cerebrovasodilation through upregulation of ERK MAPK [25]. In that injury model, the ERK antagonist U 0126 protected both Katp- and Kca-mediated cerebrovasodilation in an equivalent manner [25]. Taken together with the results of the present study, these data suggest that global and focal CNS ischemia impair cerebral hemodynamics through heterogenous and only partially overlapping pathways. From a physiological perspective, inhibition of the relaxation to K channel agonists could be due to a number of factors, such as: membrane depolarization, changes in density of K channels, membrane internalization of K channels, changes in gating, and/or changes in receptor/ effector coupling. While we were initially surprised to detect a supposedly intracellular signaling system in the CSF under sham control conditions, we make the point that this does not reflect damage or pathology. In response to peer review concerns over the years, we have reproducibly detected MAPK in CSF under control conditions and monitored its change with a range of stimuli [26–28]. In particular, we documented parallelism of change in response to hypoxia/ischemia using qualitative immunohistochemistry, semi-quantitative Western, and quantitative ELISA of CSF [28].
Many studies of cerebral ischemia have been performed in rodent models. Piglets offer a unique advantage in elucidating pathways involved in CNS ischemic injury by virtue of having a gyrencepahalic brain that contains substantial white matter similar to humans, which is more sensitive to ischemic damage than grey matter. On the basis of interspecies extrapolation of brain growth curves [29], the age of the newborn pig used in these studies roughly approximated the newborn infant time period in the human. Although the incidence of cerebral ischemic events in the pediatric population is relatively low compared to the adult [30], the clinical and social impact is amplified by the potential long-term loss of quality of life years for children with CNS ischemic disorders. Many children, however, are receiving tPA based on the assumption that studies in adults can be generalized to the pediatric population. The data from this study provide additional evidence that both safety and efficacy of tPA must be evaluated systematically in children before being widely adopted in clinical care. A potential limitation of the present study is that we did not directly address the issue of whether safety and efficacy differ in neonates and older children, indicating the need for scientific investigation using different maturational ages of pigs and/or other species.
Prior studies of the delivery of plasminogen activators by carrier RBCs [15] provide a logical framework for understanding differences in outcome and signaling from soluble tPA and the implication of these findings for the translational potential of this novel drug delivery strategy. We postulate that the protective effect of RBC-tPA arises from both prolonged duration of RBC-tPA activity, which facilitates thrombolytic reperfusion and intravascular spatial constraints. Anchoring tPA to RBCs does not inhibit its enzymatic activity [16]. However, the biodistribution of radiolabeled RBC-tPA in animals [15] and in vitro studies of binding to vascular cells [31] show that anchoring constrains tPA from exiting the vasculature and inhibits interaction with cellular receptors. These attributes prolong the intravascular activity of tPA, which enables thrombolysis to proceed at lower doses of drug and with less side effects. Moreover, differences in permeation of the vasculature and brain tissue by tPA and RBC-tPA likely account for variation in the MAPK isoforms that were upregulated. This suggests that the location or pattern of activation of these MAPK isoforms by tPA differ within endothelium, vascular smooth muscle, neurons, and glia. From the translational standpoint, it is encouraging that a single IV injection anchors a recombinant protein that fuses a plasminogen activator with single chain fragments of antibodies to circulating RBC and provides prolonged intravascular thromboprophylaxis [32, 33]. This new approach will facilitate the clinically applicability of using RBC carriage of plasminogen activators without the need for ex vivo coupling of tPA to isolated RBCs followed by reinfusion as utilized in this study.
Conclusion
In conclusion, these data indicate that photothrombosis impairs K channel-mediated cerebrovasodilation (Fig. 4). tPA exacerbates the loss of Katp channel function after injury by upregulating JNK MAPK and intensifies the loss of Kca channel-mediated vasodilation through as yet unknown mechanisms. In contrast, RBC-tPA prevents impairment of Katp-mediated cerebrovasodilation after injury through blockade of JNK and upregulation of p38 MAPK. RBC-tPA also prevents impairment of Kca-mediated cerebrovasodilation after injury through upregulation of p38. These data suggest that current thrombolytic therapy for treatment of CNS ischemic disorders dysregulates cerebrohemodynamics by impairing cation-mediated control of cerebrovascular tone (Fig. 4). RBC carriage of tPA preserves adaptive cerebrohemodynamic responses and thereby offers a novel therapeutic approach towards increasing the benefit/risk ratio of thrombolytic therapy for treatment of CNS ischemic disorders (Fig. 4).
Fig. 4.

Schematic diagram illustrating contrasting effects of tPA and RBC-tPA on mediators affecting outcome in the setting of focal ischemic stroke. This figure has been modified from its original published form [34]
Acknowledgments
This research was funded by grants from the National Institutes of Health, NS53410 and HD57355 (WMA), HL76406, CA83121, HL76206, HL07971, and HL81864 (DBC), HL77760 and HL82545 (AARH), HL66442 and HL090697 (VRM), the University of Pennsylvania Research Foundation (WMA), the University of Pennsylvania Institute for Translational Medicine and Therapeutics (DBC), and the Israeli Science Foundation (AARH).
Contributor Information
William M. Armstead, Email: armsteaw@uphs.upenn.edu, Departments of Anesthesiology and Critical Care, University of Pennsylvania, 3620 Hamilton Walk, JM3, Philadelphia, PA 19104, USA. Pharmacology, University of Pennsylvania, Philadelphia, PA 19104, USA
Kumkum Ganguly, Los Alamos National Laboratory, Los Alamos, NM, USA.
John Riley, Departments of Anesthesiology and Critical Care, University of Pennsylvania, 3620 Hamilton Walk, JM3, Philadelphia, PA 19104, USA.
Sergei Zaitsev, Pharmacology, University of Pennsylvania, Philadelphia, PA 19104, USA.
Douglas B. Cines, Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
Abd Al-Roof Higazi, Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA. Department of Clinical Biochemistry, Hebrew University-Hadassah Medical School, Jerusalem, Israel.
Vladimir R. Muzykantov, Pharmacology, University of Pennsylvania, Philadelphia, PA 19104, USA. Insttitue of Environmental Medicine University of Pennsylvania, Philadelphia, PA 19104, USA
References
- 1.Nelson KB, Lynch JK. Stroke in newborn infants. Lancet Neurol. 2004;3:150–8. doi: 10.1016/S1474-4422(04)00679-9. [DOI] [PubMed] [Google Scholar]
- 2.Gunther G, Junker R, Strater R, Schobess R, Kurnik K, Kosch A, et al. Symptomatic ischemic stroke in full-term neonates. Stroke. 2000;31:2437–41. doi: 10.1161/01.str.31.10.2437. [DOI] [PubMed] [Google Scholar]
- 3.Kraus FT, Acheen VI. Fetal thrombotic vasculopathy in the placenta: cerebral thrombi and infarcts, coagulopathies, and cerebral palsy. Hum Pathol. 1999;30:759–69. doi: 10.1016/s0046-8177(99)90136-3. [DOI] [PubMed] [Google Scholar]
- 4.DeVeber G, Andrew M. Cerebral sinovenous thrombosis in children. N Engl J Med. 2001;345:417–23. doi: 10.1056/NEJM200108093450604. [DOI] [PubMed] [Google Scholar]
- 5.Volpe JJ. Brain injury in the premature infant: overview of clinical aspects, neuropathology, and pathogenesis. Semin Pediatr Neurol. 1998;5:135–51. doi: 10.1016/s1071-9091(98)80030-2. [DOI] [PubMed] [Google Scholar]
- 6.Armstead WM, Riley J, Kiessling JW, Cines DB, Higazi AAR. Novel plasminogen activator inhibitor-1 derived peptide protects against impairment of cerebrovasodilation after photo-thrombosis through inhibition of JNK MAPK. Am J Physiol. 2010;299:R480–5. doi: 10.1152/ajpregu.00256.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armstead WM, Ganguly K, Riley J, Kiessling JW, Cines DB, Higazi AAR, Zaitsev S, Muzykantov VR. RBC-coupled tPA prevents impairment of cerebral vasodilatory responses through inhibition of JNK and potentiation of p38 MAPK after cerebral photothrombosis in the newborn pig. Ped Crit Care Med. 2011 doi: 10.1097/PCC.0b013e3181fe40a7. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faraci FM, Heistad DD. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78:53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- 9.Armstead WM. Brain injury impairs ATP-sensitive K+ channel function in piglet cerebral arteries. Stroke. 1997;28:2273–80. doi: 10.1161/01.str.28.11.2273. [DOI] [PubMed] [Google Scholar]
- 10.Bari F, Louis T, Meng W, Busija DW. Global ischemia impairs ATP-sensitive K channel function in cerebral arterioles in piglets. Stroke. 1996;27:1874–81. doi: 10.1161/01.str.27.10.1874. [DOI] [PubMed] [Google Scholar]
- 11.Armstead WM. Hypotension dilates pial arteries by KATP and Kca channel activation. Brain Res. 1999;816:158–64. doi: 10.1016/s0006-8993(98)01146-9. [DOI] [PubMed] [Google Scholar]
- 12.Kim YH, Park JH, Hong SH, Koh JY. Nonproteolytic neuroprotection by human recombinant tissue plasminogen activator. Science. 1999;284:647–50. doi: 10.1126/science.284.5414.647. [DOI] [PubMed] [Google Scholar]
- 13.Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, et al. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor mediated signaling. Nat Med. 2001;7:59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- 14.Wang YF, Tsirka SE, Strickland S, Stiege PE, Lipton SA. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4:228–31. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- 15.Murciano JC, Medinilla S, Eslin D, Atochina E, Cines DB, Muzykantov VR. Prophylactic fibrinolysis through selective dissolution of nascent clots by tPA-carrying erythrocytes. Nat Biotechnol. 2003;21:891–6. doi: 10.1038/nbt846. [DOI] [PubMed] [Google Scholar]
- 16.Ganguly K, Krasik T, Medinilla S, Bdeir K, Cines D, Muzykantov VR, et al. Blood clearance and activity of erythrocyte-coupled fibrinolytics. J Pharmacol Exp Ther. 2005;312:1106–13. doi: 10.1124/jpet.104.075770. [DOI] [PubMed] [Google Scholar]
- 17.Zaitsev S, Danielyan K, Murciano JC, Krasik T, Taylor RP, Pincus S, Muzykantov VR, et al. CR-1-directed targeting of tPA to circulating erythrocytes for prophylactic fibrinolysis. Blood. 2006;108:1895–902. doi: 10.1182/blood-2005-11-012336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Danielyan K, Ganguly K, Ding B, Atochin D, Zaitsev S, Murciano JC, et al. Cerebrovascular thromboprophylaxis by erythrocyte coupled tPA. Circulation. 2008;118:1442–9. doi: 10.1161/CIRCULATIONAHA.107.750257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stein SC, Ganguly K, Belfeld CM, Xu X, Swanson WE, Chen XH, et al. Erthrocyte bound tissue plasminogen activator (tPA) is neuroprotective in experimental traumatic brain injury. J Neurotrauma. 2009;26:1585–92. doi: 10.1089/neu.2008.0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci. 1999;96:12866–9. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi T, Sakai K, Sasaki C, Zhang WR, Warita H, Abe K. c-JUN N-terminal kinase (JNK) and JNK interacting protein response in rat brain after transient middle cerebral artery occlusion. Neurosci Lett. 2000;284:195–9. doi: 10.1016/s0304-3940(00)01024-7. [DOI] [PubMed] [Google Scholar]
- 22.Laher I, Zhang JH. Protein kinase C and cerebral vasospasm. J Cereb Blood Flow Metab. 2001;21:887–906. doi: 10.1097/00004647-200108000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Kuluz JW, Prado R, He D, Zhao W, Dietrich WD, Watson B. New pediatric model of ischemic stroke in infant piglets by photothrombosis. Stroke. 2007;38:1932–7. 120. doi: 10.1161/STROKEAHA.106.475244. Transl Stroke Res 2012, 3, 114–121. [DOI] [PubMed] [Google Scholar]
- 24.Armstead WM, Ganguly K, Kiessling JW, Chen XH, Smith DH, Higazi AAR, et al. RBC-coupled tPA prevents impairment of cerebral vasodilatory responses and tissue injury in pediatric cerebral hypoxia/ischemia through inhibition of ERK MAPK. J Cereb Blood Flow Metab. 2009;29:1463–74. doi: 10.1038/jcbfm.2009.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Armstead WM, Riley J, Cines DB, Higazi AAR. tPA contributes to impairment of ATP and calcium sensitive K channel mediated cerebrovasodilation after hypoxia/ischemia through upregulation of ERK MAPK. Brain Res. 2011;1376:88–93. doi: 10.1016/j.brainres.2010.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Armstead WM, Cines DB, Bdeir K, Bdeir Y, Stein SC, Higazi AAR. uPA modulates the age dependent effect of brain injury on cerebral hemodynamics through LRP and ERK MAPK. J Cereb Blood Flow Metab. 2009;29:524–33. doi: 10.1038/jcbfm.2008.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Armstead WM, Kiessling JW, Riley J, Cines DB, Higazi AAR. tPA contributes to impaired NMDA cerebrovasodilation after traumatic brain injury through activation of JNK MAPK. Neurol Res. 2011;33:726–33. doi: 10.1179/016164110X12807570509853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Armstead WM, Cines DB, Bdeir K, Kulikovskaya I, Stein SC, Higazi AAR. uPA impairs cerebrovasodilation after hypoxia/ ischemia through LRP and ERK MAPK. Brain Res. 2008;1231:121–31. doi: 10.1016/j.brainres.2008.06.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobbing J. The later growth of the brain and its vulnerability. Pediatrics. 1974;53:2–6. [PubMed] [Google Scholar]
- 30.Janjua N, Nasar A, Lynch JK, Qureshi AI. Thrombolysis for ischemic stroke in children. Data from the nationwide inpatient sample. Stroke. 2007;38:1850–4. doi: 10.1161/STROKEAHA.106.473983. [DOI] [PubMed] [Google Scholar]
- 31.Murciano JC, Higazi AA, Cines DB, Muzykantov VR. Soluble urokinase receptor conjugated to carrier red blood cells binds latent pro-urokinase and alters its functional profile. J Control Release. 2009;139:190–6. doi: 10.1016/j.jconrel.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zaitsev S, Spitzer D, Murciano JC, Ding BS, Tliba S, Kowalska MA, et al. Targeting of a mutant plasminogen activator to circulating red blood cells for prophylactic fibrinolysis. J Pharmacol Exp Ther. 2010;332:1022–31. doi: 10.1124/jpet.109.159194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaitsev S, Spitzer D, Murciano JC, Ding BS, Tliba S, Kowalska MA, et al. Sustained thromboprophylaxis mediated by an RBC-targeted pro-urokinase zymogen activated at the site of clot formation. Blood. 2010;115:5241–8. doi: 10.1182/blood-2010-01-261610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armstead WM, Ganguly K, Kiessling JW, Riley J, Chen XH, Smith DH, et al. Signaling, delivery, and age as emerging issues in the benefit/risk ratio outcome of tPA for treatment of CNS ischemic disorders. J Neurochem. 2010;113(2):303–12. doi: 10.1111/j.1471-4159.2010.06613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]