

Abstract
Several causative genes have been identified for both dystonia-parkinsonism and neurodegeneration with brain iron accumulation (NBIA), yet many patients do not have mutations in any of the known genes. Mutations in the ATP13A2 lead to Kufor Rakeb disease, a form of autosomal recessive juvenile parkinsonism that also features oromandibular dystonia. More recently, evidence of iron deposition in the caudate and putamen have been reported in patients with ATP13A2 mutations. We set out to determine the frequency of ATP13A2 mutations in cohorts of idiopathic NBIA and dystonia-parkinsonism. We screened for large deletions using whole genome arrays, and sequenced the entire coding region in 92 cases of NBIA and 76 cases of dystonia-parkinsonism. A number of coding and non-coding sequence variants were identified in a heterozygous state, but none were predicted to be pathogenic based on in silico analyses. Our results indicate that ATP13A2 mutations are a rare cause of both NBIA and dystonia-parkinsonism.
Keywords: Neurodegeneration, Iron, NBIA, Kufor Rakeb, ATP13A2, Dystonia, Parkinsonism
1. Introduction
Although several genes have been identified that may lead to dystonia-parkinsonism [14], many cases remain idiopathic despite extensive workup. Similarly, many patients with neurodegeneration with brain iron accumulation (NBIA) lack mutations in any of the known genes [10].
Kufor Rakeb disease (KRD) is an autosomal recessive pallido-pyramidal syndrome [11] caused by mutations in the lysosomal type 5 ATPase ATP13A2 [13]. The parkinsonism in KRD is typically responsive to levodopa, at least initially, while dystonic orofacial spasms are also frequent in the disease [12].
Schneider and colleagues recently reported a case with homozygous mutations in ATP13A2, clinical parkinsonism, and T2*MRI evidence of iron deposition [15]. Subsequently, at least one additional case with evidence of iron deposition has been identified [1], although most patients with ATP13A2 mutations do not have identifiable brain iron deposits despite being clinically symptomatic [1,4]. Given the clinical and neuroimaging overlap between KRD, dystonia-parkinsonism, and NBIA, we sought to determine if patients with idiopathic forms of NBIA or dystonia-parkinsonism harbored mutations in ATP13A2.
2. Materials and methods
2.1. Subjects
Approval for this study as part of an ongoing study of NBIA and dystonia-parkinsonism was obtained through the Oregon Health & Science University institutional review board (US) or the ethics committee of the National Hospital for Neurology & Neurosurgery (UK). Patients were referred for study by treating physicians because of clinical dystonia-parkinsonism or NBIA. This population was diverse and included patients from five continents. The majority of these patients represented sporadic, non-consanguineous cases contributed by referring physicians outside of both institutions. A diagnosis of idiopathic NBIA was made if probands had evidence of iron deposition in the basal ganglia (Fig. 1) without identifiable mutations in PANK2, PLA2G6, c19orf12, or FA2H. FTL and ACP sequence analysis was performed in select cases with evidence of iron deposition in the caudate, thalamus, or dentate nuclei. Dystonia-parkinsonism cases presented without mutations in PANK2 or PLA2G6. Written informed consent was collected for all participants. Whole blood was collected in citrate ACD or EDTA collection tubes.
Fig. 1.
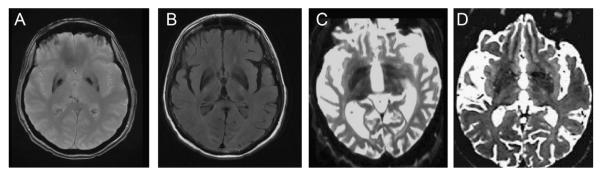
Neuroimaging features of idiopathic NBIA and Kufor Rakeb Disease. Axial T2* (A) and FLAIR (B) images from a 68 year-old woman with cognitive decline and parkinsonism diagnosed with idiopathic NBIA. (C) and (D) represent axial T2-weighted images taken from patients with compound heterozygous mutations (c.3057delC/c.130615G → A) in ATP13A2 (images C and D reprinted with permission from [1,15]).
2.2. Genotyping and sequencing
Genomic DNA was extracted from whole blood or EBV-transformed lymphoblast cultures prepared from the original blood sample using standard methods. Mutation analysis for known causes of NBIA or dystonia-parkinsonism was negative in cases selected for inclusion in the current study. Twenty-six patients had array-based genomic analyses (Illumina HumanCNV370-Quad or CytoSNP 12 Beadchips) performed per the manufacturer’s instructions which excluded duplications, deletions, and insertions larger than ~25–30 kb. Genomic copy number variants were detected using the Genome Studio CNV plugin (http://www.illumina.com/software/illumina connect.ilmn). DNA sequence analysis was then performed using Sanger sequencing. Primers were designed to amplify all 29 coding exons and at least 30 bp of the intron–exon boundary of ATP13A2 (sequences available upon request) using PrimerZ (http://genepipe.ngc.sinica.edu.tw/primerz/beginDesign.do) or ExonPrimer (http://ihg.gsf.de/ihg/ExonPrimer.html). Polymerase chain reaction (PCR) amplification was performed in a total volume of 12 ml using 10–20 ng of genomic DNA, 10 pmol transcript-specific forward and reverse primers, and 10 ml FastStart PCR Master Mix (Roche). Each purified product was sequenced in forward and reverse directions using BigDye-terminator version 3.1 (Applied Biosystems). Purified sequences were run on an ABI3730XP automated sequencer per the manufacturer’s protocol (Applied Biosystems). Sequence data were analyzed using Sequencher version 4.1.4 (GeneCodes). Sequence variants were identified via comparison to reference sequence (NM 022089.2) using Ensembl (www.ensembl.org) and Mutalyzer (Wildeman et al., 2008). In silico algorithms (SIFT (http://sift.jcvi.org/) and PolyPhen (http://genetics.bwh.harvard.edu/pph/) were used to interrogate any sequence variants identified.
3. Results
Microarray analysis demonstrated no deletions or duplications within 1 Mb of the ATP13A2 physical position (GRCh37/hg19 chr1:17,312,453–17,338,423) in any of the samples analyzed. Sequence analysis revealed several coding and noncoding sequence variants (Tables 1 and 2). Several variants (rs2076603, rs3738815, rs9435662, rs3170740) represented well-characterized high-frequency SNPs, present in ≥20% of cases, and were not considered further. Although we identified several sequence variants that had not been previously identified, none of these variants were predicted to be deleterious using in silico algorithms, and none were found in a homozygous or compound heterozygous state, indicating that they likely represent benign polymorphisms.
Table 1.
ATP13A2 sequence variants identified in dystonia-parkinsonism (a) and NBIA (b) cases.
| Location | Physical positiona | SNP ID | Variant | Frequency | Interpretation |
|---|---|---|---|---|---|
| (a) | |||||
| Exon 10 | chr1:17,326,767 | rs56367069 | c.881G>A | 1/76 (1%) | p.R294Q(het) tolerated/possibly damaging |
| Exon 10 | chr1:17,326,750 | c.898C>A | 4/76 (5%) | p.P300T (het) tolerated | |
| Exon 13 | chr1: 17,322,928 | c.1043C>T | 1/76 (1%) | p.R414W (het) tolerated | |
| Location | Physical positiona | SNP ID | Variant | Frequencyb | Interpretation |
|
| |||||
| (b) | |||||
| Exon 3 | chr1:17,332,025 | c.132A>G | 1/92 (1%) | Synonymous coding | |
| Exon 5 | chr1:17,331,192 | rs113643181 | c.472G>A | 1/92 (1%) | p.G158R (het) affect protein function/benign |
| Exon 10 | chr1:17,326,540 | rs56290406 | c.1005C>T | 1/92 (1%) | Synonymous coding |
| Exon 10 | chr1:17,326,767 | rs56367069 | c.881G>A | 3/92 (3%) | p.R294Q(het) tolerated/possibly damaging |
| Exon 12 | chr1:17,323,601 | c.1109G>A | 1/92 (1%) | p. R370Q(het) benign | |
| Exon 15 | chr1:17,322,477 | c.1536C>T | 1/92 (1%) | Synonymous coding | |
| Exon 22 | chr1:17,316,472 | c.2439C>T | 1/92 (1%) | Synonymous coding | |
| Exon 26 | chr1:17,313,654 | rs761421 | c.2970G>A | 2/92 (2%) | Synonymous coding |
| Exon 27 | chr1:17,313,343 | rs9435659 | c.3192C>T | 1/92 (1%) | Synonymous coding |
| Exon 28 | chr1:17,313,021 | rs115985012 | c.3342C>T | 2/92 (2%) | Synonymous coding |
| Exon 29 | chr1:17,312,592 | rs15786 | c.3365C>T | 1/92 (1%) | p.P1122L(het) benign |
| Exon 29 | chr1:17,312,485 | rs76298930 | c.3472G>A | 1/92 (1%) | p.V1158M (het) benign |
Physical position based upon RCh37.
Present in either a heterozygous or homozygous state.
Table 2.
ATP13A2 sequence variants identified in NBIA cases.
| Location | Physical positiona | SNP ID | Variant | Frequencyb | Interpretation |
|---|---|---|---|---|---|
| Exon 3 | chr1:17,332,025 | c.132A>G | 1/92 (1%) | Synonymous coding | |
| Exon 5 | chr1:17,331,192 | rs113643181 | c.472G>A | 1/92 (1%) | p.G158R (het) affect protein function/benign |
| Exon 10 | chr1:17,326,540 | rs56290406 | c.1005C>T | 1/92 (1%) | Synonymous coding |
| Exon 10 | chr1:17,326,767 | rs56367069 | c.881G>A | 3/92 (3%) | p.R294Q(het) tolerated/possibly damaging |
| Exon 12 | chr1:17,323,601 | c.1109G>A | 1/92 (1%) | p.R370Q(het) benign | |
| Exon 15 | chr1:17,322,477 | c.1536C>T | 1/92 (1%) | Synonymous coding | |
| Exon 22 | chr1:17,316,472 | c.2439C>T | 1/92 (1%) | Synonymous coding | |
| Exon 26 | chr1:17,313,654 | rs761421 | c.2970G>A | 2/92 (2%) | Synonymous coding |
| Exon 27 | chr1:17,313,343 | rs9435659 | c.3192C>T | 1/92 (1%) | Synonymous coding |
| Exon 28 | chr1:17,313,021 | rs115985012 | c.3342C>T | 2/92 (2%) | Synonymous coding |
| Exon 29 | chr1:17,312,592 | rs15786 | c.3365C>T | 1/92 (1%) | p.P1122L(het) benign |
| Exon 29 | chr1:17,312,485 | rs76298930 | c.3472G>A | 1/92 (1%) | p.V1158M (het) benign |
Physical position based upon RCh37.
Present in either a heterozygous or homozygous state.
3.1. Analysis of sequence variants
3.1.1. Dystonia-parkinsonism
We observed a c.881G>A polymorphism within exon 10 (p.R294Q) in a single patient with dystonia-parkinsonism and ataxia that began at age 23. This patient had T2 hyperintense signal changes in the basal ganglia on MRI. Although this variant was predicted to be ‘possibly damaging’ by one of the in silico algorithms, it was predicted to be tolerated by another, and it is published in dbSNP as rs56367069. Multiple sequence alignment demonstrated sequence conservation across mammalian species, but variability in lower organisms. This variant has previously been observed in neurologically normal individuals [16], indicating it is not pathogenic.
Also within exon 10, a novel sequence change (c.898C>A) was observed in 4 patients. Patient 1 presented at age 40 with personality change, cognitive decline, dystonia-parkinsonism, myoclonus and apraxia, and T2 hyperintensity in the basal ganglia on MRI. Patient 2 developed dystonia-parkinsonism, chorea, ataxia, and cognitive decline at age 36. Patient 3 exhibited extrapyramidal features, ataxia, and cognitive decline beginning in the late teens associated with basal ganglia T2 hyperintensity. Patient 4 developed ataxia, dystonia, and cognitive decline with frontal release signs and mutism at age 32; MRI demonstrated ex vacuo ventriculomegaly and diffuse T2 white matter hyperintensity. Predicted to lead to a heterozygous p.P300T substitution within the ATPase-associated domain, the c.898C>A sequence variant was considered unlikely to be deleterious based on in silico analysis. Sequence identity was not conserved between species.
In a patient with onset of dystonia-parkinsonism and cognitive decline at age 30, a single c.1043C>T heterozygous variant was identified in exon 13. This sequence change led to a p.R414W substitution that also fell within the ATPase-associated domain. In silico modeling indicated that this variant was unlikely to be pathogenic.
3.1.2. NBIA
A c.472G>A variant was detected within exon 5 in a single NBIA patient, who presented w/extrapyramidal symptoms and hypointensity of the globus pallidus and substantia nigra. This variant has previously been assigned SNP ID rs113643181. While the ensuing p.G158R heterozygous variant was predicted to affect protein function by one algorithm, it was predicted to be a benign change by the other. This residue is not conserved across species, and occurs outside of both the P-type ATPase and transmembrane domains of the protein. This sequence change thus most likely represents a benign variant.
The p.R294Q variant was detected in a heterozygous state in three NBIA patients, in addition to being detected in dystonia-parkinsonism.
In a patient who developed axial rigidity, resting tremor, myoclonic jerks, and foot drop at age 16 (along with neuroimaging indicating iron deposition), a heterozygous c.1109G>A variant in exon 12 was identified. The resultant p.R370Q residue change was predicted to be benign, and in fact, a glutamine is found at this site in lieu of arginine in the ATP13A2 homolog Ypk9 in S. cerevisiae.
In a patient with dystonia-parkinsonism, pyramidal tract signs, and T2 hypointensity of the basal ganglia, 2 heterozygous sequence changes were noted in exon 29 (c.3365C>T and c.3472G>A). These variants were predicted to lead to p.P1122L (rs15786) and p.V1158M (rs76298930), respectively. In addition to being previously described in unaffected individuals, both variants were predicted to represent benign polymorphisms.
Additional novel sequence changes (c.132A>G, c.1536C>T, and c.2439C>T) led to synonymous amino acid residues and are thus unlikely to be of consequence.
4. Discussion
Considerable phenotypic overlap exists between subtypes of NBIA and dystonia-parkinsonism. In particular, mutations in PLA2G6 have been associated with surprisingly diverse phenotypes that include psychomotor regression in infancy with death in early childhood at one end of the spectrum and adult-onset parkinsonism-dystonia at the other. Patients with mutations in PLA2G6 variably exhibit evidence of iron deposition in the globus pallidus and substantia nigra [10], perhaps related to differential effects of mutations on the enzyme’s catalytic activity toward phospholipids and lysophospholipids [7].
Given that select patients with mutations in ATP13A2 developed MRI evidence of iron deposition in the basal ganglia and that both dystonia and parkinsonism are features of KRD, we sought to determine the frequency of ATP13A2 mutations in large cohorts of patients with dystonia-parkinsonism with or without brain iron deposition of unknown cause. It is worth mention that although homozygous or compound heterozygous mutations are known to lead to the complicated neurological phenotype of Kufor Rakeb disease, heterozygous ATP13A2 mutations have been implicated in cases of isolated parkinsonism, often of early-onset [3,5,9]. To date, ATP13A2 mutations have not been detected in patient cohorts with typical-onset sporadic Parkinson’s disease [6,16].
Our results indicate that ATP13A2 mutations are rare in cases of both dystonia-parkinsonism and NBIA ascertained based on clinical and neuroimaging findings. These results are not surprising given that few cases of most single gene forms of these disorders have been identified worldwide [14]. These findings may help guide the clinical evaluation of parkinsonian disorders.
5. Conclusions
Mutations in ATP13A2 are associated with a complex phenotype. The nosology of this disorder is further complicated by the recent finding that both Tibetan terriers and a human family with brain accumulation of ceroid lipofuscin suggestive of Kufs’ disease bear mutations in ATP13A2 [2,8] indicating phenotypic overlap with the neuronal ceroid lipofuscinoses. The current lack of knowledge related to the function of ATP13A2 and the fact that no postmortem Kufor Rakeb samples have been reported suggests that other approaches, such as the use of mouse models, will likely be necessary to elucidate the pathophysiologic basis of this unusual parkinsonian disorder.
HIGHLIGHTS.
▶ Mutations in ATP13A2 may lead to complex, overlapping neurodegenerative phenotypes.
▶Dystonia, parkinsonism, brain iron deposition, and ceroid lipofuscinosis have been described.
▶A cause is not able to be identified for many patients with dystonia-parkinsonism (D-PD) and NBIA.
▶ We screened the largest assembled cohort to date of D-PD and NBIA patients for mutations in ATP13A2.
▶ ATP13A2 mutations are at best a rare cause of both phenotypes.
Acknowledgements
We are grateful to the patients and their families for their participation in this research. This work has been supported in part by the NBIA Disorders Association and Associazione Italiana Sindromi Neurodegenerative da Accumulo di Ferro [MCK, SJH], the American Academy of Neurology [MCK], the Oregon Medical Research Foundation [MCK], the American Philosophical Society [MCK], and the Oregon Clinical and Translational Research Institute (OCTRI) (MCK), grant number UL1 RR024140 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research.
Footnotes
Disclosures MCK receives research support from EMD-Serono. No other financial disclosures are reported.
References
- [1].Behrens MI, Brüggemann N, Chana P, Venegas P, Kägi M, Parrao T, Orel-lana P, Garrido C, Rojas CV, Hauke J, Hahnen E, González R, Seleme N, Fernández V, Schmidt A, Binkofski F, Kömpf D, Kubisch C, Hagenah J, Klein C, Ramirez A. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Movement Disorder. 2010 Sep;25(12):1929–1937. doi: 10.1002/mds.22996. [DOI] [PubMed] [Google Scholar]
- [2].Bras J, Verloes A, Schneider SA>, Mole SE, Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Human Molecular Genetics. 2012;21(12):2646–2650. doi: 10.1093/hmg/dds089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Brüggemann N, Hagenah J, Reetz K, Schmidt A, Kasten M, Buchmann I, Eckerle S, Bähre M, Münchau A, Djarmati A, van der Vegt J, Siebner H, Binkofski F, Ramirez A, Behrens MI, Klein CC. Recessively inherited parkinsonism: effect of ATP13A2 mutations on the clinical and neuroimaging phenotype. Archives of Neurology. 2010 Nov;67(11):1357–1363. doi: 10.1001/archneurol.2010.281. [DOI] [PubMed] [Google Scholar]
- [4].Chien HF, Bonifati V, Barbosa ER. ATP13A2-related neurodegeneration (PARK9) without evidence of brain iron accumulation. Movement Disorder. 2011 Jun;26(7):1364–1365. doi: 10.1002/mds.23514. [DOI] [PubMed] [Google Scholar]
- [5].Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto C, Fabbrini G, Marconi R, Fincati E, Abbruzzese G, Marini P, Squitieri F, Horstink HW, Montagna P, Libera AD, Stocchi F, Goldwurm S, Ferreira JJ, Meco G, Martignoni E, Lopiano L, Jardim LB, Oostra BA, Barbosa ER, Italian Parkinson Genetics Network. Bonifati V. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. 2007 May;68(19):1557–1562. doi: 10.1212/01.wnl.0000260963.08711.08. [DOI] [PubMed] [Google Scholar]
- [6].Djarmati A, Hagenah J, Reetz K, Winkler S, Behrens MI, Pawlack H, Lohmann K, Ramirez A, Tadić V, Brüggemann N, Berg D, Siebner HR, Lang AE, Pramstaller PP, Binkofski F, Kostić VS, Volkmann J, Gasser T, Klein C. ATP13A2 variants in early-onset Parkinson’s disease patients and controls. Movement Disorder. 2009 Oct;24(14):2104–2111. doi: 10.1002/mds.22728. [DOI] [PubMed] [Google Scholar]
- [7].Engel LA, Jing Z, O’Brien DE, Sun M, Kotzbauer PT. Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS One. 2010 Sep;5(9):e12897. doi: 10.1371/journal.pone.0012897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Farias FH, Zeng R, Johnson GS, Wininger FA, Taylor JF, Schnabel RD, McKay SD, Sanders DN, Lohi H, Seppälä EH, Wade CM, Lindblad-Toh K, O’Brien DP, Katz ML. A truncating mutation in ATP13A2 is responsible for adult-onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiology of Disease. 2011 Jun;42(3):468–474. doi: 10.1016/j.nbd.2011.02.009. [DOI] [PubMed] [Google Scholar]
- [9].Fong CY, Rolfs A, Schwarzbraun T, Klein C, O’Callaghan FJ. Juvenile parkinsonism associated with heterozygous frameshift ATP13A2 gene mutation. European Journal of Pediatrics Neurology. 2011 May;15(3):271–275. doi: 10.1016/j.ejpn.2011.01.001. [DOI] [PubMed] [Google Scholar]
- [10].Kruer MC, Boddaert N, Schneider SA, Houlden H, Bhatia KP, Gregory A, Anderson JC, Rooney WD, Hogarth P, Hayflick SJ. Neuroimaging features of neurodegeneration with brain iron accumulation. American Journal of Neuroradiology. 2012 Mar;33(3):407–414. doi: 10.3174/ajnr.A2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Najim al-Din AS, Wriekat A, Mubaidin A, Dasouki M, Hiari M. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurologica Scandinavica. 1994;89(5):347–352. doi: 10.1111/j.1600-0404.1994.tb02645.x. [DOI] [PubMed] [Google Scholar]
- [12].Ning YP, Kanai K, Tomiyama H, Li Y, Funayama M, Yoshino H, Sato S, Asahina M, Kuwabara S, Takeda A, Hattori T, Mizuno Y, Hattori N. PARK9-linked parkinsonism in eastern Asia: mutation detection in ATP13A2 and clinical phenotype. Neurology. 2008;70(16 Pt 2):1491–1493. doi: 10.1212/01.wnl.0000310427.72236.68. [DOI] [PubMed] [Google Scholar]
- [13].Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods GC, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nature Genetics. 2006;38(10):118–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- [14].Schneider SA, Bhatia KP. Rare causes of dystonia parkinsonism. Current Neurology and Neuroscience Reports. 2010;10(6):431–439. doi: 10.1007/s11910-010-0136-0. [DOI] [PubMed] [Google Scholar]
- [15].Schneider SA, Paisan-Ruiz C, Quinn NP, Lees AJ, Houlden H, Hardy J, Bhatia KP. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Movement Disorder. 2010;25(8):979–984. doi: 10.1002/mds.22947. [DOI] [PubMed] [Google Scholar]
- [16].Vilariño-Güell C, Soto AI, Lincoln SJ, Ben Yahmed S, Kefi M, Heckman MG, Hulihan MM, Chai H, Diehl NN, Amouri R, Rajput A, Mash DC, Dickson DW, Middleton LT, Gibson RA, Hentati F, Farrer MJ. ATP13A2 variability in Parkinson disease. Human Mutation. 2009 Mar;30(3):406–410. doi: 10.1002/humu.20877. [DOI] [PMC free article] [PubMed] [Google Scholar]