

Abstract
Episodic marine incursion has been a major driving force in the formation of present-day diversity. Marine incursion is considered to be one of the most productive ‘species pumps’ particularly because of its division and coalescence effects. Marine incursion events and their impacts on diversity are well documented from South America, North America and Africa; however, their history and impacts in continental East Asia largely remain unknown. Here, we propose a marine incursion scenario occurring in East Asia during the Miocene epoch, 10–17 Ma. Our molecular phylogenetic analysis of Platorchestia talitrids revealed that continental terrestrial populations (Platorchestia japonica) form a monophyletic group that is the sister group to the Northwest Pacific coastal species Platorchestia pacifica. The divergence time between the two species coincides with Middle Miocene high global sea levels. We suggest that the inland form arose as a consequence of a marine incursion event. This is the first solid case documenting the impact of marine incursion on extant biodiversity in continental East Asia. We believe that such incursion event has had major impacts on other organisms and has played an important role in the formation of biodiversity patterns in the region.
Keywords: habitat shift, biogeography, molecular phylogeny, Platorchestia
1. Introduction
Marine incursions are the inundation of continental land by ocean waters, generally resulting from rises in sea levels and regional tectonic subsidence [1]. The extensive lowland flooding produced by marine incursion has had a dramatic influence on landscape and ecosystem evolution [2]. Historical marine incursion events have exerted profound effects on the dynamics of biodiversity, as best demonstrated in Neotropical communities [2–4]. Marine incursions produce progressively desalinized habitats and episodically allow aquatic connections between freshwater systems and the marine realm [5]. These estuarine environments of varying salinity provide pathways for evolutionary habitat transition of many marine-derived organisms [6–8]. Marine incursions may have also functioned as impenetrable dispersal barriers for terrestrial species, fragmenting their previously continuous distribution ranges and leading to vicariant speciation [9]. Because its division and coalescence effects often produce bursts of diversification, incursion is considered to be one of the most productive ‘species pumps’ [2]. In addition, ecological impacts do not cease after termination of marine incursion, because soil properties and river water characteristics are permanently altered. This enhanced edaphic and ecosystem heterogeneity has sustained present-day high terrestrial diversity [5]. There is little doubt that the combined effect of Andean uplift and multiple historical marine incursions have contributed significantly to the extraordinarily high biodiversity of the Neotropics [3,4,10]. Interestingly, despite increasingly available evidence of marine incursion for other continents, such as North America and Africa [11–13], few attempts have been made to document the impact of marine incursion on the East Asian continent [7].
Compared with the Neotropics, continental East Asia experienced similar, if not even more dramatic, topographic changes in the Neogene. Prior to the end of the Palaeocene, the eastern Asian foreland was higher than central and western Asia [14]. The India–Asia collision occurring approximately 50 Ma led to the uplift of the Himalayan–Tibetan orogen [15]. During the same time period, eastern Asian sedimentary basins experienced continuous subsidence [14]. Approximately 23 Ma during the Neogene, uplift and subsidence collectively changed the landscape from an eastwest tilt to a westeast tilt [16]. This reconfiguration dramatically altered drainage systems [16]; for example, the course of the Yangtze River, which initially flowed southwest to the Paratethys Sea, switched eastwards to the Pacific Ocean [17]. These topographic changes also set the stage for marine incursions during the Early-to-Middle Miocene (23–11 Ma), a time of high global sea levels [18]. Indeed, marine and brackish foraminifer, ostracod and gastropod fossils have been reported in eastern China [19,20], suggesting the occurrence of brief large-scale episodic marine incursions during the Miocene [7,21]. Surprisingly, very few investigations have been carried out to address the potential impact of such incursions on extant biodiversity; an effect on river dolphins is currently the sole documented case [7].
Testing historical marine incursion events and their biogeographic impact on extant biodiversity ideally requires a group of organisms that currently span both coastal and inland ranges. Amphipods of the genus Platorchestia (Amphipoda: Talitridae) are predominantly intertidal forms, with many intertidal species being common and widespread along the Northwest Pacific coast. Several terrestrial species, however, occur further inland on the East Asian continent. With distributions spanning both the coast and inland areas of East Asian continent, Platorchestia talitrids can thus serve as an excellent model system to test the marine incursion hypothesis.
We reconstructed a phylogeny of all terrestrial Platorchestia talitrids from continental East Asia and their potential coastal relatives using nuclear and mitochondrial DNA sequence data. We hypothesized that historical marine incursions are probably responsible for the origin of the inland terrestrial species. During an incursion episode, ancestral marine intertidal organisms invaded the inland; upon subsequent regression, they became isolated from their marine relatives and evolved a terrestrial lifestyle. We used a molecular phylogenetic approach to test this marine incursion hypothesis and we predicted that (i) terrestrial Platorchestia talitrids are nested within coastal species, with their most closely related coastal species occurring along the Northwest Pacific coast, the source of marine incursion and (ii) temporally consistent with incursion-related scenarios, divergence between terrestrial talitrids and their closest coastal relatives occurred during the Miocene, an epoch of high global sea levels and subsidence of the eastern Asian continental margin.
2. Material and methods
(a). Sample collection
Extensive coverage, particularly of coastal species, is essential for determining the closest coastal relative of terrestrial Platorchestia species. Samples of Platorchestia were collected (see the electronic supplementary material, table S1) from 49 sites covering most known distribution ranges along the Northwest Pacific coastline and continental East Asia. A total of 64 specimens of five species were examined, including one terrestrial species (P. japonica) and four coastal species (Platorchestia pacifica, Platorchestia pachypus, Platorchestia munmui and Platorchestia joi). Two species from genera Talorchestia and Sinorchestia were collected as outgroups. Detailed sample information is presented in the electronic supplementary material, table S1. All collected samples were immediately preserved in 95 per cent ethanol and stored at −20°C.
(b). Laboratory protocols
Total genomic DNA extractions and amplifications were performed following published protocols [22], with annealing temperatures optimized at 50°C–53°C. An 864-bp mitochondrial cytochrome oxidase I (COI) fragment was amplified using species-specific primers (forward primer 5′-TCTACTAATCACAAAGATATTGG-3′ and reverse primer 5′-CCAWCTAAAAACTTTRATACCKGT-3′). An approximately 410-bp portion of mitochondrial 16S was amplified with primers Y16F (5′-GGTAATTTGACCGTGCTAAG-3′) and Y16R (5′-CCGRTTTGAACTCARATCATGT-3′). A 327-bp histone H3 region was amplified with primers H3F and H3R [23], and an approximately 1340-bp nuclear 28S fragment was amplified with primer pair 28S-3311F and 28S-4434R [22]. DNA sequencing was carried out using BigDye technology on an ABI 3730 automated sequencer (Applied Biosystems, Foster City, CA, USA).
(c). Construction of data matrices
DNA sequences were checked and edited with sequencher v. 4.1 (Gene Codes Corporation). Sequences of coding fragments COI and H3 were aligned using ClustalW [24] with default multiple alignment settings. Upon subsequent visual inspection, no insertions or deletions were found in either of these two regions. For sequences of length-variable 16S and 28S rDNA, we used two different alignment programs, ClustalW and mafft v. 5 [25], to assess sensitivity of our data to different alignment parameters [26]. The preferred alignment for each locus gave the lowest score of incongruence with the protein-coding sequences. The number of variable sites and parsimony informative sites was calculated using MEGA5 [27].
(d). Phylogenetic analyses
Both maximum-likelihood and Bayesian inference were used for phylogenetic tree reconstruction, with analyses conducted on both individual and combined datasets. COI and 16S mitochondrial DNA (mtDNA) gene regions were concatenated as a single locus but partitioned in the phylogenetic analyses. Maximum-likelihood analyses were conducted using RAxML v. 7.0.3 [28] starting with 1000 rapid bootstrap replicates followed by a thorough maximum-likelihood tree search. Each individual locus was analysed separately under the GTRCAT model of evolution; the concatenated dataset was analysed under the GTRMIX model, with each gene partition assigned a distinct model but with a joined branch length optimization. Bayesian phylogenetic analyses were conducted using MrBayes v. 3.1.2 [29]. The best-fit models for each gene partition were selected based on the Akaike information criterion using MrModeltest v. 2.3 [30]. For single locus, Bayesian analyses consisted of two runs of five million generations, each with four simultaneous Markov chains. Default priors were used and all states were considered equally probable. Tracer v. 1.4 [31] was used to plot the resulting likelihood values and to determine when the Markov chains reached convergence. Trees were sampled every 500 generations; after discarding the first 3000 sampled trees as burn-in, we used the remaining trees to estimate a consensus tree and Bayesian posterior probabilities. For the combined data analysis, the data were separated into gene partitions with each partition following its best-fit evolutionary model. MrBayes was set to estimate parameters independently and simultaneously for each partition, and default priors were used. Each analysis consisted of two independent runs of 10 million generations, with sampling carried out every 500 generations. The first 5000 sampled trees were discarded as a conservative burn-in.
(e). Divergence time estimation
To specify interspecific levels of variation when estimating ages of divergence events, single- and multiple-threshold general mixed Yule coalescent (GMYC) analyses were optimized using the SPLITS package [32] in R. An ultrametric tree was constructed from all COI haplotypes of Platorchestia samples and outgroups under an uncorrelated lognormal relaxed molecular clock model using BEAST v. 1.6.2 [33], with the substitution model selected by MrModeltest and the substitution rate under the clock parameter setting set to one. A coalescent prior with constant population size was used to invoke the GMYC null model, with default options employed for all other parameters. We ran three independent analyses of 20 million generations each, sampling every 1000 generations; results were combined using LogCombiner v. 1.6.2 [33], with the first five million generations of each run discarded as burn-in. Effective sample size (ESS) values were used to determine the Bayesian statistical significance of each parameter (ESS > 200). Tracer was used to determine run convergence and stability.
We used two different strategies to estimate divergence times. First, divergence dates were estimated under a gene tree framework from the mtDNA data. A single exemplar representing each independent entity was selected for divergence dating analysis at interspecific levels. The sparse fossil record complicated the assessment of ages of certain lineages; the arthropod mitochondrial clock of 2.3 per cent divergence per million years [34] was therefore applied as calibration. The analysis implemented with BEAST was run using separate substitution models for COI and 16S partitions under an uncorrelated lognormal relaxed molecular clock model, and the Yule process was used to describe the speciation. Given the variation in topologies derived from the nuclear DNA data, we constrained the topology to that generated from maximum-likelihood analysis of the combined mitochondrial and nuclear DNA. Four independent simultaneous runs of 30 million generations were conducted with sampling carried out every 1000 generations. The resulting files from the independent runs, with the first 10 million generations excluded as burn-in, were combined using LogCombiner.
A more accurate estimate of divergence times is achievable via a fully Bayesian implementation of the multispecies coalescent carried out using a species tree framework [35,36]. This multispecies coalescent model co-estimates multiple gene trees embedded in a shared species tree, and incorporates uncertainty of the coalescent process. It has been suggested that this method has much better estimation accuracy for species tree topology than simple gene concatenation [36]. Consequently, we also used a Bayesian Markov chain Monte Carlo method implemented in *BEAST v. 1.7.3 [36,37], to simultaneously estimate both a species tree and divergence times from our multilocus data. Because analysis in *BEAST requires a priori designation of taxonomic units, we defined the independent evolutionary entities identified by the GMYC model as distinct taxonomic units. We selected the best-fit substitution models calculated by MrModeltest, and unlinked substitution models, clock models and trees among loci. We applied Yule process for the species tree prior and specified a random starting tree for each locus. As described earlier, we used an uncorrelated relaxed clock model, with a mitochondrial rate of 2.3 per cent divergence per million years as the informative prior. We conducted four independent runs of 200 million generations each, sampling every 1000 generations and discarding the first 25 per cent as burn-in. Acceptable convergence to the stationary distribution was assessed by inspecting posterior samples using Tracer. Because all runs produced the same topology with very similar posterior probabilities, we combined the independent runs using LogCombiner and subsampled every third tree.
(f). Palaeogeographic reconstruction
Palaeogeographic reconstruction of the fluvio-deltaic marine system and episodic marine incursions was adapted from [7,21].
3. Results
(a). Sequences
A total of 262 sequences of COI, 16S, 28S and H3 regions were successfully generated from 66 specimens in this study. Because 28S sequences obtained from two samples could not be interpreted unambiguously, only 64 sequences of 28S were used in subsequent analyses. Length of aligned protein-coding sequence datasets were 864 bp for COI and 327 bp for H3. Alignment parameters for length-variable fragments were selected to reduce incongruence with protein-coding sequences (see the electronic supplementary material, table S2). The final 16S and 28S alignments contained 416 and 1346 nucleotide sites, respectively. Detailed information on sequence variation is presented in the electronic supplementary material, table S3. All sequences were deposited in GenBank (for accession numbers, see the electronic supplementary material, table S1).
(b). Phylogenetic analyses
For the mtDNA data, best-fit models of evolution were determined to be GTR + I + G for COI and HKY + G for 16S. In the topologies recovered from both maximum-likelihood and Bayesian analyses (figure 1a), terrestrial Platorchestia talitrids appear as a well-supported monophyletic group (bootstrap value = 97 and Bayesian posterior probability = 100). This P. japonica clade is the sister species to coastal P. pacifica, although with low support. Both analyses strongly support each of the remaining species as monophyletic taxa, but the relationships among them are not strongly supported (figure 1a).
Figure 1.
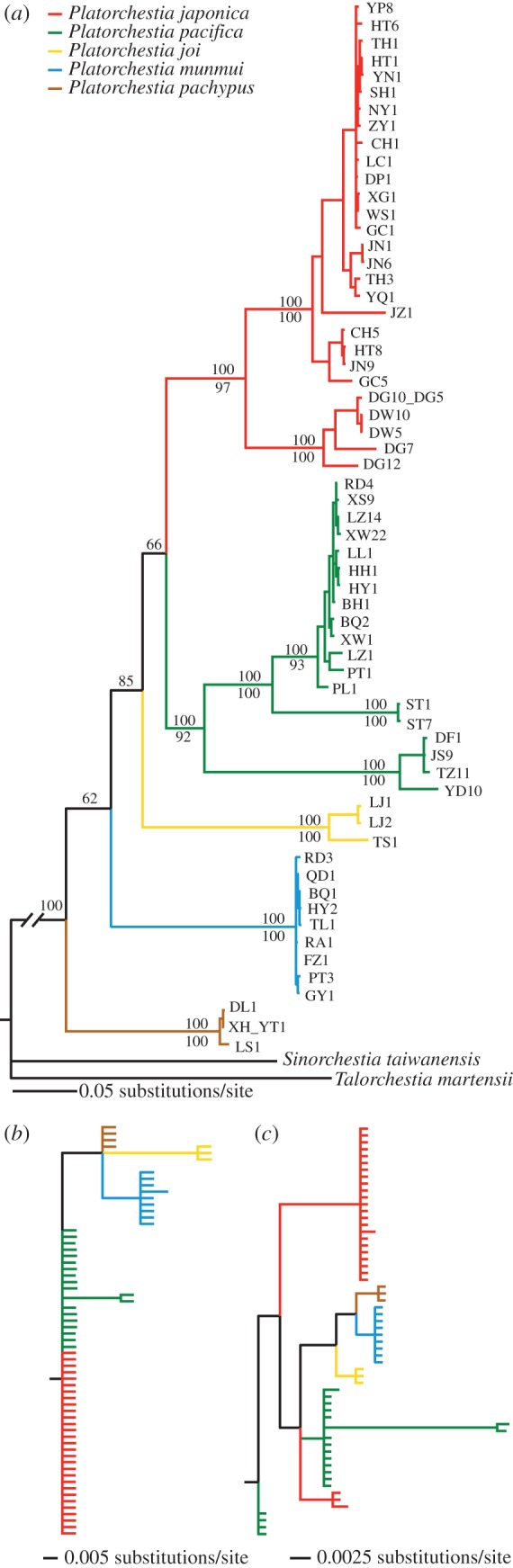
Phylogenetic trees of Platorchestia derived from Bayesian analysis of nuclear and mitochondrial DNA data. (a) Mitochondrial DNA phylogeny. Haplotype names are as in the electronic supplementary material, table S1. For major clades, Bayesian posterior probabilities and maximum-likelihood bootstrap support values greater than 50 are shown above and below branches, respectively. (b) Nuclear H3 phylogeny and (c) nuclear 28S phylogeny. The five species are colour-coded to show lack of monophyly.
For the nuclear genes, best-fit models were GTR+I for 28S and HKY+I for H3. Overall, the two nuclear loci exhibited few informative characters, leading to unresolved gene trees in many cases, particularly with regard to interspecies relationships (see figure 1b,c and electronic supplementary material, table S3). There was no well-supported conflict between mitochondrial and nuclear gene trees. With respect to observed monophyletic taxa, both 28S and H3 trees are largely congruent with the mitochondrial phylogeny, except that P. japonica and P. pacifica are weakly supported as paraphyletic in the 28S tree (figure 1c).
The combined nuclear and mitochondrial gene dataset comprised 2953 bp. Maximum-likelihood analysis and Bayesian inference both support the same phylogenetic relationships between terrestrial and coastal species. All terrestrial lineages cluster as a well-supported (bootstrap value = 98 and Bayesian posterior probability = 100) monophyletic group, the P. japonica clade, which is nested within the coastal species (figure 2a).
Figure 2.

Gene tree and species tree derived from the combined nuclear and mitochondrial dataset. (a) The maximum-likelihood tree based on concatenated data analysis. Haplotype names are as in the electronic supplementary material, table S1. Numbers above branches are nonparametric bootstrap support values greater than 50; the numbers below branches are Bayesian posterior probabilities greater than 95 for major lineages. (b) The species tree derived from *BEAST analysis. Bayesian posterior probabilities for major lineages are given above branches. Vertical bars are used to designate species. Terrestrial lineages are indicated in black; coastal lineages are shown in blue. (Online version in colour.)
The species tree estimated using *BEAST is identical to that generated from concatenated mitochondrial and nuclear data, again showing support for the monophyly of terrestrial lineages (Bayesian posterior probability = 100) and a sister relationship to coastal P. pacifica (Bayesian posterior probability = 96; figure 2b). For all nodes, support values obtained from species tree inference analysis are considerably higher than those derived from analysis of concatenated data (figure 2).
(c). Divergence times
To minimize the potential risks of mixing inter- and intraspecific levels of variation when estimating divergence times, we applied a GMYC model to specify independent entities at the interspecific level. Compared with the null model that assumes uniform branching rate for all lineages, both single- and multiple-threshold GMYC analyses provided significantly better fits to the ultrametric tree (likelihood ratio test, p < 0.05). Single- and multiple-threshold models delimited 22 (confidence interval = 18–24) and 24 (16–24) entities, respectively. Because the multiple-threshold model did not provide a significant improvement for the data (likelihood ratio test, p = 0.88), a single exemplar representing each independent entity delimited from the single-threshold model was selected for estimation of divergence times (figure 3).
Figure 3.
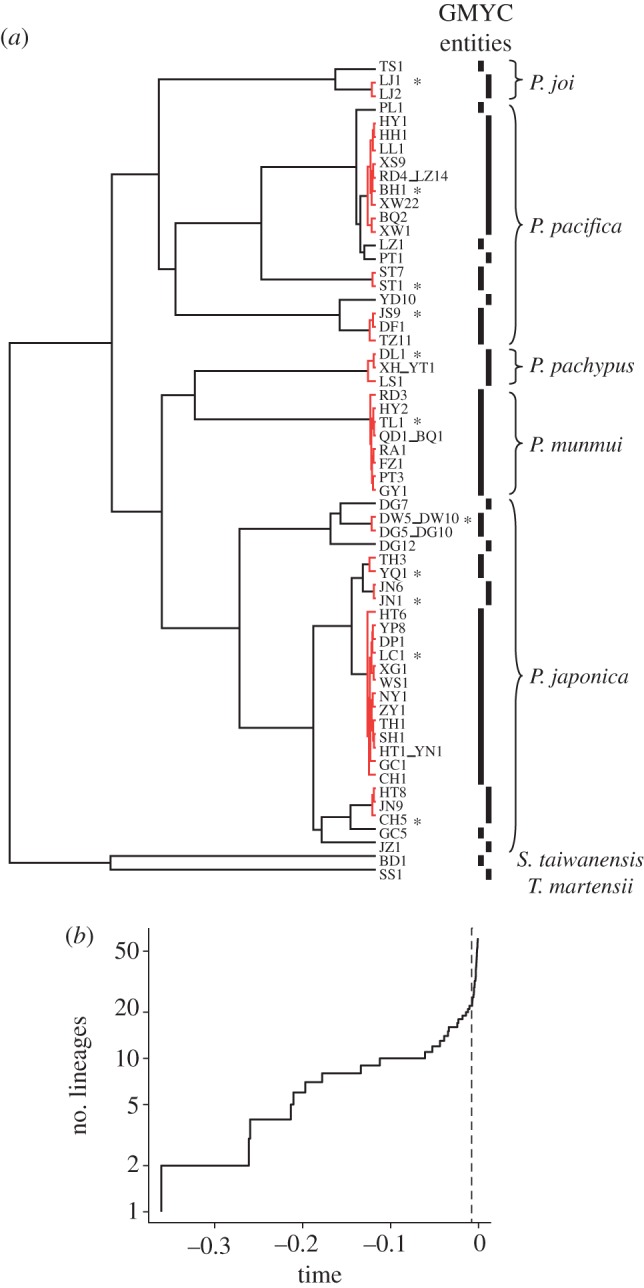
Results of single-threshold GMYC analysis of a COI ultrametric tree. (a) Interspecific entities delimitated. In distinct clusters, red clades indicate intraspecific branching and asterisks indicate samples selected for subsequent divergence time analysis. Right vertical bars show interspecific branching. (b) Lineages-through-time plot. The vertical dashed line indicates the likelihood transition from interspecific diversification to coalescent lineage branching events. (Online version in colour.)
Overall, our two dating approaches produced similar divergence time estimates. For most nodes, the 95% highest posterior density (HPD) of divergence events from the two analyses overlapped, but the means of estimates derived from the species tree analysis were slightly closer to the present (see the electronic supplementary material, figure S1). This is expected because divergence timing based on gene trees might overestimate than those derived using multilocus coalescent species tree-based approaches [35]. Divergence time estimates derived from the species tree suggest that the terrestrial P. japonica diverged from its closest coastal relative at approximately 16.9 Ma (95% HPD: 13.6–20.3 Ma; node A in figure 4b and electronic supplementary material, figure S1), with initial divergence among the terrestrial forms occurring approximately 9.7 Ma (95% HPD: 7.5–12.1 Ma; node B in figure 4b and electronic supplementary material, figure S1).
Figure 4.

Marine incursion scenario for the origin of terrestrial Platorchestia talitrids in continental East Asia. (a) Map of the Northwest Pacific showing sampling sites. Blue dots denote the terrestrial species P. japonica, red dots denote its sister species P. pacifica and black dots denote additional Platorchestia coastal species. Blue-shaded areas illustrate regions with marine ecosystems or regions into which episodic marine incursions occurred during the Miocene in East Asia (reconstructed from [7,21]). (b) Chronogram obtained using a relaxed clock model. The complete phylogeny is shown in figure 2. Purple bars indicate estimated standard deviations on selected nodes. Node A represents the divergence between terrestrial talitrids and their closest coastal relative, and node B represents the initial divergence within the terrestrial group. Coloured tree branches correspond to coloured sampling sites on the map. The smooth curve represents global sea-level fluctuations [18]. The gradual colour change over time in the schematic represents change from high to low altitude in east China.
4. Discussion
The recovered phylogenies, molecular dating, and the biogeography of endemic Platorchestia from East Asia are consistent with our predictions of the marine incursion hypothesis. The generated phylogenies strongly support the monophyly of all terrestrial Platorchestia lineages and their derived relationship to coastal species (figure 2). This phylogenetic pattern suggests that a single land colonization event by marine intertidal ancestors gave rise to all current endemic terrestrial populations (P. japonica). Platorchestia pacifica, the sister species to P. japonica, has the largest geographical distribution of all coastal relatives from the Northwest Pacific. It is therefore reasonable to suggest that the most recent common ancestor of P. japonica and P. pacifica was a widespread species in the Northwest Pacific, which would have had the greatest opportunity to participate in a marine incursion event. According to molecular clock calibrations, terrestrial P. japonica diverged from its closest coastal species, P. pacifica, at approximately 16.9 Ma. This time coincides with a period of high global sea levels during the Early-to-Middle Miocene (figure 4b). The time of initial divergence among the terrestrial forms coincides with low global sea levels during the Late Miocene (figure 4b). In summary, these results are consistent with our temporal and geographical predictions under a marine incursion hypothesis.
Considering all the evidence, we reconstructed the following marine incursion scenario. During the Early-to-Middle Miocene, a time of high global sea levels, populations of the P. pacifica-like ancestor invaded inland areas of the East Asian continent, and became isolated from their coastal cousins. These populations evolved into the current P. japonica, while the coastal populations evolved into the current P. pacifica. Subsequent low sea levels in the Late Miocene led to fragmentation of the inland populations and vicariant divergence within P. japonica.
Several potential alternative explanations exist for the origin of terrestrial Platorchestia talitrids in East Asia. The occurrence of inland species could be the result of active and direct land penetration by ‘palustral’ ancestors [38]. For instance, the land invasion may have been facilitated along coastlines by high rainfall blowing in from the sea. In such cases, invasions would be essentially opportunistic [6]. Given the wide distribution of inland talitrids, however, we would expect them to have arisen on multiple independent occasions, which is not consistent with our results. Alternatively, surface streams might have indirectly facilitated passive dispersal of estuarial forms that then gave rise to the inland species [38]. This explanation seems unlikely because talitrid amphipods exhibit direct development without a planktonic larval stage [39], and thus have limited long-distance-dispersal ability.
It has been recognized that marine incursion could play an important biogeographic role in organizing biodiversity [1]. Incursion conditions produce progressively desalinized habitats that have been proposed as possible ‘competition troughs’ that can be exploited by newly arriving marine lineages [8]. Consequently, this unique set of conditions may have facilitated ecological shifts of marine-derived taxa, such as molluscs, ostracods, fishes and dolphins [4,7,12,19]. Because marine incursion events also function as obvious dispersal and ecological barriers, particularly for resident terrestrial and freshwater organisms [9,40], congruent biogeographic patterns would reflect shared responses by different species to the same major vicariant event. Previous studies suggested that some resident organisms, including freshwater fishes, birds and frogs, may have been pushed into upland areas by elevated salinity in lowland regions [11,41,42]. Such situations would have isolated populations and over time led to allopatric speciation [1]. Sediment composition, geochemistry and water characteristics were largely altered following the termination of marine incursions. This enhanced ecological heterogeneity provided an ideal setting for divergent natural selection in terrestrial and aquatic taxa, which in turn would generate and sustain high diversity in the region [5], such as in a recent study of croaker [43]. Over the long term, the complex ecological and evolutionary trends initiated by Neogene tectonic events (e.g. topographic deformation and marine incursion) and continued by the Pleistocene climatic changes would be important for the generation and maintenance of biodiversity on the East Asian continent.
Species diversity in the terrestrial group may be much higher than currently recognized. Because of their morphological conservativity, all populations are currently circumscribed under the same name (P. japonica). Results from a recent study, however, suggest that terrestrial forms in Japan and Taiwan probably represent different species [44]. The genetic diversity of the continental forms is high; for example, COI per cent divergence ranges from 2.0 to 14.1 per cent. The recent development of new species delimitation methods using multilocus data under a coalescent framework [45] enables a more efficient and less subjective approach to species delimitation. Application of these new methods and a more thorough examination will probably reveal multiple species within the continental terrestrial forms.
In conclusion, the phylogeographic patterns exhibited by Platorchestia talitrids provide the first solid evidence of a marine incursion event onto the East Asian continent leading to diversification of extant taxa. We expect that the shared signature of this incursion event is preserved in a diverse cross-section of East Asian continental organisms. Preliminary findings that point to an important role for marine incursions suggest that further biogeographic investigations of this palaeogeographic event will likely prove fruitful. Our understanding of the impact of historical events such as glaciation and marine incursion on this region of rich biodiversity is just beginning. Our study should open a new window to the evolution of ecosystems, as well as the origin and maintenance of endemic biodiversity patterns in East Asia.
Acknowledgements
We thank Jinzhong Fu, Associate Editor and two anonymous reviewers for constructive comments which greatly improved this manuscript. We thank Bryan C. Carstens, Anna Papadopoulou and Jean-François Flot for assistance with analyses. Ruiying Zhang helped with artwork. This study was supported by the National Natural Science Foundation of China (China National Funds for Distinguished Young Sciencists-31025023 to S.L. and NSFC-31172049 to Z.H.).
References
- 1.Bloom DD, Lovejoy NR. 2011. The biogeography of marine incursion in South America. In Historical biogeography of Neotropical freshwater fishes (eds Albert JS, Reis RE.), pp. 137–144 Berkeley, CA: University of California Press [Google Scholar]
- 2.Webb SD. 1995. Biological implications of the middle Miocene Amazon seaway. Science 269, 361–362 10.1126/science.269.5222.361 (doi:10.1126/science.269.5222.361) [DOI] [PubMed] [Google Scholar]
- 3.Valentí R. 2011. Neotropical biodiversity: timing and potential drivers. Trends Ecol. Evol. 26, 508–513 10.1016/j.tree.2011.05.011 (doi:10.1016/j.tree.2011.05.011) [DOI] [PubMed] [Google Scholar]
- 4.Hoorn C, et al. 2010. Amazonia through time: andean uplift, climate change, landscape evolution, and biodiversity. Science 330, 927–931 10.1126/science.1194585 (doi:10.1126/science.1194585) [DOI] [PubMed] [Google Scholar]
- 5.Wesselingh FP, Salo JA. 2006. A Miocene perspective on the evolution of the Amazonian biota. Scripta Geologica (Leiden) 133, 439–458 [Google Scholar]
- 6.Lovejoy NR, Bermingham E, Martin AP. 1998. Marine incursion into South America. Nature 396, 421–422 10.1038/24757 (doi:10.1038/24757) [DOI] [Google Scholar]
- 7.Hamilton H, Caballero S, Collins AG, Brownell RL. 2001. Evolution of river dolphins. Proc. R. Soc. Lond. B 268, 549–556 10.1098/rspb.2000.1385 (doi:10.1098/rspb.2000.1385) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lovejoy NR, Albert JS, Crampton WGR. 2006. Miocene marine incursions and marine/freshwater transitions: evidence from Neotropical fishes. J. S. Am. Earth Sci. 21, 5–13 10.1016/j.jsames.2005.07.009 (doi:10.1016/j.jsames.2005.07.009) [DOI] [Google Scholar]
- 9.Antonelli A, Nylander JAA, Persson C, Sanmartín I. 2009. Tracing the impact of the Andean uplift on Neotropical plant evolution. Proc. Natl Acad. Sci. USA 106, 9749–9754 10.1073/pnas.0811421106 (doi:10.1073/pnas.0811421106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santos JC, Coloma LA, Summers K, Caldwell JP, Ree R, Cannatella DC. 2009. Amazonian amphibian diversity is primarily derived from late Miocene Andean lineages. PLoS Biol. 7, e1000056. 10.1371/journal.pbio.1000056 (doi:10.1371/journal.pbio.1000056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemmon EM, Lemmon AR, Cannatella DC. 2007. Geological and climatic forces driving speciation in the continentally distributed trilling chorus forgs (Pseudacris). Evolution 61, 2086–2103 10.1111/j.1558-5646.2007.00181.x (doi:10.1111/j.1558-5646.2007.00181.x) [DOI] [PubMed] [Google Scholar]
- 12.Yamanoue Y, Miya M, Doi H, Mabuchi K, Sakai H, Nishida M. 2011. Multiple invasions into freshwater by pufferfishes (Teleostei: Tetraodontidae): a mitogenomic perspective. PLoS ONE 6, e17410. 10.1371/journal.pone.0017410 (doi:10.1371/journal.pone.0017410) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson AB, Teugels GG, Meyer A. 2008. Marine incursion: the freshwater herring of Lake Tanganyika are the product of a marine invasion into West Africa. PLoS ONE 3, e1979. 10.1371/journal.pone.0001979 (doi:10.1371/journal.pone.0001979) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang P. 1998. Deformation of Asia and global cooling: searching links between climate and tectonics. Quat. Sci. 18, 213–221 [Google Scholar]
- 15.Royden LH, Burchfiel BC, van der Hilst RD. 2008. The geological evolution of the Tibetan Plateau. Science 321, 1054–1058 10.1126/science.1155371 (doi:10.1126/science.1155371) [DOI] [PubMed] [Google Scholar]
- 16.Wang P. 2004. Cenozoic deformation and the history of sea-land interactions in Asia. In Continent–Ocean interactions within East Asian marginal seas (eds Clift P, Wang P, Kuhnt W, Hayes D.), pp. 1–22 Washington, DC: American Geophysical Union [Google Scholar]
- 17.Yang D. 1985. The primary study of the dating and causes of the Changjiang (Yangtze) River flowing eastwards into the sea. J. Nanjing Univ. 21, 155–165 [Google Scholar]
- 18.Haq BU, Hardenbol J, Vail PR. 1987. Chronology of fluctuating sea levels since the Triassic. Science 235, 1156–1167 10.1126/science.235.4793.1156 (doi:10.1126/science.235.4793.1156) [DOI] [PubMed] [Google Scholar]
- 19.Chen X. 1994. The Upper Tertiary transgressive beds at Yancheng depression. J. Stratigr. 18, 119–123 [Google Scholar]
- 20.Li J, Xiang W, Fan N. 1990. Neogene biota of guantao formation in the Gudong district, Kenli, Shandong. Pet. Explor. Dev. 1990, 30–39 [Google Scholar]
- 21.Wang H. 1985. Atlas of the palaeogeography of China. Beijing, China: Cartographic Publishing House [Google Scholar]
- 22.Witt JDS, Threloff DL, Hebert PDN. 2006. DNA barcoding reveals extraordinary cryptic diversity in an amphipod genus: implications for desert spring conservation. Mol. Ecol. 15, 3073–3082 10.1111/j.1365-294X.2006.02999.x (doi:10.1111/j.1365-294X.2006.02999.x) [DOI] [PubMed] [Google Scholar]
- 23.Colgan DJ, McLauchlan A, Wilson GDF, Livingston SP, Edgecombe GD, Macaranas J, Cassis G, Gray MR. 1998. Histone H3 and U2 snRNA DNA sequences and arthropod molecular evolution. Aust. J. Zool. 46, 419–437 10.1071/ZO98048 (doi:10.1071/ZO98048) [DOI] [Google Scholar]
- 24.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 10.1093/nar/22.22.4673 (doi:10.1093/nar/22.22.4673) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katoh K, Kuma K, Toh H, Miyata T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518 10.1093/nar/gki198 (doi:10.1093/nar/gki198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papadopoulou A, Anastasiou I, Vogler AP. 2010. Revisiting the insect mitochondrial molecular clock: the mid-Aegean trench calibration. Mol. Biol. Evol. 27, 1659–1672 10.1093/molbev/msq051 (doi:10.1093/molbev/msq051) [DOI] [PubMed] [Google Scholar]
- 27.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 10.1093/molbev/msr121 (doi:10.1093/molbev/msr121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 10.1093/bioinformatics/btl446 (doi:10.1093/bioinformatics/btl446) [DOI] [PubMed] [Google Scholar]
- 29.Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 10.1093/bioinformatics/btg180 (doi:10.1093/bioinformatics/btg180) [DOI] [PubMed] [Google Scholar]
- 30.Nylander JAA. 2004. MrModeltest v2. Program distributed by the author. Evolutionary Biology Centre, Uppsala University [Google Scholar]
- 31.Rambaut A, Drummond AJ. 2007. Tracer See http://beast.bio.ed.ac.uk/Tracer [Google Scholar]
- 32.Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell S, Kamoun S, Sumlin WD, Vogler AP. 2006. Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst. Biol. 55, 595–609 10.1080/10635150600852011 (doi:10.1080/10635150600852011) [DOI] [PubMed] [Google Scholar]
- 33.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214. 10.1186/1471-2148-7-214 (doi:10.1186/1471-2148-7-214) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brower AVZ. 1994. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc. Natl Acad. Sci. USA 91, 6491–6495 10.1073/pnas.91.14.6491 (doi:10.1073/pnas.91.14.6491) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCormack JE, Heled J, Delaney KS, Peterson AT, Knowles LL. 2011. Calibrating divergence times on species trees versus gene trees: implications for speciation history of Aphelocoma jays. Evolution 65, 184–202 10.1111/j.1558-5646.2010.01097.x (doi:10.1111/j.1558-5646.2010.01097.x) [DOI] [PubMed] [Google Scholar]
- 36.Heled J, Drummond AJ. 2010. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 27, 570–580 10.1093/molbev/msp274 (doi:10.1093/molbev/msp274) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 10.1093/molbev/mss075 (doi:10.1093/molbev/mss075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friend JA, Richardson AMM. 1986. Biology of terrestrial amphipods. Annu. Rev. Entomol. 31, 25–48 10.1146/annurev.en.31.010186.000325 (doi:10.1146/annurev.en.31.010186.000325) [DOI] [Google Scholar]
- 39.Väinölä R, Witt JDS, Grabowski M, Bradbury JH, Jazdzewski K, Sket B. 2008. Global diversity of amphipods (Amphipoda; Crustacea) in freshwater. Hydrobiologia 595, 241–255 10.1007/s10750-007-9020-6 (doi:10.1007/s10750-007-9020-6) [DOI] [Google Scholar]
- 40.Delsuc F, Superina M, Tilak MK, Douzery EJP, Hassanin A. 2012. Molecular phylogenetics unveils the ancient evolutionary origins of the enigmatic fairy armadillos. Mol. Phylogenet. Evol. 62, 673–680 10.1016/j.ympev.2011.11.008 (doi:10.1016/j.ympev.2011.11.008) [DOI] [PubMed] [Google Scholar]
- 41.Aleixo A. 2004. Historical diversification of a terra-firme forest bird superspecies: a phylogeographic perspective on the role of different hypotheses of Amazonian diversification. Evolution 58, 1303–1317 (doi:10.1111/j.0014-3820.2004.tb01709.x) [DOI] [PubMed] [Google Scholar]
- 42.Hubert N, Renno JF. 2006. Historical biogeography of South American freshwater fishes. J. Biogeogr. 33, 1414–1436 10.1111/j.1365-2699.2006.01518.x (doi:10.1111/j.1365-2699.2006.01518.x) [DOI] [Google Scholar]
- 43.Cooke GM, Chao NL, Beheregaray LB. 2012. Marine incursions, cryptic species and ecological diversification in Amazonia: the biogeographic history of the croaker genus Plagioscion (Sciaenidae). J. Biogeogr. 39, 724–738 10.1111/j.1365-2699.2011.02635.x (doi:10.1111/j.1365-2699.2011.02635.x) [DOI] [Google Scholar]
- 44.Cheng YT, Nakazono K, Lin YK, Chan BKK. 2011. Cryptic diversity of the semi-terrestrial amphipod Platorchestia japonica (Tattersall, 1922) (Amphipoda: Talitrida: Talitridae) in Japan and Taiwan, with description of a new species. Zootaxa 2787, 1–18 [Google Scholar]
- 45.Ence DD, Carstens BC. 2011. SpedeSTEM: a rapid and accurate method for species delimitation. Mol. Ecol. Resour. 11, 473–480 10.1111/j.1755-0998.2010.02947.x (doi:10.1111/j.1755-0998.2010.02947.x) [DOI] [PubMed] [Google Scholar]