

Abstract
Despite its charismatic appeal to both scientists and the general public, remarkably little is known about the giant squid Architeuthis, one of the largest of the invertebrates. Although specimens of Architeuthis are becoming more readily available owing to the advancement of deep-sea fishing techniques, considerable controversy exists with regard to topics as varied as their taxonomy, biology and even behaviour. In this study, we have characterized the mitochondrial genome (mitogenome) diversity of 43 Architeuthis samples collected from across the range of the species, in order to use genetic information to provide new and otherwise difficult to obtain insights into the life of this animal. The results show no detectable phylogenetic structure at the mitochondrial level and, furthermore, that the level of nucleotide diversity is exceptionally low. These observations are consistent with the hypotheses that there is only one global species of giant squid, Architeuthis dux (Steenstrup, 1857), and that it is highly vagile, possibly dispersing through both a drifting paralarval stage and migration of larger individuals. Demographic history analyses of the genetic data suggest that there has been a recent population expansion or selective sweep, which may explain the low level of genetic diversity.
Keywords: Architeuthis, genetic diversity, giant squid, mitogenome, population structure
1. Introduction
The giant squid, Architeuthis spp., is one of the largest invertebrates. Described primarily from remains in sperm whale stomachs, from carcasses of dead or moribund animals found floating on the ocean surface or washed up on beaches, and, rarely, from fresh specimens caught by deep-sea trawling activity [1], it was not until 2004 that a live specimen was observed in its natural habitat [2] and this year that the first video footage was published [3]. Because of their huge size and elusive nature, many myths and legendary sea monsters have been based on them, including the fabled sinker of ships, the Kraken. They have also been of considerable interest and speculation both in the scientific community and in popular literature, inspiring authors such as Jules Verne and Herman Melville. However, despite this, the scarcity of preserved specimens means knowledge about their biology, distribution and taxonomy is very limited, and, indeed, often confused with fiction. For example, while claims have been made of individuals measuring up to 50 m in total length [4], a more realistic estimate is a maximum total length of 18 m for females [5], with males reaching slightly smaller sizes [4]. What is more certain is that, with the exception of the polar regions, Architeuthis are globally distributed [6,7]. Furthermore, they are probably voracious carnivores, preying upon fish, but also smaller cephalopods [1], including other giant squid [8]. Carbon and nitrogen isotope profiles obtained from analysing upper beaks suggest Architeuthis undergoes an ontogenetic diet shift early in life, abandoning smaller prey of relatively low trophic status in favour of larger prey of higher status. Isotope profiles also indicate that adult giant squid inhabit relatively small, well-defined and productive areas, where food resources have a constant carbon isotope composition [9]. It is also clear that juvenile giant squid are hunted by many other animals, including dolphins, fish and sea birds [1,10], and the adults are consumed in large quantities by whales, especially the sperm whale, Physeter macrocephalus [11]. As such it is likely (although unproved) that the giant squid population must be large, in order to sustain such levels of predation by whales. A number of other characters are more uncertain. There has been debate, for example, about the activity level and metabolism of Architeuthis. Although many scientists have concluded that giant squid must be sluggish ambush-predators, which wait quietly in the water column for their prey to stumble into them [12,13], more recent data [14] based on a comparison of citrate synthase activity between cephalopods appears to support quite the opposite, consistent with recent photographic evidence [2]; the giant squid portrayed in these images seems to be a highly active predator with considerable strength [15].
The taxonomy of the giant squid remains controversial. Since the first description as Architeuthis dux by the Danish naturalist Japetus Steenstrup in 1857, as many as 21 nominal species of Architeuthis have been described [6,16]. Given the fact that many of these were described based exclusively on location, and in some cases on incomplete remains such as single beaks, suckers or arms regurgitated by sperm whales, the majority of these have been considered likely to be synonymous, and today different opinions suggest there may be as many as eight or as few as one species (with three subspecies) [1,4,17–21]. In addition to this uncertainty, the life cycle and life history of the giant squid also remain enigmatic, as do growth rate, time of sexual maturity and longevity, with the many estimates of maximum age ranging from 1 to 46 years [9,22–24].
Given the obvious difficulty of providing answers to some of the earlier-mentioned questions using conventional monitoring/observational techniques, alternative approaches are required if more is to be understood about the giant squid. Recent advances in DNA sequencing techniques have made the sequencing of large stretches of DNA (e.g. mitochondrial genomes) both quick and economical, and mitochondrial genome-based analyses are playing an increasingly important role in phylogenetic and population genetic studies [25,26]. In order to both reassess the estimated number of species and describe the global population structure of the giant squid, we have generated a dataset of 37 complete and six partial mitochondrial genomes (mitogenomes) using samples collected across its known range.
2. Methods
(a). Samples
Forty-three Architeuthis soft tissue samples were analysed in this study. Detailed information about the tissue samples can be found in the electronic supplementary material, table S1. Architeuthis are rarely caught alive, and many tissue samples were derived from the carcasses of dead animals found stranded on beaches or floating moribund on the ocean surface, although several tissue samples were derived from animals taken as accidental by-catch on deep-sea trawlers and frozen specimens subsequently donated to local scientific institutions. Many of the tissue samples had undergone some degree of decay (or digestion), owing to the quality of the source specimens (see the electronic supplementary material, table S1).
(b). DNA extraction
DNA extraction from cephalopods is challenging owing to high concentrations of mucopolysaccharides found in their soft tissues, which tend to co-separate with DNA during extraction, and subsequently can inhibit many downstream enzymatic processes. Furthermore, the muscular tissues of Architeuthis (as well as squid belonging to 15 other families of the order Oegopsida) naturally contain high levels of ammonia, conferring additional complications to DNA extraction [27]. Tissue samples were initially digested at 56°C with agitation, in a cetyltrimethylammonium bromide (CTAB) containing buffer (100 mM Tris–HCl, pH 8.0; 1.4 M NaCl; 20 mM EDTA; 2% w/v CTAB; 0.2% w/v dithiothreitol; 500 µg ml−1 proteinase K). Subsequently, the digests underwent three purifications, each of one volume of 25 : 24 : 1 phenol : chloroform : isoamyl alcohol saturated with 10 mM Tris–HCl (pH 8.0) and 1 mM EDTA. One volume of ddH2O was subsequently added to the aqueous phase in order to dilute the salt concentration, prior to precipitation of the DNA by adding one volume of 100 per cent isopropanol and centrifugation at 20 000 r.p.m., followed by a 70 per cent ethanol wash step. The extracted DNA was eluted in 1× TE buffer pH 8.0 (10 mM Tris–HCl, pH 8.0; 1 mM EDTA). The quality of all extracts was assessed by electrophoresis on 0.7 per cent agarose gels, as well as by fluorometric quantitation on the Qubit1.0 fluorometer (Invitrogen, Carlsbad, CA).
(c). Mitogenome amplification and sequencing
An unpublished Architeuthis mitogenome sequence present in GenBank (NC_011581, hereafter referred to as the reference sequence) indicates that the mitogenome contains two duplicated regions (see the electronic supplementary material, figure S1) that show a remarkably high degree of sequence similarity of approximately 99.9 per cent [28]. To verify the accuracy of this phenomenon, we designed four pairs of primers (see the electronic supplementary material, table S2) to independently PCR amplify each duplicated region in four individuals chosen to span the geographical distribution of Architeuthis (caught in the proximity of the Falkland Islands, Florida, South Africa and New Zealand). Reactions were undertaken with Platinum Taq DNA polymerase high fidelity (Invitrogen), in 25 µl volumes containing 1× high-fidelity PCR buffer, 0.2 mM dNTPs, 2–3 mM magnesium sulphate (primer pair-specific; see electronic supplementary material, table S2), 0.4 µM each primer and 10 units of polymerase per reaction. The thermal cycling conditions were as follows: 5 min at 94°C, 40 cycles of (30 s at 94°C, 30 s at 63°C, 1 min kb−1 at 68°C), final extension of 5 min at 68°C. Sanger sequencing of these initial amplicons, using both the original PCR primers and nested sequencing primers (see the electronic supplementary material, table S2), confirmed that the conserved duplications are a general feature of the Architeuthis mitogenome, thus subsequent mitogenome amplification was performed in such a way that the two copies of each duplicate region could be kept separate.
The mitogenome dataset was generated using several strategies, depending on the quality of the DNA in each sample. For samples where DNA preservation was sufficient to allow large (more than 4.7 kb) PCR amplicons to be generated (n = 37), mitogenomes were generated using long-range PCR coupled to Roche GS FLX or Illumina HiSeq2000 sequencing [26,29]. Briefly, each mitogenome sequenced in this way was PCR amplified using a combination of primer sets, tailored to the DNA quality of the sample, which yielded amplicons spanning the entire length of the mitogenome (see the electronic supplementary material, table S2). Following PCR amplification, amplicons for each individual were purified, quantified and pooled at equimolar concentration in two pools per individual so as to keep copies of duplicated genes in separate pools. Post pooling, the DNA was sheared to approximately 200–600 bp in size using a Bioruptor (Diagenode, Liege, Belgium), then converted into either Illumina or GS FLX libraries following the manufacturers’ guidelines. GS FLX libraries incorporated MID identifiers, and Illumina libraries were indexed through library PCR. The libraries were sequenced at the Danish National High-throughput DNA Sequencing Centre.
The quality of the DNA in the remaining five samples was not sufficient for mitogenome generation using the above method. Therefore, we additionally used the ‘target capture’ principle described by Maricic et al. [30] to generate data for these samples. In this method, the DNA extracts were directly converted into indexed Illumina sequencing libraries, then denatured and incubated with biotinylated ‘bait’ generated from long-range Architeuthis mtDNA amplicons derived from the higher-quality material. Subsequent streptavidin-based capture of the biotinylated bait, now hybridized to the mitochondrial sequences from the library, enabled target sequencing of the mitogenomes on a HiSeq2000 platform. For full details on the method, see Maricic et al. [30].
(d). Assembly and sequence verification
After sequencing, the different datasets (amplicon-FLX, amplicon-HiSeq, bait-captured) were processed, using three different approaches relevant to the technology. For amplicon-FLX mitogenomes (n = 34), sequence reads were initially sorted by MID, then assembled into contigs with the publicly available Architeuthis reference sequence (NC_011581), using the software GsMapper (Roche). These contigs were aligned manually in MEGA v. 5.04 [31] and then imported into Geneious Pro v. 5.3.6 [32] for further inspection. Because minor sequencing and assembly errors are common, especially with regard to mononucleotide repeats, all sequence differences in the alignment were verified by manually inspecting the assembly of reads for the individual sequences and making sure the contig was correct. In the cases of variable monomer length, the number was standardized according to the reference sequence, to account for the commonly accepted limitation of the GS-FLX platform with regard to accurately sequencing monomers of five or more bases in length. In this regard, we note that, as all monomers are located in protein-coding genes, if real, any length fluctuation with regard to the reference sequence would lead to frame shifts, which thus suggested they were unlikely to be real. Once the contig sequences were verified, the matching halves of each individual mitogenome were joined together to construct the complete mitogenome sequence.
For amplicon-HiSeq (n = 4) and bait-captured mitogenomes (n = 5), raw sequence reads were trimmed to remove sequencing adapters as well as adapter dimmers, using the AdapterRemoval tool [33]. Sequences with average- to low-quality stretches were also filtered out. Trimmed reads were mapped to references using BWA v. 0.5.9-r26-dev [34], and duplicate reads, as well as reads with multiple hits were removed, using SAMtools v. 0.1.18 (r982:295) [35]. Alignments were then visualized using the Geneious DNA sequence analysis package. For the five bait-captured mitogenomes, the duplicated regions were excluded, because during assembly of the sequence it was not possible to assign reads to the appropriate copy with certainty. Thus, the length of these sequences was reduced to 11 655 bp in length.
(e). Population genetic analyses
Initial data analysis indicated that the level of nucleotide variation in the alignment was too low to allow generation of a well-supported phylogenetic tree of the mitogenomes. Therefore, to visualize the relationship between the samples, a haplotype network was generated, based on the concatenated protein-coding genes from the 38 complete mitogenomes, using the median-joining method implemented in Network v. 4.61 [36]. Descriptive statistics (π, h and Hd; the neutrality index Tajima's D and the dN/dS ratio) were calculated for the whole genomes, for all the protein-coding genes combined and for each gene separately (including both copies of the duplicated genes), using DnaSP v. 5.1 [37]. A small number of the mitogenomes were incomplete, thus some of the single gene statistics were calculated based on alignments of a subset of the 44 sequences, as in each case incomplete sequences were removed. FST was also computed with DnaSP. The calculation of FST values between different locations was only possible between locations that were represented by more than one individual (Florida, Japan, Australia and New Zealand), as the calculation requires a sequence diversity index.
DnaSP was also used to generate a pairwise mismatch distribution on the data, as well as the pairwise difference plot parameter τ, and the raggedness index. To reduce the amount of noise that might result from including regions with pronounced differences in their substitution patterns, the analysis was restricted to the concatenated protein-coding genes, thus excluding the tRNAs, rRNAs and the two control regions, but including both copies of duplicated genes. The haplotype network was drawn on the same alignment for the sake of consistency.
The extremely limited fossil record of coleoid cephalopods provides a significant challenge for studies that aim to use molecular clock analysis to date past events, thus resulting in the absence of a mutation rate estimate for any species closely related to Architeuthis. Therefore, we used four different mutation rates to tentatively estimate a time of expansion, based on the pairwise difference plot parameter τ (the median) [38]. Estimates of the first two mutation rates were obtained using MEGA v. 5.04, as well as the Jukes and Cantor model of substitution, based on (i) the corrected distances between sequences, (ii) estimates for the divergence time of families within Oegopsida as calculated by Strugnell et al. [39], which provide an upper and lower bound for the time of divergence of Architeuthidae from other oegopsid families, and (iii) the phylogenetic position of A. dux between Watasenia scintillans (Family Enoploteuthidae) and Sthenoteuthis oualaniensis (Family Ommastrephidae) as determined from the mitochondrial genome tree (see the electronic supplementary material, figure S2) [40].
The time-dependency hypothesis of mutation rates [41] suggests that rates estimated from relatively ancient fossil calibrations are likely to significantly underestimate the mutation rate as relevant to questions of recent demographic history. Therefore, additional mutation rates from published studies of gastropod evolution were also used. These estimates of gastropod divergence rates range from 0.67 to 2.4 per cent per million years [42], and despite deriving from a different class of molluscs, these may provide a better estimate of the Architeuthis mutation rate than those derived from very ancient divergences.
3. Results
Thirty-seven complete and six partial mitogenome sequences were generated, representing individuals from across the range of Architeuthis (see the electronic supplementary material, table S1). Sample average sequencing depth varied from 22- to 1051-fold, and successful application of the bait-capture method developed by Maricic et al. [30] confirmed the use of this method when working with challenging degraded materials. An image showing a visualization of the alignment of all the obtained sequences as well as the reference sequence is given in electronic supplementary material, figure S3. The mitogenome data are deposited in NCBI GenBank under accession numbers KC701722–KC701764 (see the electronic supplementary material, table S1).
The level of nucleotide diversity in the sequences was extremely low, with only 181 segregating sites of the 20 331 bp-long sequence alignment. Values of the nucleotide diversity index (π) and the haplotype diversity (Hd) are given in table 1 for the alignment of entire mitogenomes, for the concatenated protein-coding genes and for the first of the two control regions (CR-A). The dN/dS value for the alignment of 38 concatenated protein-coding gene sequences (including both copies of duplicated genes) was 0.0540. A complete set of genetic diversity and neutrality statistics for all individual genes is given in the electronic supplementary material, table S3.
Table 1.
Summary genetic diversity statistics. π, nucleotide diversity; Hd, haplotype diversity.
| region | length | π | s.d. | Hd | s.d. | Tajima's D | stat. sig. |
|---|---|---|---|---|---|---|---|
| whole mitogenomes | 20 331 | 0.00066 | 0.00005 | 1 | 0.006 | −2.51828 | p < 0.001 |
| all genes | 14 976 | 0.00068 | 0.00005 | 1 | 0.006 | −2.49263 | p < 0.01 |
| control region A | 554 | 0.00166 | 0.00034 | 0.613 | 0.087 | −1.66738 | 0.10 > p > 0.05 |
The observed haplotype diversity was high at the mitogenome level, with each specimen containing a unique haplotype, as can be seen in the median-joining haplotype network (figure 1). Despite this high haplotype diversity, the nucleotide diversity statistic is very low in comparison with estimates from other marine dwelling species (table 2).
Figure 1.

Median-joining haplotype network of 38 complete giant squid mitogenomes. Geographical origin is coded by colour, matching those on the map in the electronic supplementary material, figure S6.
Table 2.
Nucleotide diversity of the mitochondrial control region and cytochrome b.
| species | common name | π | region | distribution | source |
|---|---|---|---|---|---|
| Architeuthis dux | giant squid | 0.0004 | cytochrome b | global | — |
| Dosidicus gigas | Humboldt squid | 0.0177 | cytochrome b | eastern Pacific | [43] |
| Cetorhinus maximus | basking shark | 0.0013 | control region | global | [38] |
| Architeuthis dux | giant squid | 0.0017 | control region | global | — |
| Latimeria chalumnae | coelacanth | 0.0027 | control region | east Africa | [44] |
| Coregonus spp. | European whitefish | 0.0042 | control region | northern Europe | [25] |
| Orcinus orca | killer whale | 0.0052 | control region | global | [45] |
| Sepioteuthis lessoniana | oval squid | 0.0124 | control region | Taiwan | [46] |
| Xiphias gladius | swordfish | 0.0148 | control region | global | [47] |
| Carcharodon carcharias | great white shark | 0.0203 | control region | global | [48] |
| Thunnus obesus | bigeye tuna | 0.0540 | control region | global | [49] |
The dataset surprisingly exhibits no evidence of any phylogeographic structure (figure 1). This observation is supported by the low FST estimates calculated, using the whole mitogenomes for samples from Australia, Spain, Florida, New Zealand and Japan (table 3; negative FST values are likely to reflect the imprecision of the algorithm used by the software to estimate this value, and thus should be considered zero). A limitation of FST worth taking into consideration here is when it is used with mtDNA of highly variable species, as the high variability cuts down the number of shared haplotypes between any two populations. The FST values presented here should therefore not be over-interpreted, but rather seen in the context of the other analyses.
Table 3.
Pairwise FST results for population comparisons. We only considered populations of n > 2 for the analysis.
| population 1 | n | population 2 | n | FST |
|---|---|---|---|---|
| Australia | 5 | Florida | 3 | −0.011 |
| Australia | 5 | Japan | 6 | −0.006 |
| Australia | 5 | New Zealand | 15 | −0.015 |
| Australia | 5 | Spain | 3 | −0.025 |
| Spain | 3 | Florida | 3 | −0.017 |
| Spain | 3 | Japan | 6 | 0.006 |
| Spain | 3 | New Zealand | 15 | −0.031 |
| Florida | 3 | Japan | 6 | −0.012 |
| Florida | 3 | New Zealand | 15 | 0.01 |
| Japan | 6 | New Zealand | 15 | 0.001 |
The results of a pairwise-differences analysis using the 38 complete mitogenomes can be seen in figure 2. Both the shape of the distribution and the low raggedness index (r = 0.0075) provide strong evidence that Architeuthis has undergone a population expansion. By using τ, the estimated mode of the mismatch distribution, and an estimated generation time of 3 years, we estimated the time of expansion by the relationship τ = 2ut, where u = 2μk (t is the generation time, μ the mutation rate and k is the length of the DNA fragment used for the analysis; in this case 14 976 bp). A generation time of 3 years was chosen as it lies in the mid-range of suggested maximum ages of giant squid [9,22,23]. As mentioned earlier, three potential mutation rates were explored. The corrected distance between A. dux and Sthenoteuthis oualaniensis calculated with the Jukes and Cantor substitution model was 0.235. The upper bound for the Architeuthidae split, based on the Ommastrephidae split, was 33 Ma, and the lower bound, based on the enoploteuthid family split, was 220 Ma. Thus, the two new mutation rate estimates were calculated as 0.356 per cent Myr−1 and 0.053 per cent Myr−1, respectively. These provided a time estimate of Architeuthis population expansion of between 109 248 and 728 318 years ago.
Figure 2.
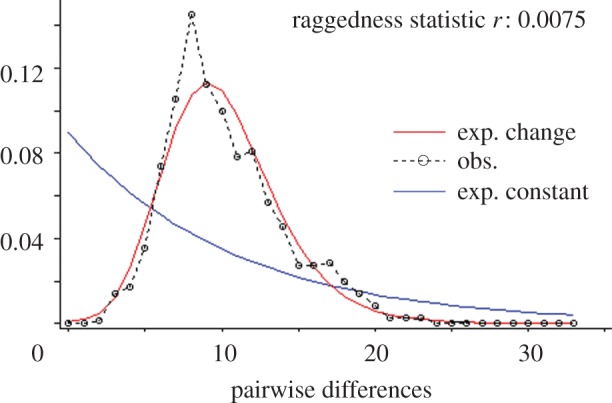
A mismatch distribution constructed from an alignment of 38 concatenated protein-coding gene sequences (including both copies of duplicated genes). The red and blue lines indicate expected shapes of the distribution after a rapid expansion and constant population size, respectively. A plot based on whole mitogenomes can be found in the electronic supplementary material, figure S4.
The calculations based on previously published gastropod mutation rates, ranging from 0.335 to 1.2 per cent Myr−1 (mutation rate is half the divergence rate) [42], yielded a more recent range for expansion time of 115 977 to 32 377 years ago.
Exploring a range of the Architeuthis generation time (which is unknown) to between 1 and 10 years, instead of 3 years, in combination with the fastest and slowest mutation rate estimates (1.2% and 0.053%, respectively), results in a wider estimated time range with an upper bound of 10 792 and a lower bound of 2 427 727 years ago.
4. Discussion
(a). Low genetic variability
The Architeuthis mitogenomes exhibit remarkably low nucleotide diversity (π), despite deriving from across its global distribution. In contrast to published records for other marine species (table 2), the diversity is lower than all except one, the basking shark Cetorhinus maximus, whose low diversity is believed due to a recent bottleneck [38]. It is notable, however, that the basking shark and coelacanth have a long generation time [50,51] and presumed small global population size, whereas Architeuthis populations are believed to be large. Architeuthis nucleotide diversity is also far lower than other squid (table 2), including 44 times lower than that of another oegopsid squid, the Humboldt squid Dosidicus gigas (based on the cytochrome b gene) [43], and seven times lower than a restricted population of oval squid Sepioteuthis lessoniana (based on the control region).
An additional curious feature of Architeuthis is that the relationship between nucleotide and haplotype diversity falls outside the 95th percentile of the positively correlated relationship between population-level estimates of haplotype (Hd) and nucleotide (π) diversity, which is reported for Cox1 by Goodall-Copestake et al. [52], with π = 0.00121 and Hd = 0.864 for one copy of Cox1, and π = 0.00073 and Hd = 0.622 for the other copy. It is interesting that even in the context of such a diverse set of animals (the study spans several phyla, including Chordata, Mollusca, Crustacea, Arachnida, Hexapoda and Porifera) Architeuthis remains an odd one out.
It is difficult to reconcile this low genetic diversity with the reasonable assumption that Architeuthis are globally distributed with relatively large population size. One explanation might be a low rate of mitochondrial DNA evolution, something observed among other marine organisms, in particular within the cnidarian class Anthozoa (sea anemones, corals and sea pens) [53], which are an order of magnitude slower than the rate observed among most other marine invertebrate species. Given that physiological and life-history traits such as metabolic rate and generation time can influence mutation rates [54], one might question whether such factors affect Architeuthis. Low metabolic rate seems unlikely, as recent data [14] demonstrate little difference between the metabolic activity of Architeuthis and that of highly active smaller squid, some of which show considerably more diversity (table 2). Long generation time is also an unlikely cause. While Architeuthis generation time remains uncertain, most published estimates based on statolith growth rings, isotope analyses and growth models suggest 1–6 years [9,22,24,55,56], and the FAO species catalogue suggests a maximum of 14 years [19], far below that needed to significantly decrease the mutation rate.
We alternatively considered whether low mutation rate might relate to the Architeuthis mitogenome duplications. Each duplication maintains near 100 per cent identity, despite their presence in all sequenced oegopsid squid mitogenomes [39], plus a non-oegopsid species Bathyteuthis abyssicola (NC_016423; indicating an origin at least 100 Ma). A low mutation rate in itself cannot explain this intraspecific sequence conservation between duplicates, as interoegopsid comparison shows that the duplicated genes in different species are indeed diverging, while somehow remaining identical within each individual—presumably by some form of gene conversion, resulting in concerted evolution of the two regions. As far as the mechanism of this concerted evolution is concerned, this study cannot present a satisfactory explanation, but it is possible that it happens via occasional recombination [57], or perhaps the duplicated sequences form stable secondary structures, which are somehow selectively beneficial, thereby causing mutations to be under negative selection. The latter, however, would be expected to lead to a decreased rate of divergence in the duplicated regions relative to the rest of the genome, which does not appear to be the case.
Alternative explanations to the idea of a low mutation rate keeping the nucleotide diversity low include a recent selective sweep, or a bottleneck coupled with expansion. While positive selection analysis using TreeSAAP [58,59] (data not shown) provides no evidence to support the former hypothesis, the ‘star-like’ shape of the network, Tajima's D, the mismatch distribution and a Bayesian skyline plot analysis (see the electronic supplementary material, figure S5) support the latter. Although application of the alternative mutation rates and a generation time of 3 years estimates the time of expansion to roughly 32 000–730 000 years ago, the validity of the mutation rates used is questionable, given the uncertainty around the parameters on which they were estimated. Nevertheless, we believe the most accurate estimate will fall closest to those calculated using the gastropod rates, owing to the effects of time-dependency rendering the other mutation rates inappropriate for such shallow-time investigations [41]. These rates also yield a younger date of expansion, fitting well with the extremely low diversity and suggesting that the causal event was quite recent.
Unfortunately, genetic data alone cannot provide a plausible explanation, whether climatic or biological, as to why this might have happened. Given that Architeuthis are cosmopolitan, with (assumed) considerable dispersal abilities, the event must have had the potential to affect the entire global population simultaneously. Given this, alternative explanations can be suggested. One such explanation, which is consistent with expansion, would be a sudden inflation of a population that was historically smaller. If the population size was originally restrained by predation and/or competition, then the cause of the inflation could have been related to a decline in the number of predator or competitor species. This would be comparable with the effect of the 1700 to late 1800s industrialized whaling, which removed almost all of their main natural predators, though this event in particular is almost certainly too recent to be the explanation. A mechanism more plausible than fisheries could be climatic effects, such as the last ice age changing the abundance or distribution of competitors such as predatory fish. The role of cephalopods in the ecosystem tends to be that of subdominant predators, which often increase radically in biomass when other species (particularly their predators and direct competitors) become depleted as a result of heavy fishing [5]. This has been observed, for example, in the sparid fishery off Sahara [5,60], the gadoid fishery in the northwest Atlantic and the trawl fishery in the Gulf of Thailand [5]. Furthermore, it has been estimated that the documented increase in tuna catches has resulted in an extra 20 million tonnes of squid in the world ocean [61]. Thus, a recent inflation is a possibility, but equally difficult to explain in terms of concrete events.
(b). Biological and taxonomic implications of the data
If the low mitochondrial diversity is indicative of variation at the nuclear genome level in Architeuthis, then the data strongly suggest that globally only a single species of Architeuthis exists, namely Architeuthis dux (Steenstrup, 1857), consistent with the suggestion of a previous study on Architeuthis beak morphometrics [17]. The genetic data also demonstrate that Architeuthis lack phylogeographic structure. This observation is consistent with several hypotheses. On the one hand, it is possible that until recently Architeuthis existed as a single, small, geographically restricted population, which has subsequently expanded globally in a non-ordered fashion (implying capability for long-distance dispersal), distributing their variation among new regional populations. This, in turn, implies either adults are nomadic (an observation inconsistent with (i) beak isotope profile data suggesting adults inhabit relatively confined areas [9] and (ii) their flexible diet [62] negating the need for adults to disperse in search of food), or dispersal happens via migrations of juveniles [63] and small pelagic paralarvae. These are capable of dispersing over long distances via currents, as is the case for many cephalopod species, as well as deep-sea foraminiferans, in which genetic analysis demonstrates similarity between Arctic and Antarctic populations of three common deep-sea species separated by distances of up to 17 000 km [64]. Other examples include surgeonfish (which show no population structure between Brazil and mid-Atlantic populations, concluded to be the result of a drifting larval stage of roughly 50–70 days duration [65]) and other cephalopods, generally large species of oceanic ommastrephids [66]. It is even hypothesized that the large size of these squids is an adaptation to living in such large current systems [67].
Ultimately, however, if Architeuthis disperse over such large areas, then it is difficult to explain why they may have been restricted to one area in the first place. Given this, we find neither scenario likely, and believe an alternative explanation is warranted. We hypothesize that a panmictic global population of Architeuthis has existed for a considerable time. This would adequately explain the lack of population structure, as it is estimated that an average of just one individual exchanged between two populations per generation will be enough to prevent genetic differentiation between them [68] and broad dispersers are, indeed, generally genetically homogeneous over large spatial scales [69]. Admittedly, this raises additional questions, including how a relatively old panmictic population has maintained such low nucleotide diversity, how the reproductive system and life cycle of this species could sustain such panmixia, and what might be the primary mode of dispersal. As discussed previously, we cannot offer a satisfactory explanation for the low diversity, and this requires future study to resolve. With regard to the biology of Architeuthis, the panmictic distribution again seems best explainable through pan-oceanic dispersal of paralarvae and juveniles, with assistance from the currents in the upper layers of the oceans. Under such a model, we hypothesize that they would prey on pelagic zooplankton and smaller animals, until reaching a sufficiently large size, after which they would descend to their closest nutrient-rich deep habitat (e.g. a continental upwelling zone [9,70]), where they would remain until maturation. The global ocean currents (see the electronic supplementary material, figure S6) powered by the thermohaline circulation, also known as the Great Ocean Conveyor, could drive the continual homogenization of the giant squid global population by transporting the juveniles.
This type of life history would be consistent with the findings from a study on δ15N and δ13C isotope profiles along giant squid beaks [9] that demonstrate ontogenetic shift in diet early in life from smaller prey of relatively low trophic status to larger prey of higher status, and fluctuations in δ13C values observed near the rostral tip, which may be associated with a greater intrinsic variability in the carbon isotope composition of relatively small prey and/or transient migratory behaviour early in life [63].
5. Conclusion
Although our results are based on a single marker, mtDNA, the sampling encompasses the known range of Architeuthis and strongly suggests that the family Architeuthidae consists of a single species of giant squid, namely Architeuthis dux (Steenstrup, 1857). If so, this species is cosmopolitan and probably has a substantial population size, yet shows unusually low levels of mitochondrial nucleotide diversity. This may be due, at least in part, to a population expansion. Architeuthis also lack discernible population structure, with no genetic differentiation between samples from regions as far apart as Florida and Japan. This suggests Architeuthis are highly migratory, probably through a pelagic paralarval stage, dispersed via the global thermohaline circulation. Full validation of our findings and interpretations will first and foremost require analyses of nuclear sequences. This would allow for distinction between demographic expansion and selective sweep, as a nuclear DNA study could incorporate several unlinked loci in the analyses. A further investigative avenue of potential interest, which arose from analysis of the selection test performed within Oegopsida, is to explore whether there are traces of positive selection between oegopsid and non-oegopsid squids in the particular genes that have been duplicated. This could account for the high degree of protein sequence similarity observed between all five oegopsid species, and possibly help explain the concerted evolution of the duplications.
Acknowledgements
The authors thank Mike Martin, Maria Ávila-Arcos, Rasmus Heller and Maanasa Raghavan for laboratory and analytical assistance, the Danish National high-throughput DNA sequencing centre for sequencing assistance, and Rute da Fonseca, Andrew Foote, Michael Møller Hansen and Rasmus Nielsen for useful discussion. The authors thank Annie Lindgren (Portland State University), Michael Vecchione (Smithsonian Institute), William Coffey and Earl Dawe (Fisheries and Oceans Canada), Jon Ablett (Natural History Museum London), Sankurie Pye (National Museums Scotland), and the National Institute of Water and Atmospheric Research, Wellington for samples used, Mote Marine Laboratory and its volunteers for their donations and time to collect and process their giant squid samples, John Field (NOAA) for provenance details on the ‘reference’ genome, and the anonymous reviewers for helpful comments, with a special thanks to Candyj. This research was supported by the Danish Basic Research Foundation ‘GeoGenetics’ grant.
References
- 1.Guerra A, González AF, Rocha F, Laria L, Garcia J. 2006. Enigmas de la ciencia: el calamar gigante. In Fundación Caja Rural de Asturiase Instituto de Investigaciones Marinas (CSIC). Luarca, Spain: CEPESMA. [Google Scholar]
- 2.Kubodera T, Mori K. 2005. First-ever observations of a live giant squid in the wild. Proc. R. Soc. B 272, 2583–2586 10.1098/rspb.2005.3158 (doi:10.1098/rspb.2005.3158) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schrope M. 2013. Giant squid filmed in its natural environment. Nature (doi:10.1038/nature.2013.12202) [Google Scholar]
- 4.Ellis R. 1998. The search for the giant squid. Guilford, CT: The Lyons Press [Google Scholar]
- 5.Roper CFE, Sweeney MJ, Nauen CE. 1984. FAO species catalogue, vol. 3. Cephalopods of the world. An annotated and illustrated catalogue of species of interest to fisheries. FAO Fish Synop. 3, 1–277 [Google Scholar]
- 6.Sweeney MJ, Roper CFE. 2001. Records of Architeuthis specimens from published reports. Washington, DC: National Museum of Natural History, Smithsonian Institution; See http://invertebrates.si.edu/cephs/archirec.pdf [Google Scholar]
- 7.Guerra Á, González ÁF, Pascual S, Dawe EG. 2011. The giant squid Architeuthis: an emblematic invertebrate that can represent concern for the conservation of marine biodiversity. Biol. Conserv. 144, 1989–1997 10.1016/j.biocon.2011.04.021 (doi:10.1016/j.biocon.2011.04.021) [DOI] [Google Scholar]
- 8.Deagle BE, Jarman SN, Pemberton D, Gales NJ. 2005. Genetic screening for prey in the gut contents from a giant squid (Architeuthis sp.). J. Hered. 96, 417–423 10.1093/jhered/esi036 (doi:10.1093/jhered/esi036) [DOI] [PubMed] [Google Scholar]
- 9.Guerra A, Rodriguez-Navarro AB, Gonzalez AF, Romanek CS, Alvarez-Lloret P, Pierce GJ. 2010. Life-history traits of the giant squid Architeuthis dux revealed from stable isotope signature recorded in beaks. ICES J. Mar. Sci. 67, 1425–1431 [Google Scholar]
- 10.Cherel Y. 2003. New records of the giant squid Architeuthis dux in the southern Indian Ocean. J. Mar. Biol. Assoc. UK 83, 1295–1296 10.1017/S0025315403008695 (doi:10.1017/S0025315403008695) [DOI] [Google Scholar]
- 11.Clarke MR. 1983. Cephalopod biomass-estimation from predation. Mem. Nat. Mus. Vict. 4, 95–107 [Google Scholar]
- 12.Seibel BA, Thuesen EV, Childress JJ. 2000. Light-limitation on predator-prey interactions: consequences for metabolism and locomotion of deep-sea cephalopods. Biol. Bull. 198, 284–298 10.2307/1542531 (doi:10.2307/1542531) [DOI] [PubMed] [Google Scholar]
- 13.Rosa R, Pereira J, Nunes ML. 2005. Biochemical composition of cephalopods with different life strategies, with special reference to a giant squid, Architeuthis sp. Mar. Biol. 146, 739–751 10.1007/s00227-004-1477-5 (doi:10.1007/s00227-004-1477-5) [DOI] [Google Scholar]
- 14.Rosa R, Seibel BA. 2010. Slow pace of life of the Antarctic colossal squid. J. Mar. Biol. Assoc. UK 90, 1375–1378 10.1017/S0025315409991494 (doi:10.1017/S0025315409991494) [DOI] [Google Scholar]
- 15.Pérez-Gándaras G, Guerra A. 1978. Nueva cita de Architeuthis (Cephalopoda: Teuthoidea): descripción y alimentación. Invest. Pesquera 42, 401–414 [Google Scholar]
- 16.Hoving HJT, Roeleveld MAC, Lipinski MR, Melo Y. 2004. Reproductive system of the giant squid Architeuthis in South African waters. J. Zool. (Lond.) 264, 153–169 10.1017/S0952836904005710 (doi:10.1017/S0952836904005710) [DOI] [Google Scholar]
- 17.Roeleveld MAC. 2000. Giant squid beaks: implications for systematics. J. Mar. Biol. Assoc. UK 80, 185–187 10.1017/S0025315499001769 (doi:10.1017/S0025315499001769) [DOI] [Google Scholar]
- 18.Förch EC. 1998. The marine fauna of New Zealand: Cephalopoda: Oegopsida: Architeuthidae (giant squid). Natl. Inst. Water Atmos. Res. Biodivers. Memoir 110, 113 [Google Scholar]
- 19.Jereb P, Roper CFE. 2010. Cephalopods of the world. An annotated and illustrated catalogue of cephalopod species known to date, vol. 2. Myopsid and oegopsid squids. FAO Species Catalogue Fishery Purposes 4, 605 [Google Scholar]
- 20.Nesis KN. 1982. Cephalopods of the world. Neptune, NJ: TFH Publisher [Google Scholar]
- 21.Clarke MR. 1966. A review of the systematics and ecology of oceanic squids. Adv. Mar. Biol. 4, 91–300 10.1016/S0065-2881(08)60314-4 (doi:10.1016/S0065-2881(08)60314-4) [DOI] [Google Scholar]
- 22.Arkhipkin IA. 2004. Diversity in growth and longevity in shortlived animals: squid of the suborder Oegopsina. J. Mar. Freshw. Res. 55, 341–355 10.1071/MF03202 (doi:10.1071/MF03202) [DOI] [Google Scholar]
- 23.Jackson GD, Lu CC, Dunning M. 1991. Growth rings within the statolith microsculpture of the giant squid Architeuthis. The Veliger 34, 331–334 [Google Scholar]
- 24.Grist EPM, Jackson G. 2007. How long would it take to become a giant squid? Rev. Fish. Biol. Fisheries 17, 385–399 10.1007/s11160-007-9046-x (doi:10.1007/s11160-007-9046-x) [DOI] [Google Scholar]
- 25.Jacobsen MW, Handen MM, Orlando L, Bekkevold D, Bernatchez L, Willerslev E, Gilbert MTP. 2012. Mitogenome sequencing reveals shallow evolutionary histories and recent divergence time between morphologically and ecologically distinct lineages of European whitefish (Coregonus spp.). Mol. Ecol. 21, 2727–2742 10.1111/j.1365-294X.2012.05561.x (doi:10.1111/j.1365-294X.2012.05561.x) [DOI] [PubMed] [Google Scholar]
- 26.Morin PA, et al. 2010. Complete mitochondrial genome phylogeographic analysis of killer whales (Orcinus orca) indicates multiple species. Genome Res. 20, 908–916 10.1101/gr.102954.109 (doi:10.1101/gr.102954.109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Voight JR, Pörtner HO, O'Dor RK. 1994. A review of ammonia-mediated buoancy in squids (Cephalopoda: Teuthoidea). Mar. Freshw. Behav. Physiol. 25, 193–203 10.1080/10236249409378917 (doi:10.1080/10236249409378917) [DOI] [Google Scholar]
- 28.Yokobori S, Fukuda N, Nakamura M, Aoyama T, Oshima T. 2004. Long-term conservation of six duplicated structural genes in cephalopod mitochondrial genomes. Mol. Biol. Evol. 21, 2034–2046 10.1093/molbev/msh227 (doi:10.1093/molbev/msh227) [DOI] [PubMed] [Google Scholar]
- 29.Vilstrup JT, et al. 2011. Mitogenomic phylogenetic analyses of the Delphinidae with an emphasis on the Globicephalinae. BMC Evol. Biol. 11, 65. 10.1186/1471-2148-11-65 (doi:10.1186/1471-2148-11-65) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maricic T, Whitten M, Pääbo S. 2010. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS ONE 5, e14004. 10.1371/journal.pone.0014004 (doi:10.1371/journal.pone.0014004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 10.1093/molbev/msr121 (doi:10.1093/molbev/msr121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drummond AJ, et al. 2010. Geneious v.v 5.3. See http://www.geneious.com. [Google Scholar]
- 33.Lindgreen S. 2012. AdapterRemoval: easy cleaning of next-generation sequencing reads. BMC Res. Notes 5, 337. 10.1186/1756-0500-5-337 (doi:10.1186/1756-0500-5-337) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 10.1093/bioinformatics/btp324 (doi:10.1093/bioinformatics/btp324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 10.1093/bioinformatics/btp352 (doi:10.1093/bioinformatics/btp352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bandelt HJ, Forster P, Sykes BC, Richards MB. 1995. Mitochondrial portraits of human populations using median networks. Genetics 141, 743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Librado P, Rozas J. 2009. DnaSP v. 5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 10.1093/bioinformatics/btp187 (doi:10.1093/bioinformatics/btp187) [DOI] [PubMed] [Google Scholar]
- 38.Hoelzel AR, Shivji MS, Magnussen J, Francis MP. 2006. Low worldwide genetic diversity in the basking shark (Cetorhinus maximus). Biol. Lett. 2, 639–642 10.1098/rsbl.2006.0513 (doi:10.1098/rsbl.2006.0513) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strugnell J, Jackson J, Drummond AJ, Cooper A. 2006. Divergence time estimates for major cephalopod groups: evidence from multiple genes. Cladistics 22, 89–96 10.1111/j.1096-0031.2006.00086.x (doi:10.1111/j.1096-0031.2006.00086.x) [DOI] [PubMed] [Google Scholar]
- 40.Allcock AL, Cooke IR, Strugnell JM. 2011. What can the mitochondrial genome reveal about higher-level phylogeny of the molluscan class Cephalopoda? Zool. J. Linn. Soc. 161, 573–586 10.1111/j.1096-3642.2010.00656.x (doi:10.1111/j.1096-3642.2010.00656.x) [DOI] [Google Scholar]
- 41.Ho SY, Larson G. 2006. Molecular clocks: when times are a-changin’. Trends Genet. 22, 79–83 10.1016/j.tig.2005.11.006 (doi:10.1016/j.tig.2005.11.006) [DOI] [PubMed] [Google Scholar]
- 42.Marko PB. 2002. Fossil calibration of molecular clocks and the divergence times of geminate species pairs separated by the Isthmus of Panama. Mol. Biol. Evol. 19, 2005–2021 10.1093/oxfordjournals.molbev.a004024 (doi:10.1093/oxfordjournals.molbev.a004024) [DOI] [PubMed] [Google Scholar]
- 43.Sandoval-Castellanos E, Uribe-Alcocer M, Diaz-Jaimes P. 2010. Population genetic structure of the Humboldt squid (Dosidicus gigas d'Orbigny, 1835) inferred by mitochondrial DNA analysis. J. Exp. Mar. Biol. Ecol. 385, 73–78 10.1016/j.jembe.2009.12.015 (doi:10.1016/j.jembe.2009.12.015) [DOI] [Google Scholar]
- 44.Lampert KP, Fricke H, Hissmann K, Schauer J, Blassmann K, Ngatunga BP, Schartl M. 2012. Population divergence in East African coelacanths. Curr. Biol. 22, R439–R440 10.1016/j.cub.2012.04.053 (doi:10.1016/j.cub.2012.04.053) [DOI] [PubMed] [Google Scholar]
- 45.Hoelzel AR, Natoli A, Dahlheim ME, Olavarria C, Baird RW, Black NA. 2002. Low worldwide genetic diversity in the killer whale (Orcinus orca): implications for demographic history. Proc. R. Soc. Lond. B 269, 1467–1473 10.1098/rspb.2002.2033 (doi:10.1098/rspb.2002.2033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aoki M, Imai H, Naruse T, Ikeda Y. 2008. Low genetic diversity of oval squid, Sepioteuthis cf. lessoniana (Cephalopoda: Loliginidae), in Japanese waters inferred from a mitochondrial DNA non-coding region. Pacific Sci. 62, 403–411 10.2984/1534-6188(2008)62[403:LGDOOS]2.0.CO;2 (doi:10.2984/1534-6188(2008)62[403:LGDOOS]2.0.CO;2) [DOI] [Google Scholar]
- 47.Lu C-L, Chen CA, Hui C-F, Tzeng T-D, Yeh S-Y. 2006. Population genetic structure of the swordfish, Xiphias gladius (Linnaeus, 1758), in the Indian Ocean and west Pacific inferred from the complete DNA sequence of the mitochondrial control region. Zool. Stud. 45, 269–279 [Google Scholar]
- 48.Pardini AT, et al. 2001. Sex-biased dispersal of great white sharks. Nature 412, 139–140 10.1038/35084125 (doi:10.1038/35084125) [DOI] [PubMed] [Google Scholar]
- 49.Martinez P, Gonzalez EG, Castilho R, Zardoya R. 2006. Genetic diversity and historical demography of Atlantic bigeye tuna (Thunnus obesus). Mol. Phylogenet. Evol. 39, 404–416 10.1016/j.ympev.2005.07.022 (doi:10.1016/j.ympev.2005.07.022) [DOI] [PubMed] [Google Scholar]
- 50.Pauly D. 1997. Growth and mortality of the basking shark Cetorhinus maximus and their implications for management of whale sharks Rhincodon typus. In Proc. Int. Seminar and Workshop, Sabah, Malaysia, July 1997 Gland, Switzerland: IUCN [Google Scholar]
- 51.Nikaido M, et al. 2011. Genetically distinct coelacanth population off the northern Tanzanian coast. Proc. Natl Acad. Sci. USA 108, 18 009–18 013 10.1073/pnas.1115675108 (doi:10.1073/pnas.1115675108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goodall-Copestake WP, Tarling GA, Murphy EJ. 2012. On the comparison of population-level estimates of haplotype and nucleotide diversity: a case study using the gene cox1 in animals. Heredity 109, 50–56 10.1038/hdy.2012.12 (doi:10.1038/hdy.2012.12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shearer TL, Van Oppen MJ, Romano SL, Worheide G. 2002. Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Mol. Ecol. 11, 2475–2487 10.1046/j.1365-294X.2002.01652.x (doi:10.1046/j.1365-294X.2002.01652.x) [DOI] [PubMed] [Google Scholar]
- 54.Martin AP, Palumbi SR. 1993. Body size, metabolic rate, generation time, and the molecular clock. Proc. Natl Acad. Sci. USA 90, 4087–4091 10.1073/pnas.90.9.4087 (doi:10.1073/pnas.90.9.4087) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lordan C, Collins MA, Perales-Raya C. 1998. Observations on morphology, age and diet of three Architeuthis caught off the west coast of ireland in 1995. J. Mar. Biol. Assoc. UK 78, 903–917 10.1017/S0025315400044866 (doi:10.1017/S0025315400044866) [DOI] [Google Scholar]
- 56.Forsythe JW, Van Heukelem WF. 1987. Growth. In Cephalopod life cycles, vol. 2 (ed. Boyle PR.), pp. 135–156 Orlando, FL: Academic Press [Google Scholar]
- 57.Rokas A, Ladoukakis E, Zouros E. 2003. Animal mitochondrial DNA recombination revisited. Trends Ecol. Evol. 18, 411–417 10.1016/S0169-5347(03)00125-3 (doi:10.1016/S0169-5347(03)00125-3) [DOI] [Google Scholar]
- 58.Woolley S, Johnson J, Smith MJ, Crandall KA, McClellan DA. 2003. TreeSAAP: selection on amino acid properties using phylogenetic trees. Bioinformatics 19, 671–672 10.1093/bioinformatics/btg043 (doi:10.1093/bioinformatics/btg043) [DOI] [PubMed] [Google Scholar]
- 59.Foote AD, Morin PA, Durban JW, Pitman RL, Wade P, Willerslev E, Gilbert MT, da Fonseca RR. 2011. Positive selection on the killer whale mitogenome. Biol. Lett. 7, 116–118 10.1098/rsbl.2010.0638 (doi:10.1098/rsbl.2010.0638) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pereiro JA, Bravo de Laguna J. 1979. Dinamica de la poblacion y evaluacion de los recursos del pulpo del Atlantico centro-oriental. Boletin del Instituto Espanol de Oceanografia 5, 71–105 [Google Scholar]
- 61.Caddy JF, Rodhouse PG. 1998. Cephalopod and groundfish landings: evidence for ecological change in global fisheries? Rev. Fish. Biol. Fisheries 8, 431–444 10.1023/A:1008807129366 (doi:10.1023/A:1008807129366) [DOI] [Google Scholar]
- 62.Bolstad KS, O'Shea S. 2004. Gut contents of a giant squid Architeuthis dux (Cephalopoda: Oegopsida) from New Zealand waters. N.Z. J. Zool. 31, 15–31 10.1080/03014223.2004.9518354 (doi:10.1080/03014223.2004.9518354) [DOI] [Google Scholar]
- 63.Cherel Y, Hobson KA. 2005. Stable isotopes, beaks and predators: a new tool to study the trophic ecology of cephalopods, including giant and colossal squids. Proc. R. Soc. B 272, 1601–1607 10.1098/rspb.2005.3115 (doi:10.1098/rspb.2005.3115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pawlowski J, Fahrni J, Lecroq B, Longet D, Cornelius N, Excoffier L, Cedhagen T, Gooday AJ. 2007. Bipolar gene flow in deep-sea benthic foraminifera. Mol. Ecol. 16, 4089–4096 10.1111/j.1365-294X.2007.03465.x (doi:10.1111/j.1365-294X.2007.03465.x) [DOI] [PubMed] [Google Scholar]
- 65.Rocha LA, Bass AL, Robertson DR, Bowen BW. 2002. Adult habitat preferences, larval dispersal, and the comparative phylogeography of three Atlantic surgeonfishes (Teleostei: Acanthuridae). Mol. Ecol. 11, 243–252 10.1046/j.0962-1083.2001.01431.x (doi:10.1046/j.0962-1083.2001.01431.x) [DOI] [PubMed] [Google Scholar]
- 66.Semmens JM, et al. 2007. Approaches to resolving cephalopod movement and migration patterns. Rev. Fish. Biol. Fisheries 17, 401–423 10.1007/s11160-007-9048-8 (doi:10.1007/s11160-007-9048-8) [DOI] [Google Scholar]
- 67.O'Dor RK, Webber DM. 1986. The constraints on cephalopods: why squid aren't fish. Can. J. Zool. 64, 1591–1605 10.1139/z86-241 (doi:10.1139/z86-241) [DOI] [Google Scholar]
- 68.Slatkin M. 1987. Gene flow and the geographic structure of natural populations. Science 236, 787–792 10.1126/science.3576198 (doi:10.1126/science.3576198) [DOI] [PubMed] [Google Scholar]
- 69.Taylor MS, Hellberg ME. 2003. Genetic evidence for local retention of pelagic larvae in a Caribbean reef fish. Science 299, 107–109 10.1126/science.1079365 (doi:10.1126/science.1079365) [DOI] [PubMed] [Google Scholar]
- 70.Guerra A, Gonzalez AF, Rocha F. 2004. A review of the records of giant squid in the north-eastern Atlantic and severe injuries in Architeuthis dux stranded after acoustic explorations. ICES CM 2004/CC:29. Singapore: ICES [Google Scholar]