

Abstract
Childhood epilepsy continues to be intractable in more than 25% of patients diagnosed with epilepsy. The introduction of new anti-epileptic drugs (AEDs) provides more options for treatment of children with epilepsy. We review the safety and tolerability of seven new AEDs (levetiracetam, lamotrigine, oxcarbazepine, rufinamide, topiramate, vigabatrin and zonisamide) focusing on their side effect profiles and safety in children and adolescents. Many considerations that are specific for children such as the impact of AEDs on the developing brain are not addressed during the development of new AEDs. They are usually approved as adjunctive therapies based upon clinical trials involving adult patients with partial epilepsy. However, 2 of the AEDs reviewed here (rufinamide and vigabatrin) have FDA approval in the U.S. for specific Pediatric epilepsy syndromes, which are discussed below. The Pediatrician or Neurologists decision on the use of a new AED is an evolutionary process largely dependent on the patient characteristics, personal/peer experiences and literature about efficacy and safety profiles of these medications. Evidence based guidelines are limited due to a lack of randomized controlled trials involving pediatric patients for many of these new AEDs.
Keywords: new AEDs, pediatric epilepsy, safety, tolerability
Introduction
About 70% of patients with partial or generalized epilepsy have their seizures controlled with anti-epileptic medications. The remaining 25%–30% of patients continue to have seizures that are intractable to AEDs. In the pediatric population the ramifications of uncontrolled seizures include impairments in the child’s development, behavioral problems and significant patient and parental anxiety that frequently lead to restriction of the child activities and independence. The introduction of newer anti-epileptic medications over the past 10–15 years has increased the treatment options for children and adolescents with refractory epilepsy. Although these new anti-epileptic medications have similar efficacy when compared to older/conventional AEDs, the tolerability and safety profile for these medication is arguably better and many have gained widespread usage as mono-therapy and adjunctive treatment of childhood epilepsies.
Levetiracetam
It is a single enantiomer, structurally similar to the prototypical drug piracetam. The chemical structure is (S)-a-ethyl-2-oxo-pyrrolidine-acetamide.1
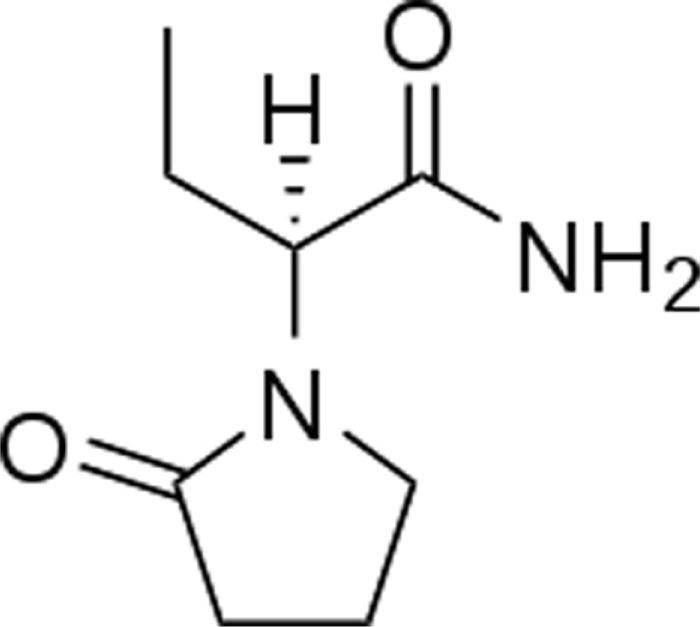
The major metabolic pathway is the enzymatic hydrolysis of the acetamide group and is not dependent on any liver cytochrome P450 isoenzymes. The mechanism of excretion is glomerular filtration. Plasma half-life of levetiracetam across studies is approximately 6–8 hours. Steady state is achieved after 2 days of twice-daily dosing. Levetiracetam and its major metabolite are less than 10% bound to plasma proteins. Clinically significant interactions with other drugs through competition for protein binding sites are therefore unlikely.
The mechanism of action of levetiracetam (LEV) is incompletely understood and does not seem to involve the known mechanisms of neurotransmission. A synaptic vesicle protein called SV2A has recently been identified as the principal target of LEV and it could mediate its effects through synaptic release mechanisms.2 Levetiracetam is an antiepileptic drug marketed in the US since 2000. In the pediatric population Levetiracetam is approved for treatment of myoclonic seizures in JME. However, there is growing evidence and widespread use of levetiracetam for a broad spectrum of pediatric epilepsies.
LEV is well tolerated and most of its adverse effects are benign: somnolence, anorexia, and tiredness. The overall incidence of levetiracetam-induced side effects ranges from 26% to 51%.3 The major adverse effect leading to a discontinuation of LEV is behavioral changes, including hostility, emotional liability, and psychotic behavior.4 These signs usually manifest in patients with various degrees of cognitive delay. According to Kugler et al,5 the incidence of behavioral disturbances is larger in children than in adults because pre-existing behavioral problems are common in children with drug-resistant epilepsy.
The studies summarized below help elucidate the tolerability profile of LEV.
Schiemann-Delgado et al6 conducted a multi-center, open-label, non-comparative 48-week extension study of adjunctive levetiracetam (mean dose of 50.2 mg/kg/d) to assess cognition and behavior in 103 children aged 4–16 years with partial-onset seizures. They found that adjunctive levetiracetam was well tolerated and the most frequently reported central nervous system-related treatment-emergent adverse events were headache (24.3%), aggression (7.8%), and irritability (7.8%). Of the patients, 4.9% patients discontinued the medication because of treatment-emergent adverse events.
Kossoff et al7 described four pediatric cases of epilepsy (ages ranged from 5 to 17 years) with levetiracetam induced psychosis. These children developed delusions and auditory or visual hallucinations within two days to three months of levetiracetam therapy. The initial dosage of levetiracetam used ranged from 15 to 25 mg/kg/day with a plan to increase the dosage in two weeks. There was no prior history of psychosis in these children. However, all of the children had a history of cognitive deficits, and some of them also had mild behavioral issues. In all of these children, psychosis was reversible within days of termination of levetiracetam therapy.
Studying a series of 155 children receiving levetiracetam, Gustafson et al8 distinguished their cases on the basis of the preexistence or absence of behavioral or emotional disturbances. Of the 63 children with previous problems, the behavior of 18 became worse, 25 had no change but 20 children improved. Of the 52 children without previous problems, 42 were unchanged, 5 were better and only 5 developed behavioral problems after LEV was started. 30% of those developing problems in this group had a history of behavioral problems with other antiepileptic treatments.
With regards to pharmacokinetics, a study by Dahlin et al9 found that LEV clearance differed significantly between age groups, younger children had a 1.7 fold higher clearance and children on enzyme inducers exhibited 1.3 fold higher clearances compared to those on mono-therapy and non-enzyme inducers. They did not find any significant drug interactions with lamotrigine, valproate, topiramate, or clonazepam.
Pyridoxine is often used to counteract the behavioral side effects of LEV in children. Miller,10 starting from a chance observation, was able to control the behavioral disturbances caused by levetiracetam completely, in 5 of 6 children aged between 2 and 10 years, by administering pyridoxine at an average dose of 7 mg/kg/day. In a study to examine the use of pyridoxine, Major et al11 analyzed 42 pediatric patients who had been treated with LEV and pyridoxine. Twenty-two patients started pyridoxine after being on LEV, due to behavioral side effects and significant behavioral improvement was observed in nine (41%), no effect in eight (36%), deterioration in four (18%), and an uncertain effect in one. The effects of pyridoxine supplementation were observed during the first week of its introduction. Evidence from basic sciences explaining the beneficial effect of pyridoxine on patients on LEV therapy is currently lacking.
Lamotrigine
It is an AED of the phenyltriazine class. Its chemical name is 3, 5-diamino-6-(2, 3-dichlorophenyl)-as-triazine.12
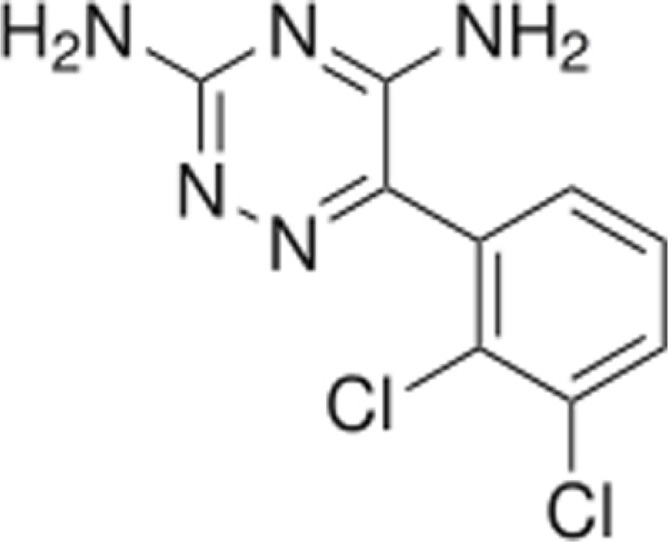
Lamotrigine is metabolized predominantly by glucuronic acid conjugation. The elimination half-life for children aged 10 months- 5 years ranges from 7 hours (patients on concomitant enzyme inducing AEDs) to 45 hours (patients on concomitant sodium valproate). The half-life for children ages 5–11 years ranges from 7–66 hours. Lamotrigine is not highly bound to plasma proteins and therefore clinically significant interactions with other drugs through competition for protein binding sites are unlikely. Lamotrigine is excreted primarily in the urine as the inactive glucuronide metabolite.
The mechanism of action of lamotrigine is related to inactivation of voltage-dependent sodium channels and inhibition of the release of excitatory neurotransmitters such as glutamate and aspartate. It was initially approved in the US in 1994 and in 1998 it received approval for use as adjunctive treatment of Lennox-Gastaut syndrome. In 2003 it was approved for use as adjunctive therapy for partial seizures in pediatric patients as young as 2 years of age.
Lamotrigine (LTG) is effective for the treatment of refractory partial and generalized epilepsy.13 It can also be used as first line therapy for childhood absence seizures14 and as add on therapy for Lennox-Gastaut syndrome. Studies have demonstrated LTG as efficacious as an adjunct AED as well as mono-therapy. In addition, it is also indicated for maintenance treatment of bipolar 1 disorder. The common adverse events are neurological, gastrointestinal and dermatological. Neuro-cognitive adverse effects include insomnia, drowsiness, dizziness, headache, somnolence, diplopia and ataxia. Rash can be seen in 10%–12% of the patients.15 Serious allergic side effects such as erythema multiforme, Steven-Johnson syndrome and toxic epidermal necrolysis occur rarely. The incidence of serious rashes including Stevens-Johnson syndrome is 0.8%–1% in pediatric patients receiving lamotrigine as adjunctive therapy for partial epilepsy as compared to 0.3% in adults.15,16
The studies summarized below illustrate the safety and tolerability profile of LTG in children and adolescents.
Valencia et al17 undertook a retrospective review of children and adolescents treated with LTG. 72 children were identified with a mean age of onset of epilepsy at 5.7 years (0–16 years). It was used as first line mono-therapy in 26.5% of the patients and as second line mono-therapy in 73.6% of the patients. Mean dose of LTG was 5.5 mg/kg/day. Mean follow up period was 33 months (3 weeks to 11 years). The most common AE was rash 6.9%. Six patients (8.3%) discontinued LTG because of side effects.
Pinea-Garza et al18 studied the tolerability and efficacy of LTG in children age 2 years and less. It was a randomized, double blind, placebo-controlled study that enrolled 204 children with partial seizures. The most common AE in patients was pyrexia (45%), upper respiratory tract infection (28%) and ear infection (22%). 5% of patients developed irritability. No cases of serious rash were reported. According to this prospective investigation, LTG was well tolerated in children younger than 2 year of age.
Aurich-Berrera et al19 compared the AEs in children and adults using post-marketing pharmacologic data. The cohort included 2457 children. Rash and Steven-Johnson were more commonly reported in children and confusion was more common in adults. A higher proportion of children (45%) stopped treatment due to lack of effectiveness.
Neurobehavioral side effects are commonly described with AEDs, however these are less common with LTG. Cardenas et al20 described 9 children average age 5 years, who developed neurobehavioral disturbances while on LTG. All 9 patients developed hyperactivity and agitation, 5 patients had violent and self-injurious behavior and 2 had insomnia. One boy developed volatile mood, threatening visual and auditory hallucinations and insomnia. The dose of LTG ranged from 0.7 to 14 mg/kg per day. All patients had dramatic improvement and/or resolution of symptoms following discontinuation or reduction of LTG.
Anticonvulsant hypersensitivity reaction although uncommon is a potentially life-threatening delayed hypersensitivity reaction. These have been described with LTG in adults and children. Ferguson et al21 described a case of an 11 year-old girl who developed fever, rash and multi-organ failure 2 weeks after initiating a transition from sodium valproate to lamotrigine therapy. LTG was discontinued and she required supportive treatment in the ICU before she recovered fully from organ failure and to baseline neurologic function. Dreesman et al22 described a similar case of a severe hypersensitivity reaction in a 6-year-old boy who presented with a triad of fever, rash and multiorgan involvement which improved 48 hours after LTG was discontinued.
Veerapandiyan et al23 reported 4 patients who developed oculogyric crisis secondary to LTG toxicity. These patients had resolution of the crisis after dose reduction. Mean plasma concentration of LTG was 15.5 microgram/ml with a mean dose of 16 mg/kg per day.
Tengstrand et al24 analyzed the number of individual case safety reports in WHO program for International Drug monitoring. LTG is suspected to be involved in alopecia in 337 patients reported from 19 countries. The age ranged between 5 months and 84 years.
Yun et al25 studied the acoustic effects of Lamotrigine in children using LTG. They picked 52 children who were recently started on LTG. They were assessed using standard speech test through a Computerized Speech Lab applied before the beginning of therapy with lamotrigine and 2 months after dosage had been stabilized. They concluded that LTG was safe for acoustic function in children.
Oxcarbazepine
Oxcarbazepine is 10, 11-dihydro-10-oxo-5H-dibenzazepine5-carboxamide.26
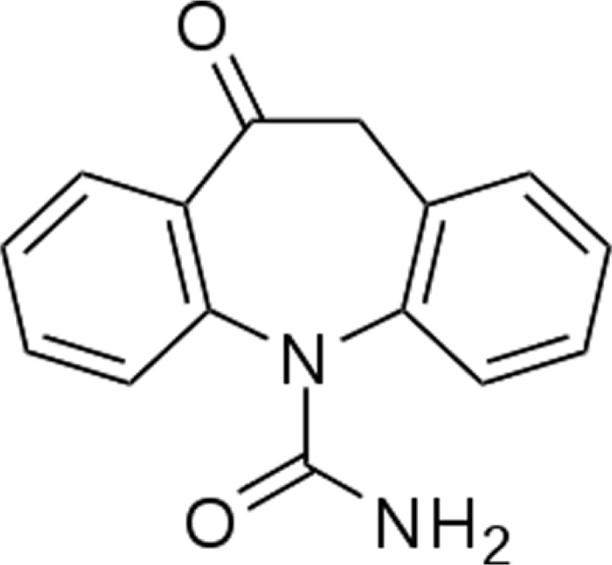
Oxcarbazepine is a prodrug, which is activated to its pharmacologically active 10-monohydroxy metabolite in the liver. The half-life of the parent drug is about two hours, while the half-life of the 10-monohydroxy metabolite is about nine hours, and this metabolite is responsible for most of the antiepileptic activity. Approximately 40% of the 10-monohydroxy metabolite is bound to serum proteins, predominantly to albumin. It is metabolized further by conjugation with glucuronic acid. It is cleared from the body in the form of metabolites, which are predominantly excreted by the kidneys.26
OXC is a structural analog of carbamazepine with a ketone in place of the carbon-carbon double-bond on the dibenzazepine ring. OXC follows a different metabolic pathway, resulting in several clinical advantages over carbamazepine, including absence of auto-induction, much less pronounced and more selective induction of the P450 enzyme system, and absence of interaction with agents, such as erythromycin, that result in excessive accumulation of carbamazepine.27
Oxcarbazepine (OXC), a homologue of carbamazepine, is a prodrug, and its 10-monohydroxy metabolite exerts an anticonvulsant effect by blocking sodium channels.28 Although it has similarities to Carbamazepine (CBZ) in its structure, efficacy, and adverse effect profiles, it has a pharmacokinetic difference in that there is no enzymatic autoinduction in OXC.28 Children younger than 8 years have clearance rates 30%–40% higher than those in older children.
It was approved in US in 2000.
Dizziness, diplopia, nausea, and ataxia are well-known adverse effects of OXC, but show more benign profiles in children.29 About 25%–33% of patients with a hypersensitivity reaction to CBZ also have an allergic reaction to OXC, a “cross-hypersensitivity reaction”. Hyponatremia is another well-known adverse effect of OXC, and its incidence is estimated to be 0.4%–1%. Sodium concentrations return to normal with dose reduction, discontinuation of the drug, or fluid restriction.30
The studies summarized below help clarify the tolerability profile of OXC
Belousova et al31 conducted a prospective non-randomized non-controlled multicenter trial that included 254 children, aged 11 months to 18 years (mean age 9.3 ± 4.5 years), with predominantly focal forms of epilepsy treated with OXC. The observation period was 31 weeks. Adverse effects were observed in 11.2% of patients but in 40% of these cases they seemed to not be related to the drug. The adverse effects were mild to moderate in severity.
2.5% and 10% of patients withdrew from well controlled pediatric trials of oxcarbazepine mono-therapy and adjunctive therapy and generally oxcarbazepine was well tolerated during mono-therapy and adjunctive therapy.32
Cansu et al33 looked at whether treatment with OXC causes weight gain in children and they concluded that OXC therapy causes neither weight change nor alterations in serum glucose, insulin, cortisol, leptin, NPY, galanin and ghrelin levels in children with epilepsy.
Tardive dyskinesia has been reported at a dose of 30 mg/kg/day in a young girl 10 days after OXC therapy was initiated.34 Her symptoms improved with diazepam and difenhydramine, 3 days after OXC was stopped.
A case report by Santucci et al35 of a 10 year old boy with intractable epilepsy who suffered OXC toxicity induced by drug-drug interaction with clarithromycin suggests that this could be explained by an increase in brain OXC concentrations due to inhibition of the blood brain barrier efflux protein, P-glycoprotein.
Rufinamide
Rufinamide is a triazole derivative.36 The chemical name is 1-[(2, 6-difluorophenyl) methyl]-1H-1, 2, 3-triazole-4 carboxamide.37
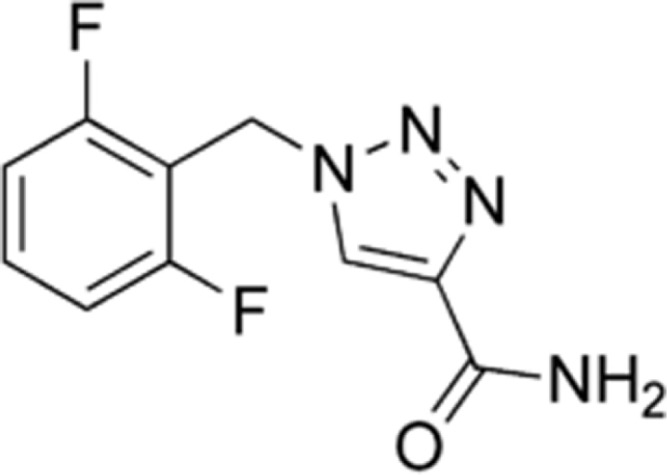
Rufinamide is extensively metabolized but has no active metabolites.
The primary biotransformation pathway is carboxyl esterase mediated hydrolysis of the carboxamide group. There is no involvement of oxidizing cytochrome P450 enzymes or glutathione in the biotransformation process. Plasma half-life of rufinamide is approximately 6–10 hours. Only a small fraction of rufinamide is bound to human serum proteins giving little risk of displacement drug-drug interactions. Renal excretion is the predominant route of elimination.37
The suspected mechanism of action is limitation of sodium-dependent action potentials, leading to a membrane stabilizing effect. It is FDA approved in the US in 2008 as adjunctive therapy for seizures associated with Lennox- Gastaut Syndrome in patients aged 4 years and older. Rufinamide is extensively metabolized in the liver by non-CYP450 enzymes with an elimination half-life of 8–12 hours. The most common adverse effects are somnolence, fatigue, dizziness, diplopia, nausea and ataxia. Rufinamide has shown promise as adjunctive treatment for Lennox-Gastaut syndrome (in particular for drop attacks) and may have some role in localization related epilepsies as well.
The safety and tolerability of rufinamide is summarized in the studies below
A recent, pooled analysis of seven clinical studies by Wheless et al38 studied the safety and tolerability of rufinamide in children with epilepsy. The data contained 212 rufinamide-treated (age range 3–16 years) and 197 placebo patients (age range 4–17 years) in the double-blind studies. 391 patients received rufinamide in the double-blind and/or open label extensions. The median dose of rufinamide was around 41 mg/kg/day and mean duration of exposure was 3 months in the double blind trial and 12–24 months in patients who were in double blind trial with open label extension. The most common AEs were somnolence (17.0% rufinamide, 8% in placebo group), vomiting (16.5% rufinamide, 7.1% placebo). No psychiatric AEs were reported with an incidence of >10%. Dizziness was reported more frequently in adolescents (12% of adolescent compared to 4.2% in children <12 years old). These AEs were similar in both patient populations. There were 5 cases of drug hypersensitivity syndrome identified retrospectively, and all the patients recovered quickly after discontinuation of rufinamide. AEs lead to discontinuation of rufinamide in 7.1% patients in double-blind population and 12.55% in the double blind plus open label extension. They also assessed laboratory changes and found that changes in thyroid, liver functions were not clinically significant between participants getting rufinamide and placebo. This analysis suggested a favorable safety and tolerability profile of rufinamide for children with intractable epilepsy.
Wier et al39 reviewed rufinamide in 138 children (average age was 12 years), as an adjunctive therapy (with an initial dosage of 10 mg/kg/day up to a target dosage of 45 mg/kg/day) in patients with Lennox-Gastaut syndrome. Rufinamide was well tolerated, with the most common adverse effects being dizziness, fatigue, nausea, vomiting, diplopia, and somnolence. They concluded that rufinamide as adjunctive therapy in the management of Lennox-Gastaut syndrome is well tolerated.
Kluger et al40 reported the most common side effects of rufinamide from a pooled safety database evaluating short and long term therapy. Headache (22.9 and 29.5%), dizziness (15.5% and 22.5%) and fatigue (13.6%and 17.7%) were reported in both groups. In the group receiving short-term therapy somnolence (11.8%) and nausea (11.4%) was also reported.
Glauser et al41 conducted a double-blind, randomized, placebo-controlled trial, for patients ranging in age from 4 to 30 years. 138 patients with Lennox-Gastaut syndrome were randomized to either rufinamide or placebo. AEs were reported in around 10% of patients receiving rufinamide. The common AEs were somnolence (24.3% with rufinamide vs. 12.5% with placebo) and vomiting (21.6% vs. 6.3%).
Rufinamide has been used in other childhood epileptic encephalopathy syndromes like Dravet syndrome. Mueller et al42 evaluated the efficacy and tolerability of rufinamide in patients with Dravet syndrome. This was a retrospective European multicenter study. The retention rate was 45% after 6 months, 15% after 18 months, and 15% after 34 months. Rufinamide treatment was stopped due to seizure aggravation in about 30% of the patients and side effects in 10% of the patients. Therefore, the authors state that rufinamide does not seem to be a suitable option for long-term treatment in patients with Dravet syndrome.
Topiramate
Topiramate is a sulfamate-substituted monosaccharide. Topiramate is designated chemically as 2, 3:4, 5-Di-O-isopropylidene-b-D-fructopyranose sulfamate.43
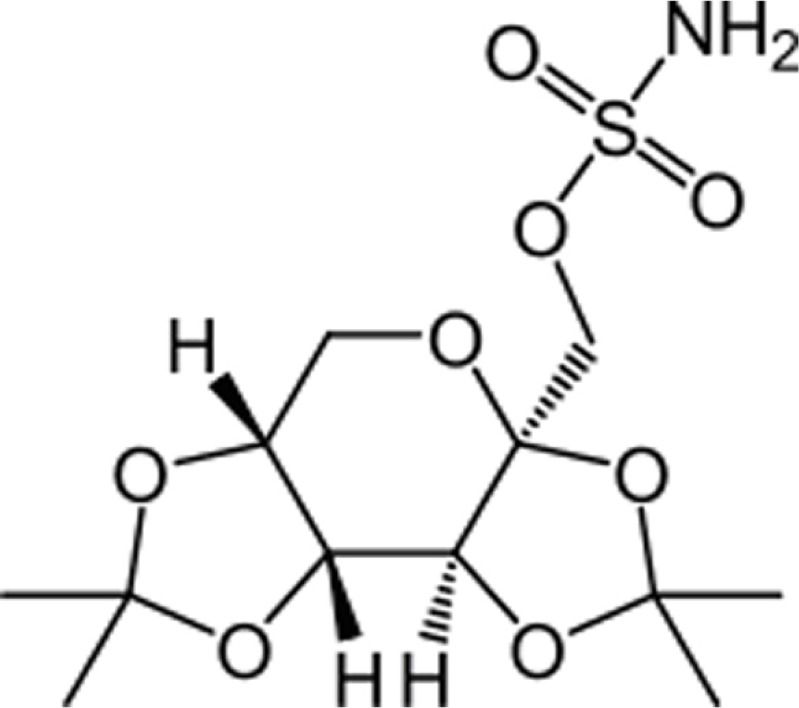
The mean plasma elimination half-life is 21 hours after single or multiple doses. Steady-state is thus reached in about 4 days in patients with normal renal function. Topiramate is not extensively metabolized and is primarily eliminated unchanged in the urine. The metabolites that have been identified are formed via hydroxylation, hydrolysis, and glucuronidation. Topiramate clearance per kg is greater in pediatric patients than in adults and in young pediatric patients (down to 2 years) than in older pediatric patients. Consequently, the plasma drug concentration for the same mg/kg/day dose is lower in pediatric patients compared to adults and also in younger pediatric patients compared to older pediatric patients. Concomitant administration of valproic acid and topiramate has been associated with hyperammonemia with and without encephalopathy and hypothermia.
The precise mechanisms by which topiramate (TPM) exerts its anticonvulsant and migraine prophylaxis effects are unknown. However, preclinical studies have revealed four properties that may contribute to TPM efficacy for epilepsy and migraine prophylaxis. Electrophysiological and biochemical evidence suggests that TPM, at pharmacologically relevant concentrations, blocks voltage-dependent sodium channels, augments the activity of the neurotransmitter gamma-aminobutyrate at some subtypes of the GABA-A receptor, antagonizes the AMPA/kainate subtype of the glutamate receptor, and inhibits the carbonic anhydrase enzyme.44
TPM was initially approved in US in 1996.
The studies summarized below elucidate the safety and tolerability of TPM.
Mohamed et al45 did a 3-year retrospective review of TPM use in 51 children, aged 3–16 years with partial and generalized epilepsy. According to this study, twenty-six children (51%) were still receiving TPM at the end of their last visit. TPM was withdrawn in 25 patients. The reason for withdrawal included adverse effects in 20, lack of effect in three and worsening of seizures in two patients. Adverse effects were noted in 29 children (57%). Majority (74%) of these adverse effects were behavioral, cognitive and neurologic in nature: 13% with agitation, 13% depression, 10% lethargy, and 3% visual hallucinations. Neurologic side effects included ataxia in 6% of patients and aphasia in 2% of patients. Anorexia and weight loss developed in three children, which persisted after a 25%–50% reduction in the dose of TPM and eventually led to drug withdrawal. No patient developed renal calculi throughout the period of evaluation.
Watanabe et al46 studied the effects of TPM in 25 children with intractable generalized epilepsy and therapy was discontinued in 5 out of 25 children but this was due to lack of efficacy. No serious side effects were observed during TPM therapy.
Reith et al47 studied the tolerability of TPM in children and adolescents. 159 patients were identified who were started on TPM and follow up data was available in 127 (80%) of the patients. After 4 years, 60% of patients discontinued the medication. Treatment limiting side effects included aggression/psychosis (n = 10), cognitive impairment (n = 6), anorexia/weight loss (n = 4) and desquamation (n = 1). Thirty percent of the patients who were started on TPM experienced a side effect that resulted in discontinuation of therapy within 2 years.
TPM is used in the treatment of neonatal and pediatric status epilepticus based on data that when seizures continue for longer than 1 hour, GABA A receptor insensitivity develops and excessive NMDA accumulates. Since TPM acts as an NMDA antagonist it may be an appropriate agent in status epilepticus. Akyildiz et al48 studied the doses and effects of TPM in treating pediatric refractory status epilepticus. They identified 32 patients, 14 of whom received TPM by naso-gastric route. The median TPM dose was 5 mg/kg/day for responders and 19 mg/kg/day for partial responders. Metabolic acidosis developed in 3 patients during PICU stay.
Glass et al49 have reported the use of TPM in neonatal seizures in a retrospective cohort study. They identified six term newborns treated with TPM for acute symptomatic seizures refractory to phenobarbital. Five children received an enteric loading dose of 10 mg/kg/day, and in one child maintenance therapy was started at 3 mg/kg/day. No serious side effects that would result in discontinuation of the medicine were reported during short or long term follow-up. However, 3 children had weight at or below 5th percentile at follow up. Follow up was at 5–11.5 months of age.
In a retrospective study, Boldyreva et al50,51 compared efficacy and tolerability of carbamazepine, valproic acid and topiramate in focal occipital lobe and frontal lobe epilepsy. Their results showed that AEs were more frequent during treatment with TPM when compared to valproic acid 17% vs. 6% in occipital lobe epilepsy and 31% vs. 6% in frontal lobe epilepsies.
Verrotti et al52 reviewed the literature on weight loss associated with TPM therapy. They concluded that TPM induces weight loss, especially in high baseline BMI patients, not strictly dependent on daily dosage and perhaps not influenced by gender of the patient. The mechanism by which TPM can induce weight loss is controversial.
In a review article for treatment of Dravet syndrome,53 Chiron identifies three studies where TPM was initiated in these patients. In all three reports half the patients had side effects: most common were anorexia and behavior problems.
Vigabatrin
It is an analog of GABA, but it is not a receptor agonist. Vigabatrin is a racemic compound, and its [S]-enantiomer is pharmacologically active. The chemical name is (±) 4-amino-5-hexenoic acid.54
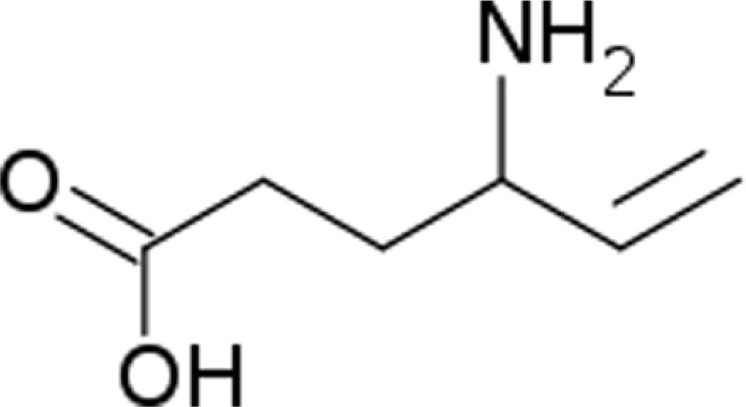
No direct correlation between plasma concentration and efficacy has been established. The duration of drug effect is presumed to be dependent on the rate of enzyme re-synthesis rather than on the rate of elimination of the drug from the systemic circulation. Vigabatrin is not significantly metabolized; it is eliminated primarily through renal excretion. The half-life of vigabatrin is about 5.7 hours in infants. Vigabatrin does not bind to plasma proteins.
Vigabatrin (VGB) is a vinyl derivative of gamma-aminobutyric acid, which has been used in Europe for treatment of infantile spasms since 1989. In 2009, VGB was approved by United States Food and Drug administration (FDA) for use as monotherapy in the treatment of infantile spasms in children aged 1 month to 2 years.55 The proposed mechanism of action is irreversible inhibition of GABA-transaminase (GABA-T). The most common adverse reactions (change of ≥5% over placebo) in addition to permanent vision loss in adult controlled trials with VGB were fatigue, somnolence, nystagmus, tremor, vision blurred, memory impairment, weight gain, arthralgia, abnormal coordination, and confusional state.
VGB is associated with a black box warning that describes the potential of permanent bilateral concentric visual field defect.55
Visual field defect is a well-known side effect of VGB. Maguire et al,56 tried to look for the prevalence of VGB induced visual field loss. They identified thirty-two studies, including 1,678 patients exposed to VGB and 406 controls and found that of the 1,678 exposed patients, 738 (44%) had visual field loss compared to just 20 out of 406 (7%) controls. The visual field loss was more common in adults (52%) and relatively lower in children at (34%). The relative risk of visual field loss in VGB exposed patients was 4.
Clayton et al57 compared the relationship between retinal nerve fiber layer (RNFL) thinning and visual field size. Two hundred and one patients who were exposed to VGB and 90 healthy controls participated in this study. Visual fields were obtained using Goldman kinetic perimetry and RNFL imaging was performed by optical coherence tomography (OCT). They concluded that 51% of patients showed VGB induced visual field deficits. Average RNFL thickness was significantly thinner in patients compared to healthy controls. They also found a strong relationship between visual field defects and average RNFL thickness. This suggests OCT can provide an estimate of visual field loss and also that VGB induced irreversible visual field loss may be related to loss of retinal cell ganglions.
Wilmore et al58 published a literature review on VGB. AEs reported included T2 hyper intensities within the brain, psychotic disorders and hallucinations. Peripheral visual field defects were also detected. The incidence of VGB induced peripheral VFD depends upon the age of the patient and the extent of exposure. The prevalence in adults was 25%–50%, while in children the prevalence was 15%. The incidence of retinal defect in infants ranged from 15%–31%. In children the first abnormal field examination was after 11 months with a mean time to onset of 5.5 years. The earliest sustained onset of retinal field defect in infants was 3.1 months. It is recommended to perform cognitive, age appropriate visual field testing at baseline and then at repeated intervals. Infants are tested at baseline and then every 3 months until 18 months of age. When patients are prescribed VGB, they are monitored closely and in cases where spasm or seizure improvement is not achieved within 12 weeks of initiation, VGB should be discontinued. There are no reliable pre- treatment predictors for children at risk for clinically significant permanent visual field defect.
Aurich-Barrera et al59 compared the adverse event profile of children and adults taking VGB. Incidence of AEs in children and adults in first month were compared to month two to six. Their results showed abnormal behavior and hyperactivity were more frequently reported in children, confusion and psychosis more frequently in adults. A higher percentage of children stopped treatment due to lack of effectiveness (57.7% vs. 47.5%).
VGB associated MRI signal changes has been described.60,61 In pediatric patients VGB can cause reversible diffusion restriction in globi pallidi, thalami, brain stem and dentate nuclei. It was observed in younger infants and patients with cryptogenic infantile spasms.
Horton et al62 described a case report of a child who developed vacuolar myelinopathy after VGB administration. This child received VGB for infantile spasms and had a history of spastic quadriparesis due to hypoxic ischemic injury at birth. He had rapid deterioration in clinical condition and died 3 weeks later. Neuropathologic examination confirmed white matter vacuolation and intramyelinic edema.
Tekgul et al63 described rapid progressive deterioration in 2 cases of early myoclonic epilepsy associated with non-ketotic hyperglycinemia. Both patients developed acute encephalopathy with respiratory failure. The symptoms improved after VGB was stopped after a few days in the first case, but not in the second case. Likewise, it is also reported to make hyperprolinemia type 1 worse and should be avoided in this condition.64
Yang et al65 described three patients who developed absence seizures after administration of carbamazepine and vigabatrin. The absence seizures subsided rapidly after the discontinuation of the drug.
Zonisamide
It is an anti-seizure drug chemically classified as a sulfonamide and is unrelated to other anti-epileptic medications. The active ingredient is 1,2-benzisoxazole-3-methanesulfonamide.
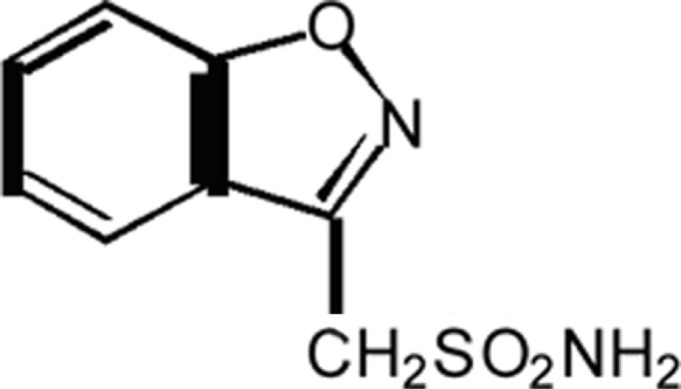
It is metabolized mostly by the CYP3A4 isoenzyme to 2-sulphamoylacetyl-phenol via reductive cleavage of the 1, 2-benzisoxazole ring.66 Zonisamide (ZNS) is excreted primarily in urine as parent drug and as the glucuronide of its metabolite. The elimination half-life of ZNS in plasma is about 63 hours. Once a stable dose is reached, steady state is achieved within 14 days.
It was introduced in the United States in 2000.
ZNS is a broad spectrum antiepileptic drug. It has a unique combination of multiple mechanisms of action that are potentially complementary with concomitant AEDs. Zonisamide has no clinically relevant effects on the pharmacokinetics of other commonly used AEDs, however, co-administration with cytochrome P450 3A4 (CYP3A4) inducers or inhibitors may change zonisamide’s pharmacokinetic profile.67 Zonisamide generally is well tolerated with the majority of adverse events being mild-to-moderate. The tolerability of zonisamide has also been shown to improve with slower drug titration and duration of drug treatment. These characteristics suggest that zonisamide may be suitable as a key adjunct in rational polytherapy. It has a broad spectrum of efficacy for pediatric partial and generalized epilepsies, especially for myoclonic epilepsies. It can also be considered a second line agent for infantile spasms, Lennox-Gastaut Syndrome and JME.28
ZNS is generally well tolerated and discontinuation of ZNS due to side effects is uncommon. The studies summarized below clarify the safety and tolerability profile of ZNS.
In a study by Kim et al68 a retrospective chart review of 68 patients with medically refractory epilepsy was performed. The age ranges of the children was 1.9–18.1 years, median maintenance dose of ZNS was 8 mg/kg/day. The seizure types included partial and generalized seizures. 22% and 78% were on mono-therapy and adjunctive therapy respectively. 16% patients discontinued ZNS but only 7% were strictly due to side effects. Common adverse effects (AEs) included behavioral/psychiatric (23%), cognitive dysfunction (12%), and sedation (10%). Based on these findings they concluded that drug discontinuation as a result of side effects was uncommon.
In another study by Tan et al69 involving 57 children with medically refractory epilepsy who were treated with ZNS at 3 UK tertiary centers and followed for at least 12 months, 44% patients reported AEs but this contributed to withdrawal of ZNS in 17% patients.
Similar findings were demonstrated in a study by Kothare et al70 in a retrospective chart review of 69 children with recently diagnosed epilepsy. The mean age of patients was 13.2 years; mean duration of follow up was 22 months on ZNS mono-therapy. ZNS was chosen as first line or second line AED in these patients. 26% (18) patients had AEs of which 6% resulted in drug discontinuation. The side effects seen with decreasing frequency were weight loss, cognitive impairment, sleepiness and dizziness.
A multicenter study by Lee et al71 looked at 163 children with intractable epilepsy who were followed for at least 6 months after initiation of ZNS, mean dosage of 8.2 mg/kg/day. AEs were documented in 9.2% children and this included somnolence, fatigue and anorexia, which were transient and successfully managed. This study agreed with published literature that adverse events reported in the first few weeks of ZNS treatment regress over time. The tolerability of ZNS improved with duration of drug treatment, and a slower drug titration reduced the incidence of adverse events. One patient (1/163, 0.6%) developed acute pancreatitis at 8 months and this led to discontinuation of ZNS and TPM while maintaining other AEDs. The patient improved from pancreatitis.
Shinnar et al72 assessed the long term safety of ZNS administered to 109 children. The mean dose received was 8.5 mg/kg/day. Of the 109 children, 48% completed 15 months treatment. Treatment-related AEs, mostly mild-to-moderate in severity, were reported by 53% patients. 6.4% patients discontinued due to treatment-related AEs. Serious AEs (pancreatitis, decreased sweating, and vertigo) were reported by three patients.
Akman et al73 describe three patients who experienced complex visual hallucinations after zonisamide treatment was begun or its dosage increased. Two of the 3 patients were girls aged 7 and 13 yrs. None of them had experienced previous psychiatric disturbances before ZNS was begun. During monitoring, visual hallucinations did not correlate with EEG readings. With either discontinuation or decreased dosage of the drug the symptoms disappeared and did not recur.
Paul et al74 looked at concurrent therapy with ketogenic diet (KD) and TPM or ZNS. 15% of 93 patients had occult hematuria or worse including 6% with urolithiasis. 3 of 6 calculi developed in KD + ZNS group of 17 patients co-treated for cumulative total of 97 months (3.1stones per 100 patient months). One of 6 calculi was in the KD + TPM group of 22 children who were co-treated for 263 months (0.4 stones per 100 patient months). All six patients had at least three of five biochemical risk factors including metabolic acidosis, concentrated urine, acidotic urine, hypercalciuria and hypocitraturia. Non-fasting KD initiation, fluid liberalization, potassium citrate prophylaxis as well as regular laboratory surveillance are indicated in this high-risk population.
Discussion
Over a dozen new AEDs for management of epilepsy have been introduced in the past 2 decades. The introduction of a new AED is always welcomed with enthusiasm by both patients and physicians. The newer AEDs however pose a specific challenge in the pediatric population, as these are usually approved as add on therapies based on clinical trials involving adults. The data when extrapolated on pediatric patients with epilepsy gives us an estimate of efficacy. However, issues specific to children such as tolerability, organ specific toxicity and the effect on development, behavior and cognition remains undetermined when the newer AEDs are first introduced and added to the armamentarium of AEDs already available.
Some of the more specific side effects become clear with time based upon individual experiences and evolving literature.
There is no specific roadmap for the use of AEDs in children. Evidence based medicine and expert opinion help provide guidelines for their use. The task of choosing an AED for a child is important and requires a thorough knowledge of the proposed mechanisms of action, pharmacokinetics in children, age specific side effects and effects on the developing brain. It also requires a tailored approach to each patient taking into account the underlying etiology/syndrome, adjunctive AEDs that the patient is receiving, age, available formulations, coexisting medical problems and behavioral profile. The purpose of this review was to summarize the literature regarding the safety and tolerability of newer AEDs over the past 15 years to help guide the process of choosing the appropriate AED.
Despite the introduction of the newer AEDs, the percentage of children with intractable epilepsy has not changed significantly since 1960. There remain 25%–30% of children with medically refractory epilepsy. This leads to recognizing an ongoing need for further research in better understanding the disease process and developing drugs with higher efficacy and minimal side effects.
Footnotes
Author Contributions
SK and DS were responsible for assimilating and reviewing literature regarding the anti-epileptic drugs reviewed in this article. SK wrote the first draft of sections of the manuscript. DS and SK Contributed equally to the writing of the manuscript and jointly developed the structure and arguments for the paper. DS made critical revisions and approved the final version. Both authors reviewed and approved of the final manuscript.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
References
- 1.2009. Levetiracetam approved label. Food and Drug Administration.
- 2.Rogawski MA. Diverse mechanisms of antiepileptic drugs in the development pipeline. Epilepsy Research. 2006 Jun;69(3):273–94. doi: 10.1016/j.eplepsyres.2006.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vigevano F. Levetiracetam in pediatrics. Journal of Child Neurology. 2005 Feb;20(2):87–93. doi: 10.1177/08830738050200020101. [DOI] [PubMed] [Google Scholar]
- 4.Sirsi D, Safdieh JE. The safety of levetiracetam. Expert Opinion on Drug Safety. 2007 May;6(3):241–50. doi: 10.1517/14740338.6.3.241. [DOI] [PubMed] [Google Scholar]
- 5.Kugler SL. Behavioral profile of levetiracetam in children. Epilepsia. 2002;43(Suppl 7):1. [Google Scholar]
- 6.Schiemann-Delgado J, Yang H, de la Loge C, et al. A Long-term open-label extension study assessing cognition and behavior, tolerability, safety, and efficacy of adjunctive levetiracetam in children aged 4 to 16 years with partial-onset seizures. Journal of Child Neurology. 2011 Aug 29; doi: 10.1177/0883073811417183. [DOI] [PubMed] [Google Scholar]
- 7.Kossoff EH, Bergey GK, Freeman JM, Vining EP. Levetiracetam psychosis in children with epilepsy. Epilepsia. 2001 Dec;42(12):1611–3. doi: 10.1046/j.1528-1157.2001.32101.x. [DOI] [PubMed] [Google Scholar]
- 8.Gustafson MC. Behavioral and emotional effects of levetiracetam in children with intractable epilepsy. Epilepsia. 2002;43(suppl 7):1. [Google Scholar]
- 9.Dahlin MG, Wide K, Ohman I. Age and comedications influence levetiracetam pharmacokinetics in children. Pediatric Neurology. 2010 Oct;43(4):231–5. doi: 10.1016/j.pediatrneurol.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 10.Miller GS. Pyridoxine ameliorates adverse behavioral effects of levetiracetam in children. Epilepsia. 2002;43(suppl 7):1. [Google Scholar]
- 11.Major P, Greenberg E, Khan A, Thiele EA. Pyridoxine supplementation for the treatment of levetiracetam-induced behavior side effects in children: Preliminary results. Epilepsy & Behavior: E&B. 2008 Oct;13(3):557–9. doi: 10.1016/j.yebeh.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 12.Lamotrigine approved label. Food and drug administration.
- 13.Celebi A, Yalnmzoglu D, Turanli G, Topaloglu H, Aysun S, Topcu M.Lamotrigine in children with refractory epilepsy The Turkish Journal of Pediatrics September–Oct2008505426–31. [PubMed] [Google Scholar]
- 14.Holmes GL, Frank LM, Sheth RD, et al. Lamotrigine monotherapy for newly diagnosed typical absence seizures in children. Epilepsy Research. 2008 Dec;82(2–3):124–32. doi: 10.1016/j.eplepsyres.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Culy CR, Goa KL.Lamotrigine. A review of its use in childhood epilepsy Paediatric drugs July–Aug200024299–330. [DOI] [PubMed] [Google Scholar]
- 16.Seo HJ, Chiesa A, Lee SJ, et al. Safety and tolerability of lamotrigine: Results from 12 placebo-controlled clinical trials and clinical implications Clinical Neuropharmacology January–Feb201134139–47. [DOI] [PubMed] [Google Scholar]
- 17.Valencia I, Pinol-Ripoll G, Khurana DS, et al. Efficacy and safety of lamotrigine monotherapy in children and adolescents with epilepsy. European Journal of Paediatric Neurology: EJPN: Official Journal of the European Paediatric Neurology Society. 2009 Mar;13(2):141–5. doi: 10.1016/j.ejpn.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Pina-Garza JE, Elterman RD, Ayala R, et al. Long-term tolerability and efficacy of lamotrigine in infants 1 to 24 months old. Journal of Child Neurology. 2008 Aug;23(8):853–61. doi: 10.1177/0883073808317348. [DOI] [PubMed] [Google Scholar]
- 19.Aurich-Barrera B, Wilton L, Brown D, Shakir S. Paediatric postmarketing pharmacovigilance using prescription-event monitoring: comparison of the adverse event profiles of lamotrigine prescribed to children and adults in England. Drug Safety: An International Journal of Medical Toxicology and Drug Experience. 2010 Sep 1;33(9):751–63. doi: 10.2165/11536830-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 20.Cardenas JF, Rho JM, Ng YT. Reversible lamotrigine-induced neurobehavioral disturbances in children with epilepsy. Journal of Child Neurology. 2010 Feb;25(2):182–7. doi: 10.1177/0883073809336874. [DOI] [PubMed] [Google Scholar]
- 21.Ferguson LP, Dargan PI, Hood JL, Tibby SM. Life-threatening organ failure after lamotrigine therapy. Pediatric Neurology. 2009 May;40(5):392–4. doi: 10.1016/j.pediatrneurol.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 22.Dreesman A, Hoorens A, Hachimi-Idrissi S. Multiple organ dysfunction syndrome: Infection or hypersensitivity reaction? European Journal of Emergency Medicine: Official Journal of the European Society for Emergency Medicine. 2010 Aug;17(4):228–9. doi: 10.1097/MEJ.0b013e3283311f04. [DOI] [PubMed] [Google Scholar]
- 23.Veerapandiyan A, Gallentine WB, Winchester SA, Baker J, Kansagra SM, Mikati MA. Oculogyric crises secondary to lamotrigine overdosage. Epilepsia. 2011 Mar;52(3):e4–6. doi: 10.1111/j.1528-1167.2010.02967.x. [DOI] [PubMed] [Google Scholar]
- 24.Tengstrand M, Star K, van Puijenbroek EP, Hill R. Alopecia in association with lamotrigine use: an analysis of individual case safety reports in a global database. Drug Safety: An International Journal of Medical Toxicology and Drug Experience. 2010 Aug 1;33(8):653–8. doi: 10.2165/11536190-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 25.Yun M, Choi YM, Eun SH, Seol IJ, Kim SJ. Acoustic effects of lamotrigine in pediatric patients with epilepsy. Brain & Development. 2011 May;33(5):374–8. doi: 10.1016/j.braindev.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Oxcarbazepine approved label. Food and Drug administration.
- 27.Khalil BWA. Oxcarbazepine and cabamazepine: Expected and unexpected differences and similarities. Epilepsy Current. 2007;7:2. doi: 10.1111/j.1535-7511.2007.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hwang H, Kim KJ. New antiepileptic drugs in pediatric epilepsy. Brain & Development. 2008 Oct;30(9):549–55. doi: 10.1016/j.braindev.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 29.Jarrar RG, Buchhalter JR. Therapeutics in pediatric epilepsy, Part 1: The new antiepileptic drugs and the ketogenic diet. Mayo Clinic proceedings. Mayo Clinic. 2003 Mar;78(3):359–70. doi: 10.4065/78.3.359. [DOI] [PubMed] [Google Scholar]
- 30.Glauser TA. Oxcarbazepine in the treatment of epilepsy. Pharmacotherapy. 2001 Aug;21(8):904–19. doi: 10.1592/phco.21.11.904.34513. [DOI] [PubMed] [Google Scholar]
- 31.Belousova ED, Mukhin K, Ermolenko NA, et al. Efficacy and safety of the monotherapy with trileptal (oxcarbazepine) in children and adolescents. Zhurnal nevrologii i psikhiatrii imeni S.S. Korsakova/Ministerstvo zdravookhraneniia i meditsinskoi promyshlennosti Rossiiskoi Federatsii, Vserossiiskoe obshchestvo nevrologov [i] Vserossiiskoe obshchestvo psikhiat. 2010;110(5 Pt 1):45–50. [PubMed] [Google Scholar]
- 32.Bang L, Goa K. Oxcarbazepine: A review of its use in children with epilepsy. Paediatric Drugs. 2003;5(8):557–73. doi: 10.2165/00148581-200305080-00006. [DOI] [PubMed] [Google Scholar]
- 33.Cansu A, Serdaroglu A, Cinaz P. Serum insulin, cortisol, leptin, neuropeptide Y, galanin and ghrelin levels in epileptic children receiving oxcarbazepine. European Journal of Paediatric Neurology: EJPN: Official Journal of the European Paediatric Neurology Society. 2011 Nov;15(6):527–31. doi: 10.1016/j.ejpn.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Herguner MO, Incecik F, Altunbasak S. Oxcarbazepine-induced tardive dyskinesia: A rare adverse reaction. Journal of Pediatric Neurosciences. 2010 Jan;5(1):85–6. doi: 10.4103/1817-1745.66664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santucci R, Fothergill H, Laugel V, et al. The onset of acute oxcarbazepine toxicity related to prescription of clarithromycin in a child with refractory epilepsy. British Journal of Clinical Pharmacology. 2010 Mar;69(3):314–6. doi: 10.1111/j.1365-2125.2009.03593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rufinamide-Food and drug administration.
- 37.Hakimian S, Cheng-Hakimian A, Anderson GD, Miller JW. Rufinamide: A new anti-epileptic medication. Expert Opinion on Pharmacotherapy. 2007 Aug;8(12):1931–40. doi: 10.1517/14656566.8.12.1931. [DOI] [PubMed] [Google Scholar]
- 38.Wheless JW, Conry J, Krauss G, Mann A, LoPresti A, Narurkar M. Safety and tolerability of rufinamide in children with epilepsy: A pooled analysis of 7 clinical studies. Journal of Child Neurology. 2009 Dec;24(12):1520–5. doi: 10.1177/0883073809350508. [DOI] [PubMed] [Google Scholar]
- 39.Wier HA, Cerna A, So TY. Rufinamide for pediatric patients with Lennox-Gastaut syndrome: A comprehensive overview. Paediatric Drugs. 2011 Apr 1;13(2):97–106. doi: 10.2165/11586920-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 40.Kluger G, Bauer B. Role of rufinamide in the management of Lennox-Gastaut syndrome (childhood epileptic encephalopathy) Neuropsychiatric Disease and Treatment. 2007 Feb;3(1):3–11. doi: 10.2147/nedt.2007.3.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glauser T, Kluger G, Sachdeo R, Krauss G, Perdomo C, Arroyo S. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008 May 20;70(21):1950–8. doi: 10.1212/01.wnl.0000303813.95800.0d. [DOI] [PubMed] [Google Scholar]
- 42.Mueller A, Boor R, Coppola G, et al. Low long-term efficacy and tolerability of add-on rufinamide in patients with Dravet syndrome. Epilepsy & Behavior: E&B. 2011 Jul;21(3):282–4. doi: 10.1016/j.yebeh.2011.04.057. [DOI] [PubMed] [Google Scholar]
- 43.Topamax approved label-Food and Drug administration.
- 44.Shank RP, Gardocki JF, Streeter AJ, Maryanoff BE. An overview of the preclinical aspects of topiramate: pharmacology, pharmacokinetics, and mechanism of action. Epilepsia. 2000;41(Suppl 1):S3–9. [PubMed] [Google Scholar]
- 45.Mohamed K, Appleton R, Rosenbloom L. Efficacy and tolerability of topiramate in childhood and adolescent epilepsy: a clinical experience. Seizure. 2000 Mar;9(2):137–41. doi: 10.1053/seiz.2000.0387. [DOI] [PubMed] [Google Scholar]
- 46.Watanabe T, Oyanagi R, Minagawa K. Short-term and long-term efficacy of topiramate in refractory generalized epilepsy of children. No to Hattatsu. Brain and Development. 2011 May;43(3):223–7. [PubMed] [Google Scholar]
- 47.Reith D, Burke C, Appleton DB, Wallace G, Pelekanos J. Tolerability of topiramate in children and adolescents. Journal of Paediatrics and Child Health. 2003 Aug;39(6):416–9. doi: 10.1046/j.1440-1754.2003.00180.x. [DOI] [PubMed] [Google Scholar]
- 48.Akyildiz BN, Kumandas S. Treatment of pediatric refractory status epilepticus with topiramate. Child’s Nervous System: ChNS: Official Journal of the International Society for Pediatric Neurosurgery. 2011 Sep;27(9):1425–30. doi: 10.1007/s00381-011-1432-y. [DOI] [PubMed] [Google Scholar]
- 49.Glass HC, Poulin C, Shevell MI. Topiramate for the treatment of neonatal seizures. Pediatric Neurology. 2011 Jun;44(6):439–42. doi: 10.1016/j.pediatrneurol.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boldyreva SR, Ermakov A. Comparative efficacy of carbamazepine, valproic acid and topiramate in symptomatic and cryptogenic frontal lobe epilepsy in children. Zhurnal nevrologii i psikhiatrii imeni S.S. Korsakova/Ministerstvo zdravookhraneniia i meditsinskoi promyshlennosti Rossiiskoi Federatsii, Vserossiiskoe obshchestvo nevrologov [i] Vserossiiskoe obshchestvo psikhiat. 2010;110(6):58–65. [PubMed] [Google Scholar]
- 51.Boldyreva SR, Ermakov A. Comparative efficacy of carbamazepine, valproic acid and topiramate in symptomatic and cryptogenic occipital lobe epilepsy in children. Zhurnal nevrologii i psikhiatrii imeni S.S. Korsakova/Ministerstvo zdravookhraneniia i meditsinskoi promyshlennosti Rossiiskoi Federatsii, Vserossiiskoe obshchestvo nevrologov [i] Vserossiiskoe obshchestvo psikhiat. 2010;110(5 Pt 1):39–44. [PubMed] [Google Scholar]
- 52.Verrotti A, Scaparrotta A, Agostinelli S, Di Pillo S, Chiarelli F, Grosso S. Topiramate-induced weight loss: a review. Epilepsy Research. 2011 Aug;95(3):189–99. doi: 10.1016/j.eplepsyres.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 53.Chiron C, Dulac O. The pharmacologic treatment of Dravet syndrome. Epilepsia. 2011 Apr;52(Suppl 2):72–5. doi: 10.1111/j.1528-1167.2011.03007.x. [DOI] [PubMed] [Google Scholar]
- 54.Sabril approved label-Food and Drug administration.
- 55.Pesaturo KA, Spooner LM, Belliveau P. Vigabatrin for infantile spasms. Pharmacotherapy. 2011 Mar;31(3):298–311. doi: 10.1592/phco.31.3.298. [DOI] [PubMed] [Google Scholar]
- 56.Maguire MJ, Hemming K, Wild JM, Hutton JL, Marson AG. Prevalence of visual field loss following exposure to vigabatrin therapy: A systematic review. Epilepsia. 2010 Dec;51(12):2423–31. doi: 10.1111/j.1528-1167.2010.02772.x. [DOI] [PubMed] [Google Scholar]
- 57.Clayton LM, Devile M, Punte T, et al. Retinal nerve fiber layer thickness in vigabatrin-exposed patients. Annals of Neurology. 2011 May;69(5):845–54. doi: 10.1002/ana.22266. [DOI] [PubMed] [Google Scholar]
- 58.Willmore LJ, Abelson MB, Ben-Menachem E, Pellock JM, Shields WD. Vigabatrin: 2008 update. Epilepsia. 2009 Feb;50(2):163–73. doi: 10.1111/j.1528-1167.2008.01988.x. [DOI] [PubMed] [Google Scholar]
- 59.Aurich-Barrera B, Wilton L, Brown D, Shakir S. Paediatric post-marketing pharmacovigilance: Comparison of the adverse event profile of vigabatrin prescribed to children and adults. Pharmacoepidemiology and Drug Safety. 2011 Jun;20(6):608–18. doi: 10.1002/pds.2105. [DOI] [PubMed] [Google Scholar]
- 60.Dracopoulos A, Widjaja E, Raybaud C, Westall CA, Snead OC., III Vigabatrin-associated reversible MRI signal changes in patients with infantile spasms. Epilepsia. 2010 Jul;51(7):1297–304. doi: 10.1111/j.1528-1167.2010.02564.x. [DOI] [PubMed] [Google Scholar]
- 61.Wheless JW, Carmant L, Bebin M, et al. Magnetic resonance imaging abnormalities associated with vigabatrin in patients with epilepsy. Epilepsia. 2009 Feb;50(2):195–205. doi: 10.1111/j.1528-1167.2008.01896.x. [DOI] [PubMed] [Google Scholar]
- 62.Horton M, Rafay M, Del Bigio MR. Pathological evidence of vacuolar myelinopathy in a child following vigabatrin administration. Journal of Child Neurology. 2009 Dec;24(12):1543–6. doi: 10.1177/0883073809348796. [DOI] [PubMed] [Google Scholar]
- 63.Tekgul H, Serdaroglu G, Karapinar B, et al. Vigabatrin caused rapidly progressive deterioration in two cases with early myoclonic encephalopathy associated with nonketotic hyperglycinemia. Journal of Child Neurology. 2006 Jan;21(1):82–4. doi: 10.1177/08830738060210011801. [DOI] [PubMed] [Google Scholar]
- 64.Humbertclaude V, Rivier F, Roubertie A, et al. Is hyperprolinemia type I actually a benign trait? Report of a case with severe neurologic involvement and vigabatrin intolerance. Journal of Child Neurology. 2001 Aug;16(8):622–3. doi: 10.1177/088307380101600820. [DOI] [PubMed] [Google Scholar]
- 65.Yang MT, Lee WT, Chu LW, Shen YZ. Anti-epileptic drugs-induced de novo absence seizures. Brain & Development. 2003 Jan;25(1):51–6. doi: 10.1016/s0387-7604(02)00120-1. [DOI] [PubMed] [Google Scholar]
- 66.Zonisamide Approved Label. Food and Drug Administration.
- 67.Baulac M, Leppik IE. Efficacy and safety of adjunctive zonisamide therapy for refractory partial seizures. Epilepsy Research. 2007 Jul;75(2–3):75–83. doi: 10.1016/j.eplepsyres.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 68.Kim HL, Aldridge J, Rho JM. Clinical experience with zonisamide mono-therapy and adjunctive therapy in children with epilepsy at a tertiary care referral center. Journal of Child Neurology. 2005 Mar;20(3):212–9. doi: 10.1177/08830738050200030801. [DOI] [PubMed] [Google Scholar]
- 69.Tan HJ, Martland TR, Appleton RE, Kneen R. Effectiveness and tolerability of zonisamide in children with epilepsy: A retrospective review. Seizure. 2010 Jan;19(1):31–5. doi: 10.1016/j.seizure.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 70.Kothare SV, Kaleyias J, Mostofi N, et al. Efficacy and safety of zonisamide monotherapy in a cohort of children with epilepsy. Pediatric Neurology. 2006 May;34(5):351–4. doi: 10.1016/j.pediatrneurol.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 71.Lee YJ, Kang HC, Seo JH, Lee JS, Kim HD. Efficacy and tolerability of adjunctive therapy with zonisamide in childhood intractable epilepsy. Brain& Development. 2010 Mar;32(3):208–12. doi: 10.1016/j.braindev.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 72.Shinnar S, Pellock JM, Conry JA. Open-label, long-term safety study of zonisamide administered to children and adolescents with epilepsy. European Journal of Paediatric Neurology: EJPN: Official Journal of the European Paediatric Neurology Society. 2009 Jan;13(1):3–9. doi: 10.1016/j.ejpn.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 73.Akman CI, Goodkin HP, Rogers DP, Riviello JJ., Jr Visual hallucinations associated with zonisamide. Pharmacotherapy. 2003 Jan;23(1):93–6. doi: 10.1592/phco.23.1.93.31911. [DOI] [PubMed] [Google Scholar]
- 74.Paul E, Conant KD, Dunne IE, et al. Urolithiasis on the ketogenic diet with concurrent topiramate or zonisamide therapy. Epilepsy Research. 2010 Jun;90(1–2):151–6. doi: 10.1016/j.eplepsyres.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]