

Abstract
Serotonergic agents have been used in the past for reduction of appetite and body weight. As reported by Zhou et al. (2007) in this issue of Cell Metabolism, they also have unexpected effects on peripheral glucose homeostasis independent of food intake and body weight.
Previous research on appetite suppression by serotonin has centered on the role of serotonin signaling in the brain’s “appetite center,” the hypothalamus. Effective weight loss has been achieved via fenfluramine and dexfenfluramine, which increase brain serotonin by interfering with its uptake and causing a reverse release of serotonin through the serotonin transporter. Of the many subtypes of serotonin (5-HT) receptors, the 5-HT2C receptor (5-HT2CR) is the proposed target mediating the anorectic effects of fenfluramine. Unfortunately, use of fenfluramine was associated with pulmonary hypertension through action on peripheral 5-HT2B receptors, leading to proscription of its use. Today, the only antiobesity drug with serotonergic action is sibutramine (Meridia), which inhibits serotonin reuptake but is not very effective. Despite these problems, the search for selective 5-HT2CR agonists devoid of 5-HT2A and 5-HT2B activity has continued, and some agents have progressed to phase III clinical trials (e.g., lorcaserin) (Halford et al., 2007). Most studies have focused on the weight-reducing properties of serotonergic agents; their role in peripheral metabolism has not been previously explored. In this issue, Zhou et al. (2007) show that subanorectic doses of mCPP, a classical 5-HT2CR agonist, also affect glucose homeostasis independent of food intake and weight loss.
Zhou et al. (2007) present convincing evidence for improved glucose and insulin tolerance in genetic and diet-induced models of obesity after both acute injections and long-term infusion of mCPP. These results, together with decreased insulin levels, are suggestive of increased insulin sensitivity and are consistent with the reported enhanced signaling downstream of the insulin receptor in mCPP-treated mice. However, glucose tolerance is influenced by the ability of pancreatic cells to release insulin and glucagon and by hepatic gluconeogenesis. Thus, the available results do not completely define the mechanism leading to improved glucose tolerance. This is underscored by the finding that the expression of two key gluconeogenic enzymes, PEPCK and G6Pase, is reduced in the liver of mCPP-treated mice: such a decrease could potentially contribute to a decline in liver glucose output. Ultimately, additional studies such as euglycemic/hyperinsulinemic clamps that carefully examine fluxes will be required to fully understand mechanisms of altered peripheral insulin sensitivity.
How might serotonin influence glucose homeostasis? As 5-HT2CRs are expressed almost exclusively in the central nervous system (CNS), the actions of mCPP on glucose homeostasis are likely to be centrally mediated. The arcuate POMC neurons are an obvious target for mCPP action, as these neurons respond to circulating hormones and nutrients such as leptin and glucose, and as loss of these signals leads to obesity and impaired glucose tolerance, respectively (Figure 1) (Balthasar et al., 2004; Parton et al., 2007). Indeed, this group suggested previously that the anorectic effects of fenfluramine are mediated by activation of the 5-HT2CR on POMC neurons (Heisler et al., 2002). In their current report (Zhou et al., 2007), they demonstrate that activation of POMC neurons is required for the improved glucose tolerance seen in mCPP-treated animals. They show that mCPP induces increased c-Fos staining, a marker of neuronal activation, in POMC neurons. Furthermore, the effects of mCPP are absent in Mc4r, but not Mc3r, knockout mice. It is worthwhile to consider why activation of POMC neurons does not result in decreased food intake and body weight. One can speculate that the smaller doses of mCPP activate only a subset of POMC neurons projecting caudally to autonomic outflow centers, but not those projecting to centers affecting appetite. Consistent with this idea, the autonomic preganglionic neurons in the spinal cord are activated by mCPP treatment, and their activation is dependent of MC4R signaling.
Figure 1. Proposed Pathway Mediating the Effect of mCPP on Peripheral Glucose Homeostasis.
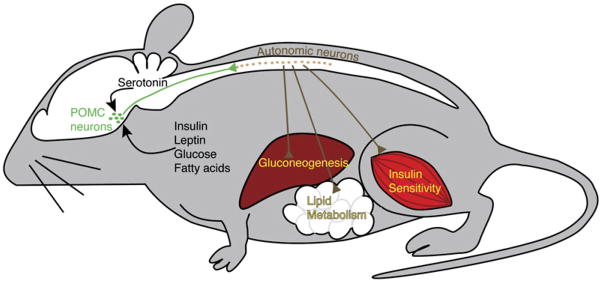
POMC neurons (green) are located in the arcuate nucleus of the hypothalamus and receive metabolic information through circulating factors such as glucose, fatty acids, leptin, and insulin as well as central inputs such as serotonin. The activity of autonomic neurons (brown) in the spinal cord depends on intact melanocortin signaling. The autonomic neurons, in turn, affect peripheral metabolism through their effects on insulin sensitivity, liver glucose output, and adipose tissue lipid synthesis and lipolysis.
Although these results are intriguing, the precise role of the melanocortin pathway in mediating serotonergic signals remains unclear. The authors correctly point out that MC4R signaling might only be permissive for the activation of the sympathetic neurons by mCPP and that the contribution of other centers cannot be excluded. Along these lines, it has been reported that mCPP causes anorexia in chronic decerebrate rats, implicating brain-stem 5-HT2CRs at least in the anorectic actions of mCPP (Kaplan et al., 1998). Definitive proof of the importance of the melanocortin system will require the selective inactivation of 5-HT2CRs in POMC neurons.
These findings also reflect an emerging literature that expands the role of the CNS in regulating metabolism. In addition to its well-established role in the control of feeding, the CNS also regulates peripheral metabolism, including energy expenditure and glucose and lipid homeostasis, through changes in autonomic sympathetic, parasympathetic, and hormonal outputs. Studies using genetic techniques have been instrumental in defining the intracellular signaling pathways critical to the expanded multiple roles of POMC neurons. Thus, glucose sensing, when impaired via a disruption of KATP channels, leads to glucose intolerance (Parton et al., 2007) and, when disrupted through targeted deletion of the α2 subunit of AMP kinase (AMPK), leads to obesity (Claret et al., 2007). Recent pharmacologic studies have extended the function of POMC neurons to include control of lipid metabolism (Nogueiras et al., 2007). Given these important outputs of the POMC system, it is not surprising that it would be the target of regulation by serotonergic inputs through 5-HT2CRs, which in turn are known to be critical to energy balance. Nevertheless, the effects on glucose sensitivity are striking. It is important to note that these expanded roles of the POMC system beyond feeding are not unique to the melanocortin system. Pharmacologic studies from as early as 1993 suggested a role for NPY in regulating insulin sensitivity (Zarjevski et al., 1993). Genetic studies of AgRP neurons reveal that insulin signaling in AgRP neurons is required for the suppression of hepatic glucose production (Konner et al., 2007). Similarly, both genetic and pharmacologic studies indicate that MCH neurons regulate energy expenditure and autonomic activity (Astrand et al., 2004).
More detailed investigation of the neuronal pathways that respond to changing hormonal and metabolite levels, the efferent pathways that relay this information, and the effects on target tissues will be required to fully understand the expanded roles of neurons whose function was previously thought to be limited to regulating feeding. More importantly, any dysregulatory mechanism in these pathways that might underlie the development of obesity and diabetes could provide a potential new target for the treatment of metabolic disorders. Along these lines, the improvement of glucose homeostasis independent of hypophagia and weight loss via 5-HT2C agonists is an important observation that validates the efforts to develop 5-HT2CR-based therapies. If this finding translates to humans as well, it could provide a novel treatment for type 2 diabetes.
References
- Astrand A, Bohlooly YM, Larsdotter S, Mahlapuu M, Andersen H, Tornell J, Ohls-son C, Snaith M, Morgan DG. Am J Physiol Regul Integr Comp Physiol. 2004;287:R749–R758. doi: 10.1152/ajpregu.00134.2004. [DOI] [PubMed] [Google Scholar]
- Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, Mc-Govern RA, Chua SC, Jr, Elmquist JK, Lowell BB. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, Clements M, Al-Qassab H, Heffron H, Xu AW, et al. J Clin Invest. 2007;117:2325–2336. doi: 10.1172/JCI31516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford JC, Harrold JA, Boyland EJ, Lawton CL, Blundell JE. Drugs. 2007;67:27–55. doi: 10.2165/00003495-200767010-00004. [DOI] [PubMed] [Google Scholar]
- Heisler LK, Cowley MA, Tecott LH, Fan W, Low MJ, Smart JL, Rubinstein M, Tatro JB, Marcus JN, Holstege H, et al. Science. 2002;297:609–611. doi: 10.1126/science.1072327. [DOI] [PubMed] [Google Scholar]
- Kaplan JM, Song S, Grill HJ. Psychopharmacology (Berl) 1998;137:43–49. doi: 10.1007/s002130050591. [DOI] [PubMed] [Google Scholar]
- Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, et al. Cell Metab. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Nogueiras R, Wiedmer P, Perez-Tilve D, Veyrat-Durebex C, Keogh JM, Sutton GM, Pfluger PT, Castaneda TR, Neschen S, Hofmann SM, et al. J Clin Invest. 2007 doi: 10.1172/JCI31743. Published online September 20, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, et al. Nature. 2007;449:228–232. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- Zarjevski N, Cusin I, Vettor R, Rohner-Jeanrenaud F, Jeanrenaud B. Endocrinology. 1993;133:1753–1758. doi: 10.1210/endo.133.4.8404618. [DOI] [PubMed] [Google Scholar]
- Zhou L, Sutton GM, Rochford JJ, Semple RK, Lam DD, Oksanen LJ, Thornton-Jones ZD, Clifton PG, Yueh C-Y, Evans ML, et al. Cell Metab. 2007;6:398–405. doi: 10.1016/j.cmet.2007.10.008. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]