

Abstract
Erdheim–Chester disease (ECD) is a rare, systemic, non-familial histiocytic disorder, first described by Jakob Erdheim and William Chester in 1930. Most patients have multiple sites of involvement at presentation. The most common site of involvement is the long bones of the axial skeleton, which is seen almost universally, followed by the nervous system, heart, lungs, orbit and retroperitoneum, which are seen in up to 50% of cases.1 Cutaneous involvement is rarely a presenting symptom of ECD, with two reported cases in the English literature.2 The diagnosis of ECD is rarely made by skin biopsy because of the relative rarity of cutaneous involvement as a presenting feature, and also perhaps because of the difficulty in distinguishing the histopathological appearance from potential mimics. The importance of distinguishing ECD from other cutaneous disorders with similar pathology lies in the implications for both treatment and prognosis. ECD is an aggressive, often fatal disorder, with death from disease occurring in greater than 50% of patients.
Keywords: dermatology, granuloma, immunohistochemistry, pathology dermatopathology
Case report
A 35-year-old White male presented with a 5-month history of a brown/red occasionally pruritic, non-painful skin rash. The rash was initially confined to the central trunk but subsequently involved the upper extremities and neck. In addition, the patient also complained of vague bilateral lower extremity pain of several months’ duration.
Magnetic resonance imaging (MRI) of the lower extremities was performed for investigation of the patient’s leg pain, which showed diffuse replacement of the fatty marrow of the long bones with abnormally diminished T1 signaling, suggestive of an infiltrative process of the bone marrow such as storage disorder, leukemoid or histiocytic infiltration. A bone marrow biopsy was performed, which showed variable cellularity estimated at 30–50% with trilineage hematopoiesis, stainable iron, no granuloma and no evidence of a leukemic disorder. In addition, no lymphoid aggregates were noted. A skin biopsy from the mid-abdominal region performed showed skin with superficial and deep xanthogranulomatous infiltrate, including multinucleate Touton-like giant cells in keeping with the radiologic findings suggesting a histiocytic infiltration.
The patient was referred to the Genetic Clinic at Brigham and Women’s Hospital for investigation of a possible storage disorder. However, the acutely progressive tempo of his clinical course, essentially normal biochemical testing [in particular, the alkaline phosphatase, liver function tests and complete blood count (CBC)] and the absence of organomegaly were considered to be inconsistent with a storage disease (Fabry or Gaucher’s disease). A 99mTc-Methyl disphosphonate (TC-MDP) bone scan of the entire body was performed, which showed bilateral symmetric uptake in the frontal skull, distal femurs, tibiae and ankles, as well as the clavicular heads (Fig. 1). The patient was referred to Dermatology for further evaluation and management.
Fig. 1.
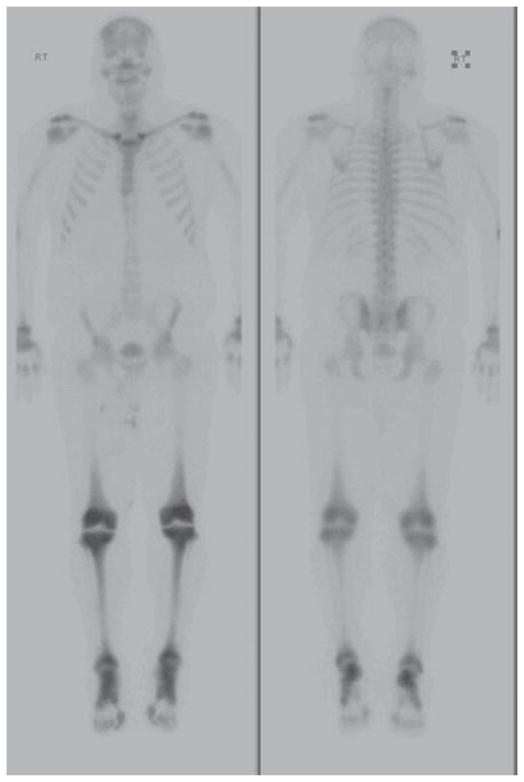
TC-MDP bone scan revealing characteristic bilateral symmetric uptake in the frontal skull, distal femurs, tibiae and ankles, as well as clavicular heads.
The patient’s past medical history was significant for obesity, sleep apnea, alcoholism and panic attacks. There was no significant family history. On physical examination, the patient was found to have diffuse brown maculopapular skin lesions on the trunk and extremities (Fig. 2A). Bilateral scleral and conjunctival brown infiltrates were noted (Fig. 2B), in addition to bony tenderness of both ankles and knees.
Fig. 2.
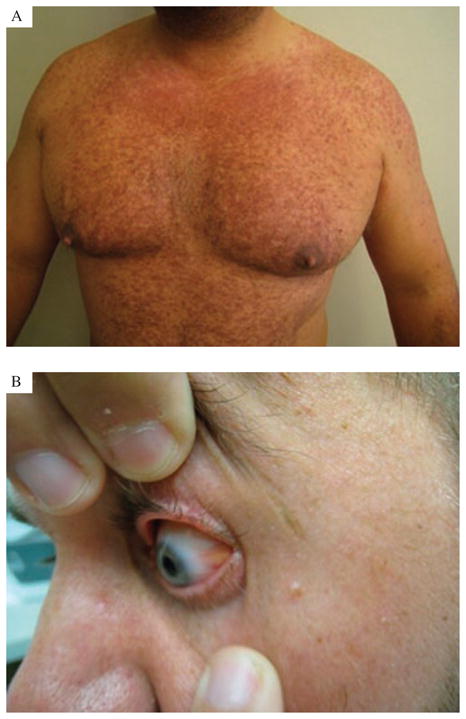
A) Photograph of the cutaneous eruption showing browney macular and indurated papular eruption in chest, upper abdomen and upper extremities. B) Photograph of the eye revealing scleral and conjunctival brown infiltrates.
Laboratory studies revealed an elevated C-reactive protein (CRP; >17.80), erythrocyte sedimentation rate (ESR; 25) and hypoalbuminemia (2.7 g/dl). CBC, urea and electrolytes, lipids, urinary studies, liver function and antinuclear antibodies were all within normal limits.
A skin biopsy of one of the maculopapular lesions was performed (Figs. 3 and 4). The 5-mm punch biopsy of the mid-back showed a dense superficial and deep dermal xanthogranulomatous infiltrate composed of histiocytes with abundant xanthomatous cytoplasm, multinucleate Touton-like giant cells and prominent intervening fibrosis. Immunohistochemistry showed the following staining profile in lesional cells: CD163 strongly positive, CD68 weakly positive and focal positivity for S-100 only in the most superficial component. HMB-45, CD1a and langerin were all negative.
Fig. 3.

A) Low-power magnification showing skin with dense superficial and deep xanthogranulomatous infiltrate and associated intervening fibrosis (H&E, original magnifications: ×2). B) Medium-power magnification showing dense superficial xanthogranulomatous infiltrate, including multinucleate Touton-like giant cells (H&E, original magnifications: ×20). C) Multinucleate Touton-like giant cells and associated intervening fibrosis (H&E, original magnifications: ×60).
Fig. 4.
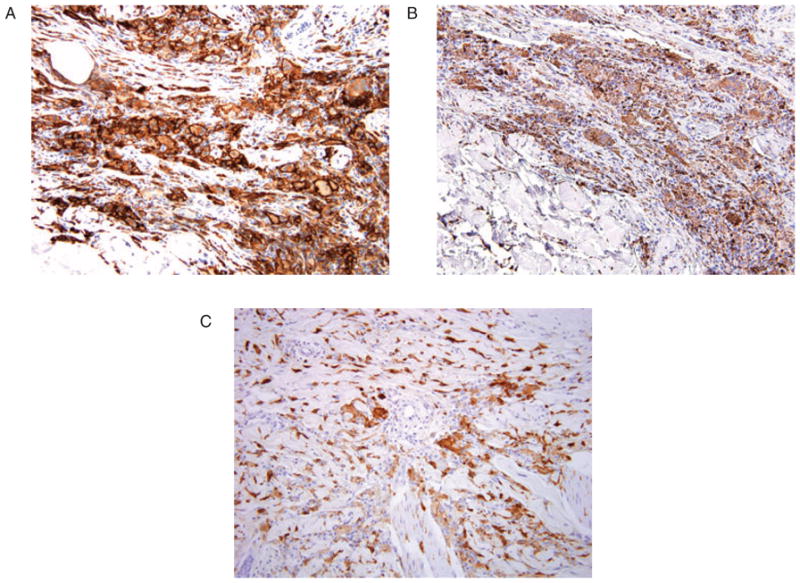
A) CD163 immunohistochemistry at ×20 magnification. B) CD68 immunohistochemistry at ×20 magnification. C) S-100 immunohistochemistry at ×20 magnification.
The subacute clinical course, lack of evidence for an inborn error of metabolism, including a normal bone marrow biopsy, the radiological findings, as well as the morphological appearance on routine H&E (hematoxylin and eosin) staining and the immunohistochemical profile led to a diagnosis of Erdheim–Chester disease (ECD) as the cause of the patient’s symptoms.
The patient received interferon therapy and is currently stable with the disease manifesting as lymphadenopathy, splenomegaly, bone disease and cutaneous and scleral involvement.
Discussion
ECD represents a rare systemic non-Langerhans cell histiocytosis. There have been roughly 350 cases reported worldwide. There is a slight male predominance, and the disease is usually diagnosed in patients in their fifth to seventh decades of life.3 The rarity of this disease, coupled with its less than specific histopathology, makes this a multidisciplinary diagnosis, requiring the combination of clinical, radiological and pathological findings. The most common clinical manifestations of this disease are outlined in Table 1. Although cutaneous involvement is rarely a presenting symptom of ECD, the histopathological and radiological appearance of ECD is usually relatively characteristic and can suggest such a diagnosis in the appropriate clinical setting. There is symmetrical sclerosis of the metaphyses and diaphyses of the long bones, usually the distal femur and proximal tibia and fibula,4 which have a pathognomonic appearance on bone scan.1 The sclerosis may be diffuse or focal, and lytic lesions may also be present, albeit much less commonly. In this case the patient showed the typical pattern of bony involvement with characteristic findings on MRI, which, in the absence of any other infiltrative systemic disease or storage disorder, is consistent with the diagnosis.
Table 1.
Clinical presentation of ECD
| System | Clinical signs and symptoms |
|---|---|
| Skeletal | Bone pain, sclerosis and lytic lesions, predominantly long bones |
| Neurological | Diabetes insipidus |
| Cardiac | Pericardial and myocardial fibrosis, heart failure |
| Pulmonary | Interstitial fibrosis, respiratory failure |
| Orbital | Exophthalmus, xanthelasma-like lesions |
| Retroperitoneal | Peri-aortic or peri-renal fibrosis |
| Cutaneous | Disseminated or localized maculopapular skin lesions, xanthelasma-like skin lesions |
Skin involvement, when present, most commonly occurs on the eyelids causing lesions clinically similar to xanthelasmas associated with dyslipidemias. In this case, the skin was the dominant site of clinical alteration and was characterized by widespread involvement of the chest, neck, and upper extremities. The skin biopsy findings, when coupled with the radiological and clinical features, allowed the diagnosis of ECD to be made. However, the pathologic findings on skin biopsy show considerable overlap with other disorders and thus a diagnosis of ECD should be made with caution. ECD is often a fatal disorder, with death occurring in greater than 50% of patients. Death often occurs within 1 year of diagnosis.5 The prognosis is worse in patients with extraskeletal involvement.5 Death is often because of cardiac or respiratory failure resulting from heart or lung involvement.6,7 Because of the small number of cases of this disease worldwide, the treatment and management of patients is mostly based on case series rather than trials. Treatment modalities include immunosuppression, high-dose chemotherapy, interferon alfa-2α therapy, radiation therapy and bone marrow transplant;6 yet, despite such aggressive therapeutic regimens, the prognosis remains extremely poor.
The pathogenesis of ECD remains elusive. Whether it represents a reactive process rather than a neoplastic process is still debated, but current evidence now favors a clonal origin for this histiocytic proliferation.3,8 In addition, the histiocytes appear to show a monocytic-macrophage origin/lineage with CD163 expression.8 A balanced chromosomal translocation t(12;15;20) has been described in one patient with osseous ECD.3 However, the use of ancillary molecular techniques for the diagnosis of ECD is not yet established.
The histopathologic findings on skin biopsy of lesions associated with ECD are similar to those seen at other sites such as lung3,9 and retroperitoneum,5,9,10 which have been more commonly reported as yielding the diagnosis of ECD than skin. The lesions comprise a dermal infiltrate of bland histiocytes with xanthomatous cytoplasm and Touton-like giant cells with intervening fibrosis. It is this fibrosis that is the most characteristic morphologic finding on routine H&E stained sections. The histiocytes express CD163, consistent with histiocytic lineage, and also CD68. Curiously, our case showed weak CD68 staining. It has been reported that the histiocytes of ECD are typically negative for S-100; however, Egan et al.9 reported that lesional tissue from 4/5 lung biopsies from patients with ECD showed S-100 positivity, two with extensive and two with limited expression. In our case, the lesional cells showed focal S-100 positivity. It is thus probable that the pattern of S-100 positivity in ECD is variable. The histiocytes should not express CD1a and langerin, in keeping with its non-Langerhans cell origin. The histiocytes of ECD do not show Birbeck granules by electron microscopy, but cytoplasmic lipid droplets can be found.11
The differential diagnosis of cutaneous ECD includes other histiocytic disorders exhibiting xanthomatous change or containing multinucleate giant cells as well as pure xanthomas. Such lesions can be broadly divided into three main groups: those with a xanthogranulomatous pattern, true xanthomas and those with xanthomatous change occurring as a secondary phenomenon within a histiocytic, dendritic, or Langerhan cell proliferation.
The differential diagnosis of ECD includes the following:
Juvenile or adult xanthogranuloma (XG)
Disseminated XGs have been reported and have been linked to leukemia, lymphoma and other hematologic malignancies. XGs are composed of small histiocytes with Touton giant cells, foamy histiocytes and a mixed inflammatory infiltrate composed of lymphocytes and neutrophils. Lesions of a long duration may show fibrosis, adding to the diagnostic difficulty in distinguishing such lesions from those of ECD. In addition they show a similar immunophenotype, as lesional histiocytes express CD68 and factor XIIIa and lack expression of CD1a and S-100. Probably the most helpful clue is the clinical history12 and one should not hesitate to survey the skeletal system and entertain the diagnosis of ECD as a possibility, especially in adults or in atypical clinical presentations. Unlike ECD, where bony involvement is most probably to be symmetric and sclerotic with almost diagnostic value on bone scan, patients with XG and bone involvement are more probable to have focal, lytic lesions.
Rosai–Dorfman syndrome
This represents a benign disorder with histiocyte accumulation in lymph nodes and also occasionally in the skin. The pathology is typified by prominent emperipolesis and a mixed infiltrate with histiocytes that avidly express S-100.13
Xanthoma disseminatum (XD)
XD represents a rare disorder, with only 100 reported cases, that most commonly affects young males. Much like ECD, lesions of bone or the central nervous system are not uncommon, and histopathologically these lesions may be indistinguishable from lesions of ECD. Immunohistochemistry is not helpful, as the histiocytes lack CD1a expression but express CD68. The most helpful tool is the clinical presentation. XD primarily affects the skin, often there is mucosal involvement and only rarely is bony involvement seen.14 In contrast, in ECD the axial skeleton is the most common site involved, and skin involvement is rare, especially at presentation. While the natural history of XD is usually benign with spontaneous resolution of cutaneous lesions over 2–40 years,15,16 lesions in critical anatomical locations may result in morbidity and mortality.
Necrobiotic xanthogranuloma (NXG)
This entity typically affects elderly patients. The lesions commonly involve the periorbital area. NXG has a strong association with paraproteinemia (IgG monoclonal gammopathy) and cryoglobulinemia. Histopathologically, the presence of necrosis with cholesterol clefts and surrounding granulomas represents a pattern that helps in differentiating NXG from ECD and other lesions within the XG family.17
True xanthomas
These are associated with dyslipidemic states. These lesions are composed of aggregates of foamy histiocytes in the upper dermis. Fibrosis can be seen secondarily in tuberous lesions. The immunophenotype is similar to that in ECD, as the constituent foamy cells express CD68, sometimes express factor XIIIa and lack expression of S-100 or CD1a. The combination of clinical features and pathology is helpful in distinguishing true xanthomas from ECD.18
Langerhans cell histiocytosis (LCH)
This disease presents with an infiltrate of histiocytes which have a characteristic reniform nuclear morphology and express the markers of Langerhans cells, namely CD1a, S-100 and langerin.19 The expression of S-100 or CD1a can be somewhat variable,20 and in such a context ECD might be difficult to distinguish from LCH. LCH is typically classified into three forms: unifocal, multifocal unisystem and multifocal multisystem. The last type occurs mostly in children less than 2 years old and carries a poor prognosis, even after aggressive chemotherapy. The prognosis is excellent for unifocal disease. With multifocal disease 60% have a chronic course, 30% achieve remission and mortality is up to 10%.21 ECD carries a worse prognosis compared to LCH.
In summary, although ECD presenting with cutaneous involvement is rare, it is necessary to consider it in the differential diagnosis of cutaneous histiocytosis and to distinguish it from its potential mimics with similar histopathology. An accurate diagnosis of ECD will guide appropriate treatment and provide a clear prognosis.
References
- 1.Spyridonidis TJ, Giannakenas C. Erdheim-Chester Disease: a rare syndrome with a characteristic bone scinitigraphy pattern. Ann Nucl Med. 2008;22:323. doi: 10.1007/s12149-007-0110-3. [DOI] [PubMed] [Google Scholar]
- 2.Garg T, Chander R, Gupta T, Mendiratta V, Jain M. Erdheim-Chester disease with cutaneous features in an Indian patient. Skinmed. 2008;7:103. doi: 10.1111/j.1751-7125.2008.07372.x. [DOI] [PubMed] [Google Scholar]
- 3.Vencio EF, Jenkins RB. Clonal cytogenetic abnormalities in Erdheim-Chester disease. Am J Surg Pathol. 2007;31:319. doi: 10.1097/01.pas.0000213366.33627.a0. [DOI] [PubMed] [Google Scholar]
- 4.Al-Quran S, Reith J, Bradley J, Rimsza L. Erdheim-Chester disease: case report, PCR-based analysis of clonality, and review of literature. Mod Pathol. 2002;15:666. doi: 10.1038/modpathol.3880583. [DOI] [PubMed] [Google Scholar]
- 5.Veyssier-Belot C, Cacoub P. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Balitimore) 1996;75:157. doi: 10.1097/00005792-199605000-00005. [DOI] [PubMed] [Google Scholar]
- 6.Mills JA, Gilberto Gonzalez R. Case 25-2008-A 43 year old man with fatigue and lesions in the pituitary and cerebellum. NEJM. 2008;359:736. doi: 10.1056/NEJMcpc0804623. [DOI] [PubMed] [Google Scholar]
- 7.Rush WL, Andriko JAW. Pulmonary pathology of Erdheim-Chester disease. Mod Pathol. 2000;13:747. doi: 10.1038/modpathol.3880130. [DOI] [PubMed] [Google Scholar]
- 8.Dickson BC, Pethe V, Chung CT, et al. Systemic Erdheim-Chester disease. Virchows Arch. 2008;452:221. doi: 10.1007/s00428-007-0538-9. [DOI] [PubMed] [Google Scholar]
- 9.Egan AJ, Boardman LA, Tazelaar HD, et al. Erdheim-Chester disease: clinical, radiologic and histopathologic findings in five patients with interstitial lung disease. Am J Surg Pathol. 1999;23:17. doi: 10.1097/00000478-199901000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Chester W. Uber lipoidgranulomatose. Virchows Arch Pathol Anat. 1930;279:561. [Google Scholar]
- 11.Ono K, Oshiro M. Erdheim-Chester disease: A case report with immunohistochemical and biochemical examination. Human Pathol. 1996;27:91. doi: 10.1016/s0046-8177(96)90145-8. [DOI] [PubMed] [Google Scholar]
- 12.Kraus MD, Haley JC, Ruiz R, et al. “Juvenile” xanthogranuloma: an immunophenotypic study with a reappraisal of histogenesis. Am J Dermatopathol. 2001;23:104. doi: 10.1097/00000372-200104000-00004. [DOI] [PubMed] [Google Scholar]
- 13.Middel P, Hemmerlein B, Fayyazi A, et al. Sinus histiocytosis with massive lymphadenopathy: evidence for its relationship to macrophages and for a cytokine-related disorder. Histopathology. 1999;35:525. doi: 10.1046/j.1365-2559.1999.00746.x. [DOI] [PubMed] [Google Scholar]
- 14.Zelger B, Cerio R, Orchard G, et al. Histologic and immunohistochemical study comparing xanthoma disseminatum and histiocytosis X. Arch Dermatol. 1992;128:1207. [PubMed] [Google Scholar]
- 15.Caputo R, Veraldi S, Grimalt R, Gianotti R, Tosti A, Varotti C, et al. The various clinical patterns of xanthoma disseminatum: consideration of 7 cases and review of the literature. Dermatology. 1995;190:19. doi: 10.1159/000246628. [DOI] [PubMed] [Google Scholar]
- 16.Flach DB, Winkleman RK. Juvenile xanthogranuloma with CNS lesions. J Am Acad Dermatol. 1986;14:405. doi: 10.1016/s0190-9622(86)70049-2. [DOI] [PubMed] [Google Scholar]
- 17.Finan MC, Winkelmann RK. Necrobiotic xanthogranuloma with paraproteinemia. A review of 22 cases. Medicine (Baltimore) 1986;65:376. doi: 10.1097/00005792-198611000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Braun-Falco O, Eckert F. Macroscopic and microscopic structure of xanthomatous eruptions. Curr Probl Dermatol. 1991;20:54. doi: 10.1159/000420008. [DOI] [PubMed] [Google Scholar]
- 19.Hage C, Willman CL, Favara BE, et al. Langerhans’ cell histiocytosis (histiocytosis X): immunophenotype and growth fraction. Hum Pathol. 1993;24:840. doi: 10.1016/0046-8177(93)90133-2. [DOI] [PubMed] [Google Scholar]
- 20.Tomaszewski MM, Lupton GP. Unusual expression of S-100 protein in histiocytic neoplasms. J Cutan Pathol. 1998;25:129. doi: 10.1111/j.1600-0560.1998.tb01704.x. [DOI] [PubMed] [Google Scholar]
- 21.Komp D, El Mahdi A, Starling K, et al. Quality of survival in histiocytosis X: a Southwest Oncology Group study. Med Pediatr Oncol. 1980;8:35. doi: 10.1002/mpo.2950080106. [DOI] [PubMed] [Google Scholar]