

Abstract

The zinc finger protein EVI1 is causally associated with acute myeloid leukemogenesis, and inhibition of its function with a small molecule therapeutic may provide effective therapy for EVI1-expressing leukemias. In this paper we describe the development of a pyrrole–imidazole polyamide to specifically block EVI1 binding to DNA. We first identify essential domains for leukemogenesis through structure–function studies on both EVI1 and the t(3;21)(q26;q22)-derived RUNX1-MDS1-EVI1 (RME) protein, which revealed that DNA binding to the cognate motif GACAAGATA via the first of two zinc finger domains (ZF1, encompassing fingers 1–7) is essential transforming activity. To inhibit DNA binding via ZF1, we synthesized a pyrrole–imidazole polyamide 1, designed to bind to a subsite within the GACAAGATA motif and thereby block EVI1 binding. DNase I footprinting and electromobility shift assays revealed a specific and high affinity interaction between polyamide 1 and the GACAAGATA motif. In an in vivo CAT reporter assay using NIH-3T3-derived cell line with a chromosome-embedded tetinducible EVI1-VP16 as well as an EVI1-responsive reporter, polyamide 1 completely blocked EVI1-responsive reporter activity. Growth of a leukemic cell line bearing overexpressed EVI1 was also inhibited by treatment with polyamide 1, while a control cell line lacking EVI1 was not. Finally, colony formation by RME was attenuated by polyamide 1 in a serial replating assay. These studies provide evidence that a cell permeable small molecule may effectively block the activity of a leukemogenic transcription factor and provide a valuable tool to dissect critical functions of EVI1 in leukemogenesis.
The EVI1 gene was first identified as a site of proviral insertion in retrovirally induced murine myeloid leukemias, which resulted in activation of the gene with overexpression of at least three different isoforms, p135, p123, and p103, that possess varying numbers of C2H2-type zinc fingers. The p135 and p123 isoforms possess 10 zinc finger motifs in two domains separated by 480 amino acids, suggesting a role in transcriptional regulation. The N-terminally located zinc finger domain 1 (termed ZF1, and encompassing fingers 1–7; amino acids 1–249) binds specifically to GACAAGATA-like sequences;1,2 domain 2 binds GAAGATGAG motif.3 Transformation assays have shown that the Nterminal, but not the C-terminal, region is required for transforming activity.4 Transfection assays for transcriptional activation by EVI1 using synthetic reporters bearing EVI1 binding sites indicate that EVI1 can act as either a transcription repressor5-7 or an activator.8-10 Indeed, a repression domain that binds the corepressor CtBP was identified by Palmer et al. and is present in the p135, p123, and p103 isoforms.7,11,12 The overlap between the binding motif for domain 1 (GACAAGATA) and the GATA protein binding site (WGATAR) suggested that EVI1 may bind to GATA sites in the genome and block the action of differentiationpromoting GATA proteins.5,6 This possibility was pursued in a series of in vitro and in vivo studies, from which it was concluded that high affinity binding by EVI1 to DNA via domain 1 required a longer sequence than WGATAR,5 specifically GACAAGATA.5
Studies by Delwel et al. indicated that zinc fingers 4–7 within zinc finger domain 1 (ZF1) of EVI1 are necessary for that domain to bind to DNA with sequence specificity.2 Through in vitro and in vivo structure–function studies, we determined that amino acids Q199 and R205 residing in zinc finger six are critical for sequencespecific DNA binding. This analysis allowed for the generation of single amino acid missense mutations (R205N and Q199D) that are essentially devoid of DNA binding ability.13 Furthermore, through a series of in vitro and in vivo studies, it was determined that the nature of EVI1 zinc fingers 1–7 binding to its cognate DNA motif is high affinity and highly specific.5 To identify in vivo targets for EVI1, we devised a tetracycline-regulated EVI1-VP16 chimera that had the binding specificity of EVI1 zinc finger domain 1 fused to the potent transcriptional activation domain of VP16. We expressed this in NIH3T3 fibroblasts under tetracycline regulation and used gene expression profiling on oligonucleotide microarrays to identify upregulated genes. This allowed the identification of 16 genes that were specifically induced by EVI1-VP16 but not by the R205N mutant that lacked DNA binding.13 The EVI1-VP16-responsive genes include genes encoding transcription factors (e.g., GATA2), signaling molecule (e.g., Nik) and extracellular matrix components (e.g., decorin). For many of these genes, EVI1 binding sites were identified in cis by a combination of in silico analysis of evolutionary conserved binding motifs and chromatin immunoprecipitation. Importantly, most of these sites were occupied by EVI1 both in fibroblasts and in myeloid leukemia cells overexpressing EVI1.13 These data suggest that these genes were indeed direct targets of EVI1 in leukemic cells and may contribute to leukemogenesis.
The development of small molecules that block the binding of EVI1 to DNA may be potential therapeutic strategy for treatment of acute myeloid leukemia. Hairpin pyrrole–imidazole (Py-Im) polyamides are a class of synthetic cell permeable ligands that can be programmed to bind DNA with high affinity and sequence specificity.14,15 The molecular recognition properties of these compounds for specific DNA sequences are encoded by the side-by-side arrangement of N-methylpyrrole (Py), N-methylimidazole (Im), and N-methyl-3-hydroxypyrrole (Hp) carboxamides that form distinct hydrogen bonds to the four Watson–Crick base pairs in the minor groove. Im/Py pairs distinguish G·C from C·G, whereas Hp/Py specifies T·A from A·T and Py/Py pairs are degenerate for T·A and A·T.16-18 These small molecules achieve affinities and specificities comparable to those of DNA binding proteins,19 have been designed to target a broad repertoire of DNA sequences,20 inhibit the DNA binding of a wide range of transcription factors, bind to chromatin,21 are cell permeable,22,23 and have been shown to downregulate endogenous gene expression in cell culture.24-27
The exact role of DNA binding of EVI1 in leukemogenesis remains elusive. In this paper, we demonstrate that DNA binding via ZF1 is essential for malignant transformation. We also describe our efforts to directly target the EVI1–DNA binding interaction using a pyrrole–imidazole polyamide. Our results indicate that polyamide 1 significantly blocked the ZF1-mediated interaction between EVI1 and DNA and partially inhibited leukemic cell growth. This study thus describes a first attempt at inhibiting the leukemogenic activity of EVI1 with a targeted therapy; this polyamide provides a valuable tool for dissecting critical ZF1–DNA direct interaction.
EXPERIMENTAL PROCEDURES
Polyamide Synthesis
Py-Im polyamides 1–2 were synthesized on Kaiser oxime resin utilizing solid-phase synthesis protocols.28 Polyamides were cleaved from resin by aminolysis with 3,3′-diamino-N-methyldipropylamine and HPLC purified, yielding compounds 5 and 6 (Figure S1). C-terminal isophthalic acid (IPA) conjugates (polyamides 1 and 2) were synthesized from 5 or 6 utilizing previously reported conditions.26 Detailed experimental protocols to cleave the benzyl carbamate (Cbz) protecting group on the chiral β-NH2 turn and transform the resulting amine to the β-NHAc functionality have been reported elsewhere.29 The benzyl carbamate (Cbz) protecting group on the chiral β-NH2 turn was cleaved by our standard conditions.29 Polyamides were purified by preparative HPLC on an Agilent 1200 Series instrument equipped with a Phenomenex Gemini preparative column (250 × 21.2 mm, 5 μm) with the mobile phase consisting of a gradient of acetonitrile (MeCN) in 0.1% CF3CO2H (aqueous). Polyamide purity was evaluated by analytical HPLC analysis on a Beckman Gold instrument equipped with a Phenomenex Gemini analytical column (250 × 4.6 mm, 5 μm), a diode array detector, and the mobile phase consisting of a gradient of MeCN in 0.1% CF3CO2H (aqueous). Matrix-assisted, LASER desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) was performed on an Applied Biosystems Voyager DE-Pro spectrometer using α-cyano-4-hydroxycinnamic acid as matrix. Electrospray ionization (ESI+) MS was performed on a Waters Acquity UPLC-LCT Premiere XE TOF-MS system. Polyamide 1 MALDI-TOF MS calculated for C65H77N22O12 [M + H]+ 1357.6, found 1357.2; polyamide 2 MALDI-TOF calculated for C65H77N22O12 [M + H]+ 1357.6, found 1357.4.
Thermal melting temperature analysis/UV absorption spectrophotometry was performed as previously described.29,30 Results from this study are shown in Table 1.
Table 1.
Thermal Stability Data for Polyamides 1 and 2 Complexes with DNAa
|
| |||||
|---|---|---|---|---|---|
| Match Polyamide | Tm/°C | ΔTm/ °C | Mismatch Polyamide | Tm/ °C | ΔTm/ °C |
|
|
67.6 (±0.2) | 17.3 (±0.9) |
|
57.1 (±0.2) | 6.8 (±0.9) |
All values reported are derived from at least three melting temperature experiments with standard deviations indicated in parentheses. ΔTm values are given as Tm(DNA/polyamide) − Tm(DNA). The propagated error in Tm measurements is the square root of the sum of the square of the standard deviations for the Tm values.
DNase I Footprinting
DNase I footprinting was performed on a 5′-32P-labeled PCR amplicon essentially as previously described.31 Primer oligonucleotides (Integrated DNA Technologies) 5′-AGGCGATTAAGTTGGGTAACG-3′ (forward) and 5′-TCGGCCTCTGCATAAATAAAA-3′ (reverse) were designed to amplify a 223 basepair region of p406 surrounding an insert (5′-AGATCTGACAAGATAAATTAAGAGATCT-3′) containing the EVI1 binding site (underlined). The forward primer was radiolabeled using [α-32P]-dATP (MP Biomedicals) and polynucleotide kinase (Roche). The PCR product was generated using the primer pair and Expand High Fidelity PCR Core Kit (Roche) following the manufacturer’s protocol, and an unradiolabeled PCR amplicon was confirmed by sequencing (Laragen). The PCR product was purified on a 7% nondenaturing preparatory polyacrylamide gel (5% crosslink) and visualized by autoradiography. The labeled band was excised, crushed, and soaked overnight (14 h) in 2 M NaCl. The gel pieces were removed by centrifugal filtration, and the DNA was precipitated with 2-propanol (1.5 volumes). The pellet was washed with 75% ethanol, lyophilized to dryness, and then resuspended in 1 mL of RNase-free H2O. Footprinting was performed on 1 μM, 300 nM, 100 nM, 30 nM, 10 nM, 3 nM, 1 nM, 300 pM, 100 pM, 30 pM, and 10 pM solutions of compounds polyamide 1 (match polyamide) and polyamide 2 (mismatch polyamide), where polyamide solutions were quantitated using ε = 69 500 M−1 cm−1. All reactions were carried out in a volume of 400 μL according to the published procedures.31 Quantitation by storage phosphor autoradiography and determination of equilibrium association constants (Table 2) were as previously described.31 Chemical sequencing reactions were performed according to published protocols.32,33
Table 2.
Equilibrium Association Constants (M−1) Determined for Polyamides 1 and 2
| 1 | 2 | |
|---|---|---|
| site I: TGACAA | 1.7 (±0.3) × 1010 | 1.0 (±0.3) × 108 |
| site II: TGTAAA | 3.1 (±0.6) × 109 | 5.5 (±1.0) × 108 |
Retroviral Constructs
The EVI1R769C mutation was created with mutagenic oligonucleotides 5′ GTTGCGCACATGACATTGCAGGTTGGAAG 3′ using the dut-ung system34 with single-stranded p39413 serving as a template. The resultant plasmid, p684, was confirmed by DNA sequencing and transferred to pBabe-Puro35 as a BamHI fragment to create p692. To create the RME fusion, 1.63 kb EcoR1-BglII fragment from a human pBSRME plasmid (p495; gift of G. Nucifora) was inserted into EcoR1-BglII-cut plasmids p394, p620, and p684 to create p798, gs475, and gs477, respectively. In each of these, the NotI site was replaced with an EcoR1 site, and the 3.6 kb EcoR1 product was inserted into pMIGR1 to generate p854, p855, and p856, respectively. The creation of EVI1R205N mutation has been described previously.13
Production of Virus
Retroviral stocks were prepared by calcium phosphate/DNA precipitate-mediated gene transfer into BOSC cells in the presence of 25 μM chloroquine. Retroviral plasmid constructs (5 μg per 60 mm plate) were coprecipitated with pNCA, a plasmid containing a full-length, functional clone of Mo-MuLV. DNA was allowed to remain on the cells for 12–18 h, and the medium was then changed. Viral supernatants were harvested at 48 and 72 h post-transfection. Viral titers were determined by infecting BAF3 cells and determining the percent GFP+ at 48 h postinfection.
Transformation Assays
Rat1 fibroblast cells were transduced with pBabe-Puro-based retroviral constructs via retroviral infection in the presence of 8 μM Polybrene. Cells were selected in 2 μg/mL puromycin, and population of resistant cells was assayed by Western blot for protein expression and by colony formation for malignant transformation. Assays were performed in triplicate in 0.6% soft agar/DMEM supplemented with 20% FBS, seeding 1 × 103 cells per 60 mm plate in 3 mL total volume with a 0.3% agar overlay, also containing DMEM and 20% FBS. Colonies in the three plates were totalled and reported in Figure 1. The entire experiment was repeated once. RME mutants were assayed by serial replating:36 Bone marrow was harvested from male Balb/c mice 5d following injection with 5-fluorouracil. Cells were flushed from the marrow with Tissue Dissociation Medium (Life Sciences) at room temperature. Mononuclear cells were purified on a metrizamide cushion (Histopaque, Sigma-Aldrich), washed in phosphate buffered saline, and then cultured in bone marrow culture medium (DMEM/10% FBS/pen/strep/amphotericinB/6 ng/mL IL-3, 10 ng/mL IL-6, and 100 ng/mL SCF). On the day following bone marrow harvest, cells were infected with retrovirus by “spinfection” as described.37 The serial replating assay was performed as described.36
Figure 1.
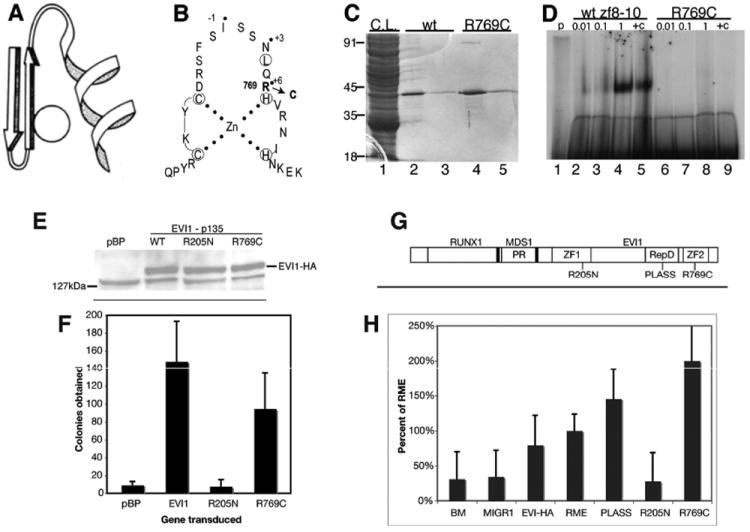
Analysis of zinc finger mutants of EVI1. (A) Ribbon structure of a C2H2 Krüppel-type zinc finger. (B) Amino acid sequence of zinc finger 9 of EVI1, showing the EVI1R769C mutation. Dotted residues are those on the exposed face of the helix and interacting with DNA. (C) Purification of GST-tagged EVI1. 1. Crude lysates; 2 and 3, glutathione eluant from affinity resin of wildtype protein, fractions 1 and 2; 4 and 5, eluant of EVI1R769C mutant. (D) Electromobility shift assay using proteins shown in lanes 3 and 5 of panel C, and a 32P-radiolabeled probe containing the GAAGATGAG motif. Lane 1, probe alone; lanes 2–5, dilutions of wildtype protein as indicated; lanes 6–9, dilutions of mutant protein as indicated. (E) Upper panel, Western blot analysis of pools of drug-selected retrovirally transduced Rat1cells, infected with the constructs indicated. To the left, location of molecular weight marker. Band representing hemagglutinin (HA)-tagged EVI1 is indicated at right. The 127 kDa band seen in all lanes represents background signal. (F) Colonies in soft agar formed after plating transduced Rat1 cells in soft agar. (G) Diagram of the structure of the RUNX-MDS1-EVI1 (RME) fusion protein product of t(3;21), with mutations R205N and R769C as indicated. Not to scale. (H) Serial replating assay of transduced primary bone marrow cells from 5-FU-treated C57BL/6 mice. Retrovirally transduced mouse bone marrow cells were plated in cytokine-supplemented semisolid methylcellulose medium, allowed to form colonies, and then passaged onto secondary and then tertiary plates of the same medium. The x-axis presents the number of colonies formed on tertiary plating, which represent malignantly transformed cells; each bar indicates the average of colonies in triplicate plating. Student t test showed the R205N significantly different than vector-transduced control (p < 0.001). This experiment was repeated with essentially the same results.
Electromobility Shift Assay
ZF2 of EVI1 from N710 to E82138 was inserted as a PCR product into the EcoR1 site of plasmid pGEX1N; the PCR product was generated with primers 5′ GATGAATTCAACACCCTGCCAGAG 3′ and 5′ GACGAATTCTAGCTCTGAGTGAGG 3′. This construct was used to produce GST-tagged protein which was purified as described.39
Hexahistidine-tagged amino-terminal fragment of EVI1 (amino acids 1–249, encompassing ZF1) was purified from IPTGinduced E. coli Rosetta strain harboring a pET-22b+-EVI1 plasmid by nickel affinity chromatography as described.13 The elute was dialyzed against 12.5% glycerol, 5 mM MgCl2, 0.01 mM ZnSO4, 50 mM NaCl, 10 mM DTT, 50 mM HEPES, pH 7.9. Purity and concentration of the protein were determined with a Coomassie Blue stained SDS-PAGE. Oligonucleotides containing the EVI1 binding site (5′-TCGACTTGACAAGATAAGCATAG-3′ and 3′-AGCTGAACTGTTCTATTCGTATC-5′) were annealed and end-labeled with [γ32P]ATP to a specific activity of ~4300 cpm/fmol. Binding reactions (50 μL, containing 10 000 cpm of labeled oligonucleotides, 5 mM MgCl2, 0.01 mM ZnSO4, 50 mM NaCl, 10 mM DTT, 50 mM HEPES, pH 7.9, 100 μg/mL BSA, 50 μg/mL, poly(dI-dC) (Sigma-Aldrich, St. Louis, MO), and 12.5% glycerol) were incubated at 25 °C for 30 min prior to fractionation on 4% nondenaturing PAGE (39:1 polyacrylamide–bisacrylamide, 0.5× TBE, and 2.5% glycerol). Active protein was determined by DNA titration experiments as described.40 Polyamides were preincubated at room temperature for 30 min with 32P-labeled oligonucleotides in a 20 μL reaction containing 5 mM MgCl2, 0.01 mM ZnSO4, 50 mM NaCl, 10 mM DTT, 50 mM HEPES, pH 7.9, 10 μg/mL BSA, 50 ug/mL, poly(dI-dC), and 12.5% glycerol. The amount of oligonulceotides (labeled and unlabeled) and protein varied in the femtomole range with each experiment, as described under Results. 10 000 cpm were used for each reaction, and the appropriate amount of unlabeled (cold) DNA was added to give a total amount of 1250 fmol DNA per reaction. Protein was added, and the binding reaction was performed as described above. All dilutions were performed in binding buffer in the presence of 100 μg/mL BSA. Dried gels were exposed to BioMax MR film at −80 °C. Quantitation was performed with a Molecular Dynamics Storm 840 phosphor imager using Quantity One software. All reactions were performed in triplicate; results were adjusted to normalize total counts in bound and free oligonucleotides. The protein:DNA Ka was determined as described,40 with the assumption that the stoichiometry of protein/DNA binding is 1:1. From the plot of protein/DNA complex [DP] against total concentration of protein [PT], the slope can be determined and this can be used to determine Keq.40
CAT Assay and Target Gene Quantitation
Cell line 6D-AP1713 was cultured in DMEM supplemented with 10% calf serum, glutamine, penicillin, streptomycin, and 0.5 μg/mL tetracycline, and plated on 10 cm plates at 2 × 105 per plate, with three plates for each condition. The following day, cells were washed and plated in medium devoid of tetracycline and containing various concentrations of polyamide. After 48 h, the cells were harvested and assayed for CAT activity and for protein concentration as described.13
Inhibition Studies
Leukemic lines were plated in 96-well plates at 3000 cells per well in 100 μL. 24 h later, polyamide was added to various concentrations. At various times after polyamide addition, 1 μC 3H-labeled thymidine was added, and the cells were incubated for an additional 16 h. Cells were harvested onto a glass filter paper sheet with a Tomtek cell harvester; the sheet was saturated with scintillation fluid and counted. For the colony formation assay, 10 000 cells were plated in IL-3-supplemented mixed with 1.5 mL of methylcellulose supplemented with 15% of WEHI condition media and 1% glutamine and were plated on 30 mm plates (three plates per sample). Cells were incubated in 37 °C for 1–2 weeks, and colonies were counted.
RESULTS
DNA Binding via First Set of Zinc Fingers Is Required for Transformation
Toward the development of agents to inhibit EVI1 action, we performed structure–function studies to identify domains required for malignant transformation. We previously described an R205N mutation, located at the +6 position of finger 6 in ZF1 that results in loss of sequencespecific binding via ZF1 to the GACAAGATA motif.13 Here we describe the development of EVI1 mutants that fail to bind DNA via ZF2 (fingers 8–10). ZF2 binds with high affinity to GAAGATGAG motif.3 On the basis of this understanding of zinc finger structure41 (Figure 1A) and analysis of non-DNA binding mutants in other systems,42,43 we predicted that finger 9, with N and R at positions +3 and +6 (Figure 1B), would contribute considerable strength to binding and that R769 in particular might be critical for domain 2 binding to DNA. To test this, we mutated residue 769 to C, mimicking a mutation found in NGFI-A that resulted in loss of DNA binding.43 We fused EVI1 comprising zinc fingers 8, 9, and 10 (from N710 to E82138) both with and without the R769C mutation to glutathione S-transferase (GST), purified by affinity chromatography (Figure 1C), and tested for ability to bind to a doublestranded radiolabeled oligonucleotide containing the GAAGATGAG motif (Figure 1D). This revealed that the R769C mutant was essentially incapable of high affinity binding.
Next, we wanted to assess if DNA binding via ZF1 or ZF2 is required for EVI1-mediated malignant transformation; for this we employed mutants R205N and R769C, respectively. Rat1 cells were transduced with pBabe-puro-based retroviruses bearing HA-tagged EVI1, either wt, EVI1R205N, or EVI1R769C. Pools transduced cells were shown to express EVI1 by Western blot, with equal expression of the three constructs (Figure 1E). These cells were seeded in soft agar, and colonies (>1 mm) were enumerated after 10 days (Figure 1F). Rat1 cells transduced with wildtype EVI1 yielded over 140 colonies/plate in soft agar; the R769C mutant was nearly as transforming, yielding over 90 colonies. Remarkably, the R205N mutation was devoid of all transforming ability, having essentially the same number of colonies per plate as empty vector, pBabe-Puro (Figure 1F).
To further assess the effect of the R205N and R769C mutations, we determined their effects on transformation of primary bone marrow cells by the RUNX1-MDS1-EVI1 fusion protein (termed RME), which is the product of the leukemia-associated t(3;21) (Figure 1G). We employed a serial replating assay in semisolid medium supplemented with growth factors.44 In two separate experiments, bone marrow was harvested 5 days after treatment of C57BL/6 mice with 5-fluorouracil (5FU) and was transduced with high titer MIGR1-based retrovirus containing either wildtype EVI1p135, RME, RMER205N, RMER769C, or RMED586A/L587S or no insert. As expected, vector-transduced and untransduced bone marrow cells formed very few colonies on the fourth plating (Figure 1H). RME proved to be transforming, yielding significantly more colonies than the vector-transduced or untransduced bone marrow cells (Figure 1H). Strikingly, however, the R205N mutant was essentially devoid of transforming ability (p < 0.001 relative to wildtype RME, Student t test; Figure 1H). However, the R769C mutation in ZF2 and the RMED586A/L587S mutant in the CtBP binding domain were no different than wildtype RME protein, as was EVI1p135 (Figure 1H). These findings are consistent with the data obtained from the Rat1 transformation assay (Figure 1F).
Design of a Py-Im Polyamide To Block EVI1 Binding via ZF1
The results of these two transformation assays clearly indicated that the ability of EVI1 to transform cells is dependent on its ability to bind to the GACAAGATA motif via ZF1. This suggested that blocking the ability of the protein to bind to this DNA sequence motif with a small molecule inhibitor such as a polyamide would abrogate EVI1-induced transformation. We thus designed a Py-Im polyamide to specifically interact with the GACAA half of the motif and thereby block EVI1 binding to DNA. We chose the GACAA half motif rather than the AGATA portion due to the role of the latter in GATA protein function. Based on the basis of the pairing rules, polyamide 1, which consists of a sequence of the aromatic rings of N-methylpyrrole (Py) and N-methylimidazole (Im) amino acids [ImPyPyPy-(R)β-H2N-γ-PyImPyPy-(+)-IPA (Figure 2)], should bind to the sequence 5′-WGWCWW-3′, where W = A or T.15,45 As a control, a “mismatch” polyamide 2 (ImPyPyPy-(R)β-H2N-γ-PyPyImPy-(+)-IPA; Figure 2) was synthesized, which targets the sequence 5′-WGCWWW-3′. Versions of polyamide were synthesized conjugated to isophthalic acid (IPA; Figure 2).
Figure 2.
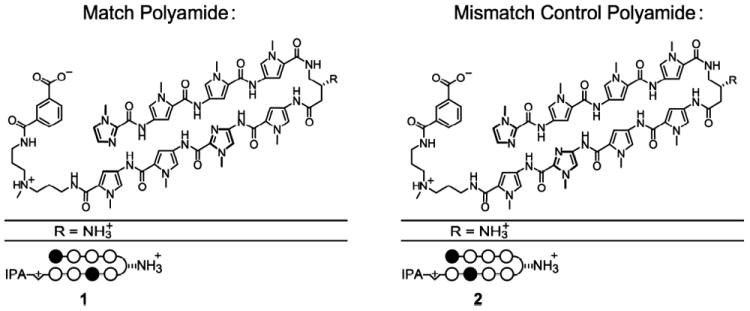
Structure and binding of polyamide. Match (1) and mismatch (2) polyamides, with modifications as shown. Dark circles represent imidazole (Im) and white circles pyrrole (Py). IPA indicates isophthalic acid.
To assess whether polyamide 1 binds specifically to the GACA motif, the magnitude of DNA thermal stabilization (ΔTm) of DNA–polyamide complexes was determined by melting temperature analysis (Table 1). A ΔTm reflects stronger stabilization of the helix, indicating tighter binding of the polyamide to the duplex. Relative to naked DNA (Tm = 50.2 °C), addition of match polyamides 1 yielded the greatest increase in Tm (11.5–17.3 °C). The value for IPA-conjugated polyamide (polyamide 1; ΔTm = 17.3) is similar to that obtained for polyamides targeting the androgen receptor response element.30 The mismatch polyamides had smaller ΔTm values, indicative of weaker binding (Table 1).
To assess the DNA binding specificity of polyamide 1 and determine the affinity of polyamide–DNA complexes, DNase I footprinting was performed using as a template a 5′-32P end-labeled 223 bp DNA fragment that contains a single GACAAGATA motif.5 Binding was performed in parallel with the mismatch polyamide 2. This revealed the specificity of DNA binding for the polyamides and the affinity of these interactions (Figure 3 and Table 2): polyamide 1 showed high affinity binding over the entire GACAAGATA EVI1 binding motif that was observed down to 300 pM, reflecting a Ka of 1.7 (±0.3) × 1010 M−1 (Table 2). Given that the polyamide was designed to bind 5′-WGWCWW-3′, this extent of binding, over the whole GACAAGATA motif, was expected. The binding affinity obtained is in the same range as that obtained with other polyamides designed to bind specific motifs (e.g., ref 24). Polyamide 2 binds to the GATA motif but not GACAA, and only at the highest concentrations of polyamide (Figure 3B, right, lanes 14 and 15), corresponding to a Ka of binding 2 orders of magnitude lower than the match polyamide (Ka = 1.0 (±0.3) × 108 M−1, Table 2). Interestingly, there is a second binding site on the plasmid (bracketed area II, TGTAA), with a binding affinity much lower than that for the GACAAGATA motif (Table 2). The differential in binding affinities between the desired interaction (between polyamide 1 and site I, the GACAA motif) and the second site (site II: TGTAA motif) is over 5-fold, possibly providing a therapeutic window between specific and nonspecific effects.
Figure 3.

DNase I footprint of polyamide on DNA template. (A) Schematic of the 223 bp double-stranded DNA template used for DNase I footprint, derived by PCR from plasmid p406,5 with labeling of the top strand as depicted. Below, DNA sequence showing the two regions footprinted: EVI1 binding motif (sequence I) and the TGTAAA motif (sequence II). (B) DNase I footprint with polyamides 1 (left) and 2 (right). Lanes 1, 2: Maxam–Gilbert sequencing reactions for G and A, respectively; lane 3: no DNase I; lane 4: DNase I, no polyamide; lanes 5–15: titration of polyamide from 10 pM to 1 μM. At bottom, graphic depiction of binding data to sequences I and II, for polyamides 1 and 2. X-axis, polyamide concentration in moles; Y-axis, fraction of maximal occupancy.
Polyamide 1 Reduces EVI1 Binding Affinity
The ability of polyamide 1 to inhibit EVI1 binding to its DNA recognition site in vitro was assessed by electromobility shift assay (EMSA). We determined the equilibrium constant (Keq) for EVI1 binding to its cognate recognition site without polyamide and with polyamide. In the first experiment, we used only the match polyamide (polyamide 1) to see if it is capable of inhibiting EVI1 binding at all. A protein titration was done at a constant concentration of DNA, with and without polyamide 1 at a 4:1 ratio of polyamide to DNA (Figure 4A). For a total DNA, 1250 fmol was used as previously described;13 polyamide concentration was 5000 fmol in a 50 μL reaction. In the absence of polyamide, the titration revealed a strong linear relationship between input DNA and the amount shifted (Figure 4, panel B) with a Keq of 3.1 × 107 M−1 (Table 3). Addition of polyamide 1 caused a significant drop in affinity of DNA for protein, to 1.7% of control (Table 3). With polyamide present, at the highest concentration of protein (700 fmol), there is a slight shift, roughly equivalent to that obtained with 10 fmol of protein in the absence of polyamide; this represents a remarkable inhibition of binding.
Figure 4.
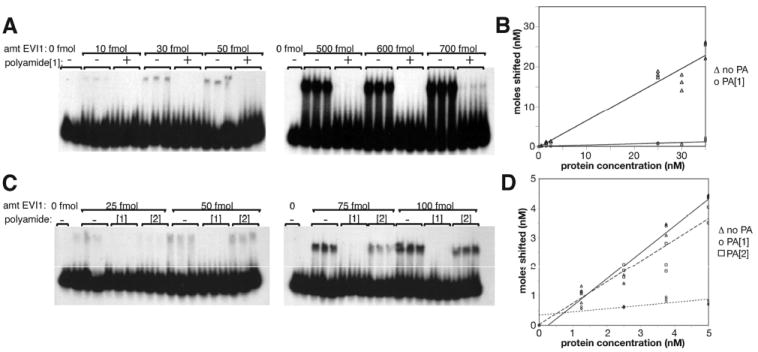
Electromobility shift assay (EMSA). (A) EVI1 protein titration from 0 to 700 fmol protein, as indicated, with 1250 fmol DNA template, in the absence and presence of a 4-fold excess (relative to DNA; 5000 fmol) of match polyamide 1, as indicated. The probe is a double-stranded 20-mer containing the GACAAGATA EVI1 binding motif. (B) Quantitation of EMSA data in panel A, displaying concentration DNA shifted relative to protein concentration. The slope of the plot is used to calculate the Keq values in Table 3 as described in the Experimental Procedures. (C) EMSA experiment using same probe as in panel A (1250 fmol); protein at various amounts as indicated are incubated with probe in the absence or presence of match (polyamide 1) or mismatch (polyamide 2) polyamide at a 1:1 molar ratio relative to DNA probe. (D) Quantitation of EMSA data in panel C, as in panel B.
Table 3.
Decrease in Affinity of EVI1 for Cognate DNA Binding Site Due to Polyamide
| polyamide | experiment 1
|
experiment 2
|
||||
|---|---|---|---|---|---|---|
| slope | Keq (M−1) | % of control | slope | Keq (M−1) | % of control | |
| no polyamide (control) | 0.66 | 3.1 × 107 | 100% | 0.91 | 1.62 × 108 | 100% |
| polyamide 1 (match) | 0.03 | 5.3 × 105 | 1.7% | 0.108 | 1.94 × 106 | 1.19% |
| polyamide 2 (mismatch) | nd | nd | nd | 0.724 | 4.20 × 107 | 25.9% |
A second protein titration was done at a polyamide:DNA ratio of 1:1. In this experiment, the mismatch polyamide control was employed to assess the specificity of the inhibitory effect exhibited by polyamide 1 (Figure 4C). Again, in the absence of polyamide, protein titration results in a near linear increase in DNA bound (Figure 4D). As in the first experiment, pretreatment of the DNA with polyamide 1 yields a dramatic inhibition of binding, giving a Keq 1.2% of that without polyamide (Table 3). While the mismatch polyamide 2 did cause a minor inhibition, likely due to nonspecific binding (Table 3), it was not nearly as effective as the match polyamide, indicating that polyamide 1 is more specific.
In these EMSA assays, we did not observe any retardation of the radioactive probe by the polyamide, most likely due to the relatively small molecular mass of polyamide 1.
Polyamide 1 Causes Inhibition of EVI1-Dependent Transcriptional Activity
We next investigated the effect of polyamide 1 on EVI1-dependent transcriptional regulation. Since the transcriptional effects of native EVI1 isoforms on transcription are not readily assayed, we initially employed a synthetic reporter system we have designed and previously described.13 This system involves the assaying transcriptional activity of a chimeric EVI1-derived activator composed of ZF1 domain of EVI1 (zinc fingers 1–7) fused to the activation domain of viral protein 16 (VP16) from herpes simplex virus 1 (HSV1). This protein has the binding specificity of ZF1 domain of EVI1 to the GACAAGATA motif but functions as a potent transcriptional activator. Importantly, this activation activity is completely dependent on the interaction between EVI1 ZF1 and the GACAAGATA motif. We have shown previously that this chimeric activator binds to and transactivates a chloramphenicol acetyl transferase (CAT) reporter we have made bearing EVI1 binding sites within the promoter. In addition, we have shown by chromatin immunoprecipitation and gene expression analyses that this protein binds to the promoter regions of endogenous genes that are candidate EVI1 targets.13 We have developed an NIH-3T3-derived cell line, termed 6D, in which the EVI1-VP16 chimera is under the control of a tetracycline operon;13 in the same cell line, there is an autoregulatory tetracycline repressor-VP16 construct.46 Thus, in the presence of tetracycline, EVI1-VP16 is off but is turned on within 6–10 h following removal of tetracycline. We have further added to the 6D cell line a chromosomeembedded CAT reporter harboring EVI1 binding sites within the promoter region. This cell line, termed 6D+AP17, has inducible CAT activity upon tetracycline removal that is dependent on interaction between EVI1 zinc fingers and its cognate binding site within the CAT promoter.
To test the ability of polyamide 1 to inhibit ZF1 binding to the GACAAGATA motif in the CAT promoter, we removed tetracycline from 6D+AP17 cells and simultaneously added vehicle, polyamide 1, or the mismatched polyamide 2. We incubated the cells for 48 h and then assayed CAT activity (Figure 5). Doses of polyamide ranged from 0.1 to 1 μM. These doses of polyamide 1 significantly suppressed EVI1-VP16-mediated activation of the CAT reporter: activity was decreased 68 to 80% relative to minus Tet/no polyamide (all p values <0.01). The suppression caused by the mismatched polyamide was not as great—20–58% reduction—with p values >0.01. However, only at 1 μM concentration of polyamide was the match polyamide significantly lower than the mismatch control (Figure 5).
Figure 5.

Polyamide 1 inhibits EVI1-VP16-mediated activation of EVI1 target. Y-axis shows normalized values of acetylated chloramphenicol. Error bars denote standard error. Significance (p values) is calculated with Student t test. Cells are washed of tetracycline to induce EVI1-VP16, and polyamide, either match or mismatch, is added at concentrations indicated. Cells are incubated 48 h and harvested for CAT assay.
Polyamide 1 Partially Inhibits Growth of DA-1 and NFS-60 Cells
To assess the growth inhibitory effect of polyamide 1 on myeloid cells, we tested the growth rate of two different immortalized lines after incubation with match polyamide. One of the two cell lines, NFS-60, has a high level of EVI1 expression due to proviral insertional activation, and the control cell line, FDCP, does not express EVI1.13 We incubated the two cell lines with 20 μM match polyamide 1 for 1 week and measured 3H-thymidine incorporation. The result showed a 35.8% (Figure 6A, p = 0.0006) decrease of incorporation in NFS-60, but no significant inhibition of thymidine uptake was observed in FDCP. Next, we examined whether the inhibition is dose-dependent; in this experiment we also compared match (1) to mismatch (2) polyamide. We treated NSF-60 cells for 1 week with polyamides 1 and 2 at different doses, ranging from 0 to 20 μM, and tested the growth. Our data demonstrated that with match polyamide 1 3H-thymidine incorporation was effectively decreased by 52.3% (p = 0.000; Figure 6B), whereas it was slightly increased by 13.7% when treated with mismatch polyamide 2 (p = 0.0007). Together, these data suggest that the EVI1-targeted polyamide specifically inhibits EVI1-expressing cells in a dose-dependent manner. However, polyamide treatment did not completely inhibit leukemic cell growth.
Figure 6.

Growth assessment of murine leukemic cells following treatment with polyamide. (A) Tritiated thymidine incorporation in myeloid cell lines that do (NFS-60) or do not (FDCP) express EVI1, with or without 1 week incubation with 20 μM match polyamide, in triplicate. Bars denote standard deviation. The decrease in thymidine uptake is significant for NFS-60 (p = 0.0006, Student t test), but not for FDCP cells. (B) NFS-60 cells grown for 1 week with either match or mismatch polyamide at the doses indicated, followed by assessment of growth by 3H-thymidine incorporation. Assays were performed in triplicate. Student t test revealed a significant decrease in incorporation at 20 μM of match relative to untreated (p = 0.000). (C) Serial replating assay in cytokine-supplemented methylcellulose, of mouse bone marrow cells transduced with either RME or NUP98-HoxA9, as shown, and treated with either match or mismatch polyamide (20 μM) as indicated. p value was calculated using Student t test. Data are expressed as a percentage of the “control”, which is the number of colonies obtained with the specified oncogene at each round with polyamide 2.
Polyamide Significantly but Partially Inhibits RME-Induced Transformation
In Figure 1H, we showed that transformation by the RME leukemogenic fusion protein is dependent on the ZF1 domain to transform primary bone marrow cells in a serial replating assay. Given this dependence, we predicted that polyamide 1 would be able to inhibit RME-induced transformation of primary bone marrow cells, but not marrow cells transformed by NUP98-HoxA9, an oncogene that is independent of RME or EVI1. We thus transduced primary bone marrow cells with either RME or NUP98-HoxA9 and plated them in cytokine-supplemented methylcellulose that contained either the match polyamide 1 or mismatch polyamide 2. Colonies were then serially replated to three platings. The results (Figure 6C) indicate a significant yet incomplete inhibition of colony formation by RME with the match polyamide 1, but not for NUP98-HoxA9-induced colony formation. These data support the data described above that the match polyamide 1 has a significant but partial ability to inhibit EVI1 or RME-induced leukemic cell growth.
DISCUSSION
The diagnosis of acute myeloid leukemia portends a particularly poor prognosis. With current therapies, 5 year survival rates range from 5 to 30%, depending on the subtype: leukemias with t(8;21), inv(16), and t(15;17) have a significantly better prognosis than those in the poor-risk category. This latter category includes leukemias with abnormalities at 3q26, which results in deregulated expression of EVI1. We have shown previously that shRNA-mediated suppression of EVI1 results in slowing of leukemic cell expansion in culture due to an increase in apoptotic cell death via the intrinsic pathway.47 In the studies described herein, we provide a first attempt at developing a small molecule therapeutic specifically designed to block transformation–critical EVI1 molecular interactions. Given that EVI1 lacks enzymatic activity, the design of a small molecule inhibitor is challenging. Since EVI1 is a nuclear protein with sequence-specific DNA binding and transcriptional regulatory activities, polyamides represent a logical choice as potential therapeutic. In this paper, we demonstrate that the ability of the EVI1 protein to transform cells is dependent on its binding to DNA; this argues that blocking DNA binding with a polyamide should block leukemic growth.
Polyamides hold significant promise in the treatment of disease processes that are driven by aberrant transcriptional regulation. Previous studies have reported efficacy of polyamides in suppressing gene expression in vivo.24-27,48,49 Lai et al. reported the development of a polyamide directed against Fat Specific Element 2 (FSE2) in the TGF-β promoter; this polyamide significantly but not completely suppresses the expression of TGF-β in cell culture.48 Yao et al. developed a polyamide to target the AP-1 binding site in the lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1) and show that it suppresses expression of the target in damaged vessels and significantly ameliorates narrowing of arteries in damaged vessels, a process that depends on LOX-1 expression. Their data clearly show that polyamides can penetrate to the nucleus of endothelial cells lining vessel walls following intravascular injection of the polyamide into rats.49
Our polyamide design is based on binding rules previously established by the Dervan lab for hairpin-configuration polyamides such as the ones employed here: Im/Py binds to G:A; Py/Py binds to A:T, and Py:Im binds to C:G. Thus, for sequence GACA, we chose a polyamide with sequence (ImPyPyPy-(R)β-H2N-γ-PyImPyPy; polyamide 1); this folds into a hairpin structure with resides paired as follows: Im/Py-Py/Py-Py/Im-Py/Py. To test the specificity and affinity of polyamide 1 for the GACA sequence, we first perform DNase I footprinting that this polyamide binds to the GACAA motif at very high affinity (association constant = 1.7 × 1010 M−1). The specificity of binding is good, but not perfect: another site on our DNase I protection probe (TGTAAA or TTTACA) also bound, but at a 5-fold lower affinity. This binding can likely be explained by the presence of an ACA motif within the TTTACA sequence. Since the highest affinity is observed for the GACAA motif, it may be possible to only bind the desired sites through dose adjustment. Alternatively, it may be possible to increase the specificity of binding by modifying the structure of polyamide 1, perhaps incorporating derivatives of the standard imidazole/pyrrole moieties. For instance, Py/Hp has been shown to bind more specifically to A:T pairs than the Py/Py used in polyamide 1.15 However, hydroxypyrrole is unstable and can degrade over time; more stable alternative structures has been discussed.
Given the high affinity of polyamide 1 for the GACAA motif, it is not surprising that it effectively blocks the ability of EVI1 bind to this site in vitro, as demonstrated by EMSA assay (Figure 4). We were also able to demonstrate inhibition of EVI1 binding in vivo, using our 6D+AP17 cell line (Figure 5). Importantly, the CAT reporter we are using is embedded in the chromosome, so it is chromatinized, unlike transient transfection constructs used by other investigators.48
Finally, we assess the ability of polyamide 1 to inhibit leukemic cell growth. While no effect on growth is seen in cell lines that lack EVI1 expression, a small but significant effect is seen on leukemic cells that have provirally activated EVI1 allele. These results raise the hopeful possibility that with further optimization of administration strategies or in drug design, a highly specific and effective novel anti-EVI1 therapy can be developed. One issue to be addressed is the efficacy of cellular uptake: while a fluorescently tagged polyamide was taken into cells and localized to the nucleus (data not shown), its intranuclear concentration may have been low, since the intensity of fluorescent staining was lower that that seen in polyamide-treated permeabilized cells, into which the drug freely enters (data not shown). Leukemic cells are known to possess effective P1 glycoprotein-type multidrug resistance export pumps;50 we assessed the importance of these to the low entry of polyamide into cells by coadministering verapimil, an effective inhibitor of these pumps, and no marked improvement of uptake was observed (data not shown). Other options exist for increasing cellular uptake. These include manipulating the charge on the γ-turn of the hairpin: Previous studies by the Dervan laboratory have noted that α-NHAc substituted γ-turns often facilitate better cellular uptake compared to unsubstituted (lacking the NHAc moiety) γ-turns.23 In this study we are employing β-substituted turns, which is a recent advance in polyamide technology.29 Nonetheless, changing the −NH3 + moiety (see Figure 2) to an −NHAc moiety may increase cellular uptake. This is the focus of current experimentation.
Specific Role of ZF1 and Relevant Target Genes for Malignant Transformation
To eliminate DNA binding via ZF1 and ZF2, we created point mutations rather than large deletions, thus minimizing the possibility that other functions of the zinc fingers, such as protein:protein interactions (e.g., with SMAD proteins51,52) are disrupted. Previous studies have suggested that DNA binding via ZF1 is required for EVI1-mediated malignant transformation.4 However, the deletions used also abrogated binding to SMAD proteins,51 making a definitive conclusion difficult. To address this, and to assess the role of DNA binding via ZF2 in transformation, we tested the ability of two different DNA binding-deficient mutants, one in ZF1 (EVI1R205N13) and one in ZF2 (EVI1R769C), for their ability to transform cells using the Rat1 transformation assay for EVI1.7 The results of these experiments clearly indicate that EVI1-induced malignant transformation is dependent on DNA binding via ZF1; however, they do not indicate what are the relevant and critical DNA targets. Recent chromatin immunoprecipitation-sequencing (ChIP-Seq) experiments we have performed have shed new light on the issue of leukemiarelevant EVI1 target genes and suggest that one reason for the limited efficacy of polyamide 1 in inhibiting the growth of EVI1-expressing leukemias is that we have not targeted the correct sequence. Thus, further advances in targeting EVI1 with polyamides will likely require more information concerning the identity of key transcriptional targets for EVI1 and the exact nature of the DNA motif by which EVI1 regulates these genes.
The identification of critical target genes that are essential for leukemogenesis has been difficult. The GACAAGATA motif to which ZF1 binds was first identified by an iterative siteselection strategy,1 selecting for the highest affinity binding under EMSA conditions; selection of EVI1 binding sites in mouse genomic DNA using an in vitro filter binding assay yielded the same motif.53 These strategies for binding site identification are flawed in that they select for the highest affinity binders using purified EVI1 protein under artificial conditions; native conditions within the nucleus differ markedly due to the presence of other proteins and chromatinized DNA packed within higher order structures. In addition, it is possible that bona fide DNA binding sites for EVI1 do not have the highest affinity, since too low an off-rate might not allow for fine regulation of gene expression.
To further refine our understanding of true EVI1 target genes, we have recently performed ChIP-Seq experiments using mouse leukemic cell lines that overexpress EVI1.47 This approach identified nearly 5000 different EVI1 binding sites within the genome, and some of these sites suggest compelling mechanisms for leukemogenesis. Interestingly, only a small minority of these contained the DNA motifs identified by site selection,1,3 and none was the same as the genomic fragments we selected using an in vitro filter binding approach.53 Even the sites identified by our in vivo selection strategy13 were only rarely occupied in vivo within leukemic cells. We are in the process of confirming the location of EVI1 binding that we obtained from ChIP-Seq experiments.
The discrepancy between our previous selection schemes and the data from ChIP-Seq experiments indicates that bona fide EVI1 binding sites and bona fide EVI1 target gene still need to be more thoroughly delineated and characterized. The identification of these leukemia-critical targets, and the site to which EVI1 binds within these genes, remains a critical goal. Once these are identified, it will likely be possible to block these interactions with polyamides and thereby inhibit the growth of EVI1-driven myeloid leukemias. The data presented here clearly indicate that EVI1 binding to DNA can be effectively blocked both in vivo and in vitro with polyamides. With further improvements in nuclear localization of polyamides and knowledge of leukemia-critical targets, we contend that these agents can become very effective therapeutics for the treatment of myeloid leukemia.
Supplementary Material
Acknowledgments
We thank Giuseppina Nucifora for providing the pBS-AML1-MDS1-EVI1 plasmid.
Funding
Support for this work comes from NIH [R01 CA-112188 (A.S.P.), R01 GM51747 (P.B.D.)], the AA-MDS Foundation, and the Dobranski Foundation. D.A.H. thanks the California Tobacco-Related Disease Research Program (16FT-0055) for a postdoctoral fellowship. The National Science Foundation Chemistry Research Instrumentation and Facilities Program (CHE-0541745) is acknowledged for providing the UPLC-MS instrument.
ABBREVIATIONS
- 5-FU
5-fluorouracil
- CAT
chloramphenicol acetyl transferase
- Cbz
benzyl carbamate
- ChIP-Seq
chromatin immunoprecipitation-sequencing
- DIC
differential interference contrast
- dIdC
deoxyinosine–deoxycytosine
- DIEA
diisopropylethylamine
- DMEM
Dulbecco’s modified Eagle’s medium
- DMF
N,N-dimethylformamide
- EMSA
electromobility shift assay
- ESI+
electrospray ionization
- FDCP
factor-dependent cell – pluripotent
- fmol
femtomole
- FSE2
fat-specific element 2
- GST
glutathione S-transferase
- HA
hemagglutinin
- Hp
N-methyl-3-hydroxypyrrole
- Im
N-methylimidazole
- IPA
isophthalic acid
- Ka
association constant
- Keq
equilibrium constant
- LOX-1
lectin-like oxidized low-density lipoprotein receptor 1
- MALDI-TOF MS
matrix-assisted, LASER desorption/ionization time-of-flight mass spectrometry
- MeCN
acetonitrile
- NUP98
nuclear pore complex protein Nup98
- Py
pyrrole
- Q-PCR
quantitative polymerase chain reaction
- RME
RUNX-MDS1-EVI1
- SMAD
mothers against decapentaplegic
- Tm
melting temperature
- VP16
viral protein 16, herpes simplex virus
- ZF1
zinc finger domain 1
- ZF2
zinc finger domain 2
Footnotes
Supporting Information
Figure S1: intermediate compounds in polyamide synthesis. Shown is the base polyamide structure from which the IPA and FITC derivatives were synthesized; compounds 5 and 6 correspond to the base polyamide with the side chain modifications indicated. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Perkins AS, Fishel R, Jenkins NA, Copeland NG. Evi-1, a murine zinc finger proto-oncogene, encodes a sequence-specific DNA-binding protein. Mol Cell Biol. 1991;11:2665–2674. doi: 10.1128/mcb.11.5.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Delwel R, Funabiki T, Kreider B, Morishita K, Ihle J. Four of the seven zinc fingers of the Evi-1 myeloid transforming gene are required for sequence-specific binding to GA(C/T)AAGA(T/C)AAGATAA. Mol Cell Biol. 1993;13:4291–4300. doi: 10.1128/mcb.13.7.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Funabiki T, Kreider BL, Ihle JN. The carboxyl domain of zinc fingers of the Evi-1 myeloid transforming gene binds a consensus sequence GAAGATGAG. Oncogene. 1994;9:1575–1581. [PubMed] [Google Scholar]
- 4.Kilbey A, Bartholomew C. Evi-1 ZF1 DNA binding activity and a second distinct transcriptional repressor region are both required for optimal transformation of Rat1 fibroblasts. Oncogene. 1998;16:2287–2291. doi: 10.1038/sj.onc.1201732. [DOI] [PubMed] [Google Scholar]
- 5.Perkins A, Kim J. Zinc fingers 1–7 of EVI1 fail to bind to the GATA motif by itself but require the core site GACAAGATA for binding. J Biol Chem. 1996;271:1104–1110. doi: 10.1074/jbc.271.2.1104. [DOI] [PubMed] [Google Scholar]
- 6.Kreider B, Orkin S, Ihle J. Loss of erythropoietin responsiveness in erythroid progenitors due to expression of the Evi-1 myeloid transforming gene. Proc Natl Acad Sci U S A. 1993;90:6454–6458. doi: 10.1073/pnas.90.14.6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartholomew C, Kilbey A, Clark A, Walker M. The Evi-1 proto-oncogene encodes a transcriptional repressor activity associated with transformation. Oncogene. 1997;14:569–577. doi: 10.1038/sj.onc.1200864. [DOI] [PubMed] [Google Scholar]
- 8.Yuasa H, Oike Y, Iwama A, Nishikata I, Sugiyama D, Perkins A, Mucenski M, Suda T, Morishita K. Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. EMBO J. 2005;24:1976–1987. doi: 10.1038/sj.emboj.7600679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morishita K, Suzukawa K, Taki T, Ihle JN, Yokota J. EVI-1 zinc finger protein works as a transcriptional activator via binding to a consensus sequence of GACAAGATAAGATAAN1–28CTCATCTTC. Oncogene. 1995;10:1961–1967. [PubMed] [Google Scholar]
- 10.Tanaka T, Nishida J, Mitani K, Ogawa S, Yazaki Y, Hirai H. Evi-1 raises AP-1 activity and stimulates c-fos promoter transactivation with dependence on the second zinc finger domain. J Biol Chem. 1994;269:24020–24026. [PubMed] [Google Scholar]
- 11.Izutsu K, Kurokawa M, Imai Y, Maki K, Mitani K, Hirai H. The corepressor CtBP interacts with Evi-1 to repress transforming growth factor beta signaling. Blood. 2001;97:2815–2822. doi: 10.1182/blood.v97.9.2815. [DOI] [PubMed] [Google Scholar]
- 12.Palmer S, Brouillet J, Kilbey A, Fulton R, Walker M, Crossley M, Bartholomew C. Evi-1 transforming and repressor activities are mediated by CtBP co-repressor proteins. J Biol Chem. 2001;276:25834–25840. doi: 10.1074/jbc.M102343200. [DOI] [PubMed] [Google Scholar]
- 13.Yatsula B, Lin S, Read A, Poholek A, Yates K, Yue D, Hui P, Perkins A. Identification of binding sites of EVI1 in mammalian cells. J Biol Chem. 2005;280:30712–30722. doi: 10.1074/jbc.M504293200. [DOI] [PubMed] [Google Scholar]
- 14.Dervan P. Molecular recognition of DNA by small molecules. Bioorg Med Chem. 2001;9:2215–2235. doi: 10.1016/s0968-0896(01)00262-0. [DOI] [PubMed] [Google Scholar]
- 15.Dervan P, Edelson B. Recognition of the DNA minor groove by pyrrole-imidazole polyamides. Curr Opin Struct Biol. 2003;13:284–299. doi: 10.1016/s0959-440x(03)00081-2. [DOI] [PubMed] [Google Scholar]
- 16.Kielkopf C, Bermer R, White S, Szewczyk J, Turner J, Baird E, Dervan P, Rees D. Structural effects of DNA sequence on T·A recognition by hydroxypyrrole/pyrrole pairs in the minor groove. J Mol Biol. 2000;295:557–567. doi: 10.1006/jmbi.1999.3364. [DOI] [PubMed] [Google Scholar]
- 17.Kielkopf CL, Baird EE, Dervan PB, Rees DC. Nat Struct Biol. 1998;5:104–109. doi: 10.1038/nsb0298-104. [DOI] [PubMed] [Google Scholar]
- 18.Kielkopf CL, White S, Szewczyk JW, Turner JM, Baird EE, Dervan PB, Rees DC. Science. 1998;282:111–115. doi: 10.1126/science.282.5386.111. [DOI] [PubMed] [Google Scholar]
- 19.Trauger JW, Baird EE, Dervan PB. Nature. 1996;382:559–561. doi: 10.1038/382559a0. [DOI] [PubMed] [Google Scholar]
- 20.Hsu CF, Phillips JW, Trauger JW, Farkas ME, Belitsky JM, Heckel A, Olenyuk BZ, Puckett JW, Wang CC, Dervan PB. Tetrahedron. 2007;63:6146–6151. doi: 10.1016/j.tet.2007.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dudouet B, Burnett R, Dickinson LA, Wood MR, Melander C, Belitsky JM, Edelson B, Wurtz N, Briehn C, Dervan PB, Gottesfeld JM. Chem Biol. 2003;10:859–867. doi: 10.1016/j.chembiol.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 22.Best T, Edelson B, Nickols N, Dervan P. Nuclear localization of pyrrole-imidazole polyamide-fluorescein conjugates in cell culture. Proc Natl Acad Sci U S A. 2003;100:12063. doi: 10.1073/pnas.2035074100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edelson B, Best T, Olenyuk B, Nickols N, Doss R, Foister S, Heckel A, Dervan P. Influence of structural variation on nuclear localization of DNA-binding polyamide-fluorophore conjugates. Nucleic Acids Res. 2004;32:2802. doi: 10.1093/nar/gkh609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olenyuk B, Zhang G, Klco J, Nikols N, Kaelin W, Dervan P. Inhibition of vascular endothelial growth factor with a sequence-specific hypoxia response element antagonist. Proc Natl Acad Sci U S A. 2004;101:16768–16773. doi: 10.1073/pnas.0407617101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nickols N, Jacobs C, Farkas M, Dervan P. Modulating Hypoxia-Inducible Transcription by Disrupting the HIF-1-DNA Interface. ACS Chem Biol. 2007;2:561. doi: 10.1021/cb700110z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nickols N, Jacobs C, Farkas M, Dervan P. Improved nuclear localization of DNA-binding polyamides. Nucleic Acids Res. 2007;35:363–370. doi: 10.1093/nar/gkl1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muzikar KA, Nickols NG, Dervan PB. Proc Natl Acad Sci U S A. 2009;106:16598–16603. doi: 10.1073/pnas.0909192106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belitsky J, Nguyen D, Wurtz N, Dervan P. Solid-phase synthesis of DNA binding polyamides on oxim resin. Bioorg Med Chem. 2002;10:2767–2774. doi: 10.1016/s0968-0896(02)00133-5. [DOI] [PubMed] [Google Scholar]
- 29.Dose C, Farkas ME, Chenoweth DM, Dervan PB. Next generation hairpin polyamides with (R)-3,4-diaminobutyric acid turn unit. J Am Chem Soc. 2008;130:6859–6866. doi: 10.1021/ja800888d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chenoweth D, Harki D, Phillips J, Dose C, Dervan P. Cyclic pyrrole-imidazole polyamides targeted to the androgen response element. J Am Chem Soc. 2009;131:7182–7188. doi: 10.1021/ja901309z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trauger J, Dervan P. Footprinting methods for analysis of pyrrole-imidazole polyamide/DNA complexes. Methods Enzymol. 2001;340:450–466. doi: 10.1016/s0076-6879(01)40436-8. [DOI] [PubMed] [Google Scholar]
- 32.Iverson B, Dervan P. Adenine specific DNA chemical sequencing reaction. Nucleic Acids Res. 1987;15:7823–7830. doi: 10.1093/nar/15.19.7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maxam A, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 34.Kunkel T. Oligonucleotide-directed mutagenesis without phenotypic selection. In: Ausubel F, Brent R, Kingston R, Moore D, Seidman J, Smith J, Struhl K, editors. Current Protocols in Molecular Biology. John Wiley and Sons; New York: 1993. pp. 8.1.1–8.1.6. [DOI] [PubMed] [Google Scholar]
- 35.Morgenstern J, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lavau C, Szilvassy S, Slany R, Cleary M. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J. 1997;16:4226–4237. doi: 10.1093/emboj/16.14.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ausubel F, Brent R, Kingston R, Moore D, Seidman J, Smith J, Struhl K. In: Chanda V, editor. John Wiley & Sons, Inc.; Hoboken, NJ: 1997. [Google Scholar]
- 38.Morishita K, Parker DS, Mucenski ML, Jenkins NA, Copeland NG, Ihle JN. Retroviral activation of a novel gene encoding a zinc finger protein in IL3-dependent myeloid leukemia cell lines. Cell. 1988;54:831–840. doi: 10.1016/s0092-8674(88)91175-0. [DOI] [PubMed] [Google Scholar]
- 39.Smith D. Purification of glutathione-S-transferase fusion proteins. Methods Mol Cell Biol. 1993;4:220–229. [Google Scholar]
- 40.Chodosh LA, Carthew RW, Sharp PA. A single polypeptide possesses the binding and transcription activities of the adenovirus major late transcription factor. Mol Cell Biol. 1986;6:4723–4733. doi: 10.1128/mcb.6.12.4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pavletich N, Pabo C. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science. 1991;252:809–817. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- 42.Blumberg H, Eisen A, Sledziewski A, Bader D, Young E. Two zinc fingers of a yeast regulatory protein shown by genetic evidence to be essential for function. Nature. 1987;328:443–445. doi: 10.1038/328443a0. [DOI] [PubMed] [Google Scholar]
- 43.Wilson T, Day M, Pexton T, Padgett K, Johnston M, Milbrandt J. In vivo mutational analysis of the NGFI-A zinc fingers. J Biol Chem. 1992;267:3718–3724. [PubMed] [Google Scholar]
- 44.Luo R, Lavau C, Du C, Simone F, Polak P, Kawamata S, Thirman M. The elongation domain of ELL is dispensible but its ELL-associated factor 1 interaction domain is essential for MLL-ELL-induced leukemogenesis. Mol Cell Biol. 2001;21:5678–5687. doi: 10.1128/MCB.21.16.5678-5687.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White S, Szewczyk J, Turner J, Baird E, Dervan P. Recognition of the four Watson-Crick base pairs in the DNA minor groove by synthetic ligands. Nature. 1998;391:468–471. doi: 10.1038/35106. [DOI] [PubMed] [Google Scholar]
- 46.Shockett P, Difilippantonio M, Hellman N, Schatz D. A modified tetracycline-regulated system provides autoregulatory, inducible gene expression in cultured cells and transgenic mice. Proc Natl Acad Sci U S A. 1995;92:6522–6. doi: 10.1073/pnas.92.14.6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.del Campo J, Wuertzer C, Xiao Y-Y, Zhang Y, Lin S, Dudley J, Tuck D, Yatsula B, Perkins A. EVI1 blocks apoptosis in myeloid leukemia cells via enhanced transcription of the prosurvival gene Bcl2a1 (A1) Blood. in revision. [Google Scholar]
- 48.Lai Y, Fukuda N, Ueno T, Matsuda H, Saito S, Matsumoto K, Ayame H, Bando T, Sugiyama H, Mugishima H, Serie K. Synthetic pyrrole-imidazole polyamide inhibits expression of the human transforming growth factor-β1 gene. J Pharm Exp Ther. 2005;315:571–575. doi: 10.1124/jpet.105.089086. [DOI] [PubMed] [Google Scholar]
- 49.Yao E, Fukuda N, Ueno T, Matsuda H, Matsumoto K, Nagase H, Matsumoto Y, Takasaka A, Serie K, Sugiyama H, Sawamura T. Novel Gene Silencer Pyrrole-Imidazole Polyamide Targeting Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 Attenuates Restenosis of the Artery After Injury. Hypertension. 2008;52:86–92. doi: 10.1161/HYPERTENSIONAHA.108.112797. [DOI] [PubMed] [Google Scholar]
- 50.Campos L, Guyotat D, Archimbaud E, Calmard-Oriol P, Tsuruo T, Troncy JDT, Fiere D. Clinical significance of multidrug resistance P-glycoprotein expression on acute non-lymphoblastic leukemia cells at diagnosis. Blood. 1992;79:473–476. [PubMed] [Google Scholar]
- 51.Kurokawa M, Mitani K, Irie K, Matsuyama T, Takahashi T, Chiba S, Yazaki Y, Matsumoto K, Hirai H. The oncoprotein Evi-1 represses TGF-beta signalling by inhibiting Smad3. Nature. 1998;394:92–96. doi: 10.1038/27945. [DOI] [PubMed] [Google Scholar]
- 52.Alliston T, Ko T, Cao Y, Liang Y-Y, Feng X-H, Chang C, Derynck R. Repression of bone morphogenetic protein and activin-inducible transcription by Evi-1. J Biol Chem. 2005;280:24227–24237. doi: 10.1074/jbc.M414305200. [DOI] [PubMed] [Google Scholar]
- 53.Kim J, Hui P, Yue D, Aycock J, Leclerc C, Bjoring A, Perkins A. Identification of candidate target genes for EVI1, a zinc finger oncoprotein, using a novel selection strategy. Oncogene. 1998;17:1527–1538. doi: 10.1038/sj.onc.1202331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.