

Abstract
T cell Ig domain and mucin domain (TIM)-3 has previously been established as a central regulator of Th1 responses and immune tolerance. In this study, we examined its functions in allograft rejection in a murine model of vascularized cardiac transplantation. TIM-3 was constitutively expressed on dendritic cells and natural regulatory T cells (Tregs) but only detected on CD4+FoxP3− and CD8+ T cells in acutely rejecting graft recipients. A blocking anti–TIM-3 mAb accelerated allograft rejection only in the presence of host CD4+ T cells. Accelerated rejection was accompanied by increased frequencies of alloreactive IFN-γ–, IL-6–, and IL-17–producing splenocytes, enhanced CD8+ cytotoxicity against alloantigen, increased alloantibody production, and a decline in peripheral and intragraft Treg/effector T cell ratio. Enhanced IL-6 production by CD4+ T cells after TIM-3 blockade plays a central role in acceleration of rejection. Using an established alloreactivity TCR transgenic model, blockade of TIM-3 increased allospecific effector T cells, enhanced Th1 and Th17 polarization, and resulted in a decreased frequency of overall number of allospecific Tregs. The latter is due to inhibition in induction of adaptive Tregs rather than prevention of expansion of allospecific natural Tregs. In vitro, targeting TIM-3 did not inhibit nTreg-mediated suppression of Th1 alloreactive cells but increased IL-17 production by effector T cells. In summary, TIM-3 is a key regulatory molecule of alloimmunity through its ability to broadly modulate CD4+ T cell differentiation, thus recalibrating the effector and regulatory arms of the alloimmune response.
T cell-dependent immune responses play a central role for allograft rejection, and the balance between the complex mechanisms of T cell activation and inhibition determine the ultimate fate of allografts (1, 2). The combination of “signal one,” provided by the engagement of the alloantigen-specific TCR with the MHC–Ag complex, and costimulatory “signal two” leads to complete T cell activation, which results in T cell proliferation, differentiation into effector and memory T cells, causes cytokine release, and might finally lead to allograft rejection (2). In contrast, abrogation of T cell activation via programmed or activation-induced cell death, regulatory cells, cytokines, and inhibitory signals via negative costimulatory pathways can prevent T cell-mediated immune responses and lead to allograft tolerance (2, 3). As the net effect of positive (activating) and negative (inhibiting) signals to T cells determines the definitive outcome of T cell-dependent immune reactions and thus the fate of allografts, exploring and manipulating activating or inhibiting signals to T cells represents a potential strategy to promote long-term allograft acceptance and patient survival.
Effector CD4+ T cells were traditionally thought to segregate into Th1 and Th2 subsets (4), and acute allograft rejection has been associated with Th1 differentiation, as rejection often correlates with expression of IFN-γ in allografts and production of IFN-γ upon restimulation of peripheral T cells with alloantigen (4). More recently, two other pathways of differentiation have been described, namely induced regulatory CD4+ T cells (iTregs) generated upon TCR stimulation during TGF-β/IL-2 signaling, and Th17 cells, murine T cells activated in the presence of TGF-β/IL-6 (5). IL-17 was found to play a major role in cardiac allograft rejection by T-bet–deficient mice that have reduced IFN-γ production and type 1 T cell differentiation (6–8). A recent paper demonstrated that TLR signals can promote IL-6/IL-17–dependent transplant rejection in wild-type recipients (9). Production of proinflammatory cytokines by various innate and adaptive immune cells can also promote the differentiation of alloreactive T cells into Th1 and Th17 cells and allow their escape from regulatory T cell (Treg) suppression (9–11). Indeed, there seems to be reciprocal control of Th17 and iTreg differentiation, with both subsets requiring TFG-β signaling and the concomitant presence of inflammatory cytokines. IL-6, in particular, has been shown to inhibit iTreg while promoting Th17 differentiation, while IL-2 exerts inverse effects (12–14). Whether this equilibrium occurs during physiologic rejection in in vivo transplantation models and by which regulatory mechanisms it can be skewed in one direction or the other remain to be determined.
The T cell Ig domain and mucin domain (TIM) family is a novel group of molecules with a conserved structure and important immunologic functions, including T cell activation, induction of T cell apoptosis, T cell tolerance, and the clearance of apoptotic cells (15–17). TIM-3 protein is not expressed by naive T cells but is up-regulated as they differentiate into Th1 cells (18–21). Administration of TIM-3–Ig to immunized mice caused hyperproliferation of Th1 cells and Th1 cytokine release (19). Galectin-9 has been identified as the ligand for TIM-3 (22). Intracellular calcium flux and aggregation and death of Th1 cells induced by galectin-9 were at least partially TIM-3 dependent in vitro, and administration of galectin-9 in vivo resulted in selective loss of IFN-γ–producing cells and suppression of Th1 autoimmunity. These data suggest that the TIM-3:galectin-9 pathway may have evolved to ensure effective termination of effector Th1 cells. Recent data also suggest that TIM-3 may be a key molecule involved in T cell exhaustion in chronic viral infections (23). In addition, TIM-3 has also been implicated in establishing tolerance (19, 20). TIM-3–Ig abrogated tolerance induction in Th1 cells, and TIM-3–deficient mice were refractory to the induction of high-dose tolerance (19). It has been shown that TIM-3 is necessary for the induction of acquired tolerance with CTLA4-Ig or anti-CD154 plus donor-specific transfusion in an islet transplantation model (20). Although the exact mechanisms of action of TIM-3 in acquired tolerance remain unknown, it is suggested that TIM-3 may modulate the graft-prolonging effects of anti-CD154/donor-specific transfusion by affecting generation of donor-specific Tregs. Several recent studies revealing TIM-3 expression on innate immune cells appear to indicate a more complex role for this molecule in regulating both innate and adaptive immunity (23–26).
Whereas there is evidence for a regulatory function of the TIM-3:galectin-9 pathway in autoimmune diseases and allergy, its role in alloimmunity is unclear. Several studies, mainly investigating graft survival, explored the role of administration of recombinant galectin-9 at pharmacological doses in experimental transplant models (27–30). However, these studies are limited by several concerns: 1) the phenotypes observed with administration of pharmacological doses of recombinant protein may not reflect the biological effects mediated by endogenous galectin-9; 2) lack of in-depth mechanistic studies, particularly in regard to the role of Th17/Tregs in vivo; and 3) the fact that galectin-9 is relatively promiscuous in its ability to bind, and identification of several other glycans aside from TIM-3 as binding partners. The purpose of our study was to explore the mechanisms of action of the TIM-3 molecule in the physiological process of allograft rejection by studying the effects of targeting this molecule on effector T cells and Tregs. Our data establish that TIM-3 plays a key immunoregulatory role in the physiologic rejection process and point to a broader role of this molecule in downregulating inflammatory responses in alloimmunity that involves Th1/Th17/Treg branches of adaptive immunity.
Materials and Methods
Mice
Wild-type (WT), CD4−/−, CD8−/−, CD28−/−, CD80−/−CD86−/− (B7 double-KO), IL-6−/− C57BL/6J (B6, H-2b), BALB/c (H-2d), B6.C-H-2bm1/By (bm1), B6.C-H-2bm12/KhEg (bm12), and DBA/2J mice were obtained at the age of 6–8 wk from The Jackson Laboratory (Bar Harbor, ME) and housed in accordance with institutional guidelines. CD4−/−CD28−/−, ABM Tg and ABM Tg.FoxP3-GFP mice on B6 background were generated and maintained as a breeding colony in our own facility.
Heart transplantation
WT, CD4−/−, CD8−/−, CD4−/−CD28−/−, CD28−/−, CD80−/−CD86−/−, and IL-6−/− B6 animals were used as recipients of heart allografts from BALB/c, bm1, or bm12 mice that were heterotopically transplanted using microsurgical techniques as described previously (16). Graft recipients were treated with either RMT3-23 (21, 31), a blocking Ab against TIM-3, or an appropriate control IgG (500 μg i.p. on day 0; 250 μg i.p. on days 2, 4, 6, 8, and 10). To block IL-6, a neutralizing anti–IL-6 mAb (MP5-20F3) was used at different dosages as indicated. Graft function was assessed daily by palpation, and allograft rejection was defined as the complete cessation of palpable contractility. Graft survival is shown as the median survival time (MST) in days.
Flow cytometry
Splenocytes from transplanted animals were stained for CD4+ and CD8+ effector/memory cells (CD44highCD62Llow phenotype) and CD4+ Tregs (CD25+FoxP3+) using fluorochrome-labeled mAbs against CD4 (clone RM4-5), CD8 (53-6.7), CD44 (IM7), CD62L (MEL-14), CD25 (PC61), ICOS (7E.17G9), CXCR5 (2G8), and FoxP3 (FJK-16s). Intracellular FoxP3 staining was performed after overnight permeabilization using a FixPerm Kit (eBioscience, San Diego, CA). Frequencies were given as percentage of effector/memory or regulatory cells of total CD4+ or CD8+ T cells. For intracellular cytokine staining, cells were restimulated for 4 h with ionomycin (Sigma-Aldrich, St. Louis, MO), PMA (Sigma-Aldrich), and Golgi-Stop (BD Pharmingen) before staining for surface markers. Cells were permeabilized with the Cytofix-Cytoperm Kit (BD Pharmingen, San Diego, CA), washed twice, and stained with mAbs against IFN-γ (XMG1.2), IL-6 (MP5-20F3), and IL-17 (eBio17E7). Graft-infiltrating cells were stained similarly, after digestion of the heart tissue with collagenase D and subsequent isolation of graft-infiltrating lymphocytes by a standard Ficoll density gradient centrifugation.
Staining of apoptotic 7-AAD− annexin-V+ cells was performed using the E Annexin V Apoptosis Detection Kit (BD Pharmingen) according to the manufacturer’s instructions.
Expression of TIM-3 on various immune cells was investigated by co-staining splenocytes from naive or transplanted mice with subset-typical surface markers (CD3 [17A2], CD4, CD8, FoxP3, CD19 [1D3], CD11b [M1/70], CD11c [HL3], NK1.1 [PK136]) and a mAb against TIM-3 (clone 215008, R&D Systems, Minneapolis, MN). All other FACS Abs were purchased from BD Pharmingen or eBioscience. Appropriate isotype controls were used in each set of experiments.
Enzyme-linked immunospot assays
ELISPOT assays were used to assess the frequency of alloreactive IFN-γ–producing, IL-6–producing, IL-17–producing, and granzyme B-producing cells according to manufacturers’ instructions (ELISPOT Kits for IFN-γ and IL-6 from BD Bioscience; ELISPOT Development Module for IL-17 and Granzyme B from R&D Systems). Generally, 0.5 × 106 recipient-derived splenocytes were stimulated with 0.5 × 106 irradiated donor-type stimulator cells for 12 h (granzyme B) or 24 h (IFN-γ, IL-6, IL-17) at 37°C and 5% CO2. To assess the main source of IL-6, we used either unselected splenocytes or splenocytes that were either enriched or depleted of CD4+ T cells, CD8+ T cells, or MHC class II+ APCs using appropriate MACS microbeads (Miltenyi, Bergisch-Gladbach, Germany). The mitogen Con A was used as stimulus for positive controls. The resulting red spots were counted on a computer-assisted ELISPOT image analyzer (Cellular Technology, Shaker Heights, OH), and frequencies were expressed as the number of cytokine-producing spots per 0.5 × 106 responder cells.
Detection of donor-specific Abs
To detect donor-specific Abs (DSAs) in the serum of transplanted animals, donor-type cells from naive BALB/c mice were incubated with diluted serum from naive (as negative control) and transplanted animals at various dilution steps (1:8, 1:16, 1:32, 1:64, 1:128, 1:256) for 30 min. Cells were washed twice with FACS medium and then stained with anti-mouse IgG1, anti-mouse IgG2a, and the appropriate isotype controls for another 30 min. After another two washing steps, cells were analyzed by flow cytometry as described earlier. DSAs were calculated as percentage of IgG1-positive or IgG2a-positive cells of total lymphocytes.
Graft-versus-host experiments
Graft-versus-host experiments were performed to investigate the effects of RMT3-23 on the polyclonal expansion of alloreactive CD4+ and CD8+ T cells. Briefly, 60 × 106 CFSE-labeled splenocytes from either WT, CD28−/−, or IL-6−/− B6 mice were adoptively transferred into sublethally irradiated (1000 rad), fully MHC-mismatched host DBA/2 mice. Animals were then treated with either RMT3-23 or an appropriate control IgG (500 μg i.p. on days 0 and 2). Animals were sacrificed on day 3, and splenocytes were assessed for proliferation of CD4+ and CD8+ T cells by analyzing CFSE dilution.
ABM Tg experiments
ABM transgenic (Tg) experiments were performed to investigate the influence of RMT3-23 on the fate of allospecific effector T cells or Tregs. In the classical ABM Tg model (32, 33), 2 × 106 CD4+ T cells from ABM Tg mice (B6 Thy1.2+) with Tg TCR and consequent defined specificity against bm12 were adoptively transferred into B6 Thy1.1+ mice simultaneously transplanted with a skin graft from a bm12 donor mouse. Animals were then treated with either RMT3-23 or control IgG (500 μg i.p. on day 0; 250 μg i.p. on days 2, 4, and 6). Mice were sacrificed on day 7, and splenocytes were analyzed for absolute numbers of total allospecific Thy1.2+ cells (to assess the influence of RMT3-23 on the proliferation/expansion of these cells) and allospecific effector/memory cells using flow cytometry. In a modified ABM model, Thy1.2+ FoxP3-GFP− T cells from ABM Tg.FoxP3-GFP mice, generated in our laboratory by mating ABM Tg mice with Rudensky reporter mice (B6 FoxP3-GFP knock-in), were used in the same manner, allowing us to assess the effects of RMT3-23 on effector/memory cells and induction of Tregs. To increase the conversion of Thy1.2+ FoxP3-GFP− T cells into Thy1.2+ FoxP3-GFP+ Tregs, recipients were concomitantly treated with murine anti-thymocyte globulin (mATG; 0.5 mg i.p. on days 0 and 4).
Moreover, transfer and similar analysis of sorted Thy1.2+ FoxP3-GFP+ allowed us to investigate the effect of RMT3-23 on the expansion of natural Tregs (nTregs).
Histological assessment
Cardiac allograft samples were fixed in 10% formalin, embedded in paraffin, coronally sectioned, stained with H&E, Elastica-van Gieson’s and Masson’s trichrome dyes, and analyzed by light microscopy. The degree of rejection was determined according to International Society of Heart and Lung Transplantation guidelines. The severity of vasculopathy was graded according to the percentage of luminal occlusion by intimal thickening with a scoring system described previously (0 = normal artery; 1 = <10% occlusion; 2 = 20–50% occlusion; 3 = >50% occlusion). To evaluate the proportion of interstitial fibrosis, the whole area of the coronal section of the upper ventricle and the area of fibrosis (green area of Masson’s trichrome staining) of the same section were measured, and then fibrosis rate was calculated.
Immunohistochemistry was performed on 5-μm-thick, formalin-fixed, paraffin-embedded tissue sections with anti-CD3 (CMC363, Cell Marque, Rocklin, CA; 1:1500, EDTA) and anti-FoxP3 (no. 14-5773, eBioscience; 1:25, citrate) primary Abs. All stained slides were scanned using an Aperio ScanScope XT (Aperio Technologies, Vista, CA). Images were analyzed using ImageScope software (version 10.0.35.1800; Aperio Technologies) and a standard analysis algorithm (nuclear version 9.0; Aperio Technologies) with appropriate modifications for cell and nuclear sizes.
Treg suppression assays
The influence of RMT3-23 on the regulatory capacity of natural Tregs was assessed in vitro by means of a Treg suppression assay: 0.125 × 106 CD4+ CD25+ Tregs from naive B6 animals were sorted by MACS and added to IFN-γ ELISPOT assays containing 0.125 × 106 responder cells from B6 mice previously sensitized with grafts from fully MHC-mismatched BALB/c mice and 0.5 × 106 irradiated (3000 rad) donor-type stimulator cells from naive BALB/c mice. RMT3-23 or appropriate control IgG (10 μg/ml) were then added to these cultures. The effect of RMT3-23 on the suppressive properties of Tregs was assessed by analyzing the frequency of IFN-γ–producing alloreactive T cells when cocultured with Tregs compared with the unmodified reaction of restimulated responder cells without Tregs and by measurement of cytokines in the cell culture supernatants using a LUMINEX 21-plex kit (Millipore, Billerica, MA; see below).
Assessment of the cell source of IL-17 production was performed using a mouse IL-17 secretion assay (Miltenyi), according to the manufacturer’s instructions.
In vitro T cell differentiation
CD4+CD25− T cells from naive B6 mice were isolated using appropriate MACS microbeads (purity 90–93%) and used to investigate the effect of RMT3-23 on the differentiation of naive CD4+ T cells into FoxP3+ Tregs, IFN-γ–producing Th1 cells, or IL-17–producing Th17 cells. CD4+CD25− T cells (1 × 106 cells/well) were incubated for 96 h in 24-well cell culture plates precoated with anti-CD3 (4 μg/ml; clone 145-2C11, BD Pharmingen). Cell culture medium contained anti-CD28 (2 μg/ml; clone 37.51, BD Pharmingen), anti–IL-4 mAb (10 μg/ml; BD Pharmingen), and IL-12 (10 ng/ml; R&D Systems) for Th1 differentiation, anti-CD28 (2 μg/ml), IL-6 (30 ng/ml; R&D Systems), and TGF-β (3 ng/ml; R&D Systems) for Th17 differentiation, and anti-CD28 (1 μg/ml) and TGF-β (3 ng/ml) for Treg differentiation. Either RMT3-23 or appropriate control IgG (10 μg/ml) was then added to the cultures. Th1, Th17, and Treg differentiation was assessed by intracellular staining for IFN-γ (Th1), IL-17 (Th17), or FoxP3.
Cytokine analysis by LUMINEX assays
We used multiplexed cytokine bead-based immunoassays (Millipore, Billerica, MA) to assess the cytokine profile either in serum from transplanted animals or in the supernatants of MLR cell cultures, according to the manufacturer’s instructions. MLR cultures were composed in the same way as ELISPOT assays and incubated at 37°C and 5% CO2 for 48 h.
IL-21 ELISA
IL-21 was assessed in the supernatants of MLR cell cultures, composed as described above, using a mouse IL-21 ELISA Kit (eBioscience), according to the manufacturer’s instructions.
Statistical analysis
Kaplan–Meier survival graphs were constructed, and the log-rank comparisons of the groups were used to calculate p values. Student t test was used for comparison of means between two groups. Data were expressed as mean ± SEM.
Results
Analysis of TIM-3 expression on recipient immune cells during acute allograft rejection
The expression of TIM-3 on various immune cell subsets was examined in naive C57BL/6 mice and in recipients of either isografts or fully MHC-mismatched heart allografts 7 d after transplantation (time of rejection) using flow cytometry. TIM-3 expression on CD4+ and CD8+ T cells was minimal; however, it was constitutively expressed by CD4+FoxP3+ Tregs and at even higher levels by CD11b+ and CD11c+ cells in naive B6 mice (Fig. 1). Whereas TIM-3 expression in recipients of cardiac isografts was comparable overall with that seen in naive mice, it was markedly upregulated on CD4+, CD8+ T cells and CD4+FoxP3+ Tregs 7 d after transplantation of fully MHC-mismatched cardiac allografts, which were acutely rejected. In contrast, TIM-3 expression was downregulated on CD11b+ cells in rejecting mice. TIM-3 was not detected on B cells and NK cells in any groups of animals studied (Fig. 1).
FIGURE 1.

TIM-3 expression during acute allograft rejection. TIM-3 expression on various subsets of immune cells derived from splenocytes of naive B6 and B6 recipients of isografts (B6 donors) or allografts (BALB/c donors) 7 d after transplantation (time point of allograft rejection). Data are shown as percentage of TIM-3+ cells of the respective cell type and are representative of three independent experiments using at least n = 3 mice per group. *p < 0.05; **p < 0.01; ***p < 0.001.
Blockade of TIM-3 enhances T cell alloreactivity and promotes allospecific IL-6 production by CD4+ T cells
Blockade of TIM-3 by RMT3-23 did not alter the tempo of cardiac allograft rejection in WT recipients of BALB/c allografts (MST = 7 versus 7 d; n = 6/14; NS). However, rejection in RMT3-23–treated WT mice was accompanied by an enhanced alloimmune response characterized by increased frequencies of alloreactive IFN-γ–producing, IL-6–producing, IL-17–producing, and granzyme B-producing splenocytes (Fig. 2A). Additionally, the frequencies of IL-17+ graft-infiltrating cells isolated from BALB/c cardiac allografts transplanted into WT B6 recipients at the time of rejection significantly increased after TIM-3 blockade compared with that in controls (Fig. 2D).
FIGURE 2.

Targeting TIM-3 molecule in WT recipients of a fully MHC-mismatched vascularized cardiac allograft. A, RMT3-23 increases the frequency of IFN-γ–producing, IL-6–producing, IL-17–producing, and granzyme B-producing alloreactive splenocytes compared with that in controls (ELISPOT; spot numbers per a total number of 500.000 responder splenocytes on day 4 posttransplantation). B, Splenocytes from RMT3-23–treated or control animals were separated into MHC class II (MHC-II)–positive (= APC) and MHC-II–negative (= non-APC) cells using appropriate MACS microbeads. Unselected, MHC-II+ and MHC-II− splenocytes were used as responder cells in an IL-6 ELISPOT as described earlier. MHC-II− (non-APC) splenocytes are the main source of RMT3-23–enhanced IL-6 production. C, CD4+ and CD8+ T cells from RMT3-23–treated and control animals were isolated using appropriate MACS microbeads and used as responder cells in an IL-6 ELISPOT as described earlier. CD4+ but not CD8+ T cells from RMT3-23–treated animals show significantly increased frequency of IL-6–producing cells compared with that in control animals. Data are representative of three or more independent experiments using at least n = 3 mice per group. All measurements were done in triplicate. *p < 0.05; **p < 0.01; ***p < 0.001. D, Intracellular staining of graft-infiltrating cells (flow cytometry, live lymphocyte gate). Grafts from RMT3-23–treated recipients show increased frequencies of IL-17+ lymphocytes compared with that in untreated controls. Data are representative of two experiments using n = 3 mice per group. *p < 0.05.
Administration of RMT3-23 also resulted in a rise of IL-6 in sera of treated mice on day 4 compared with that in controls (287 ± 192 versus 59.5 ± 23 pg/ml; n = 10/10, p < 0.01). IL-6 is known to be produced by various types of lymphoid cells such as T cells, B cells, monocytes, and dendritic cells (DCs) (34). To explore the source of IL-6 in the above experiments, the IL-6 ELISPOT assay was repeated after removal of MHC class II-expressing splenocytes (= APCs). We found that IL-6 was mainly produced by T cells but not APCs, and similar to our data with unselected splenocytes, targeting TIM-3 significantly enhanced IL-6 production by T cells (Fig. 2B). Interestingly, CD4+ but not CD8+ T cells were mainly responsible for enhanced IL-6 production after TIM-3 blockade (Fig. 2C). Taken together, these data suggest that targeting TIM-3 may alter the differentiation of T cells exposed to alloantigen.
Immunoregulatory functions of TIM-3 are dependent on the presence of CD4+ T cells in cardiac allograft recipients
Next we investigated the effect of TIM-3 blockade in both CD4−/− and CD8−/− B6 recipients of BALB/c allografts. RMT3-23 did not affect the long-term survival of BALB/c allografts in CD4−/− animals (MST > 100 d; n = 6/6; NS), and pathology examinations of both controls and treated animals showed only very discrete lymphocytic infiltration of the grafts (data not shown). In contrast, CD8−/− animals acutely rejected BALB/c allografts without significant effect of RMT3-23 on the tempo of allograft rejection (MST 10.5 versus 10 d; n = 6/6; NS). Surprisingly, in addition to strong lymphocytic infiltration observed in cardiac allografts obtained from both groups, grafts of RMT3-23–treated animals revealed extensive necrotic areas (Fig. 3A), raising the possibility of an Ab-mediated rejection. Indeed, determination of the levels of donor-specific alloantibodies showed significantly higher levels of both donor-specific IgG1 and IgG2a in RMT3-23–treated CD8−/− recipients compared with that in controls (Fig. 3B). Together, these data demonstrate an essential role of CD4+ T cells and suggest that the TIM-3 molecule may exhibit its immunoregulatory functions by affecting CD4+ T cell differentiation into Th1/Th17/Tregs and/or modulating its ability to provide helper functions to CD8+ and B lymphocytes.
FIGURE 3.

Targeting TIM-3 molecule in CD8-deficient recipients of a fully MHC-mismatched vascularized cardiac allograft. A, BALB/c allografts of RMT3-23–treated CD8−/− B6 recipients display extended necrotic areas suggestive of Ab-mediated rejection (n = 6/6). H&E, original magnification ×100. B, Levels of IgG1-and IgG2a-type DSAs are increased in RMT3-23–treated CD8−/− recipients of BALB/c allografts (n = 6/6). Significance levels are shown per dilution step (1:8 to 1:256). *p < 0.05; **p < 0.01; ***p < 0.001.
Administration of anti–TIM-3 Ab accelerates allograft rejection in the absence of CD28:B7 costimulation
We then investigated the functions of RMT3-23 in the absence of CD28 and B7 costimulation. CD28-deficient and CD80/CD86 (B7-1/2)–double-deficient recipients of BALB/c cardiac allografts display prolonged allograft survival compared with that in WT recipients (35, 36). In CD28−/− animals, administration of RMT3-23 significantly accelerated allograft rejection compared with that in untreated animals (Fig. 4A). Accelerated allograft rejection was accompanied by decreased frequency of CD4+CD25+FoxP3+ Tregs and a trend toward higher CD4+ effector T cells, thus altering the Treg/effector T cell ratio (Fig. 4C). Furthermore, targeting TIM-3 molecule significantly increased the frequencies of IFN-γ–producing, IL-6–producing, IL-17–producing, and granzyme B-producing alloreactive host splenocytes (Fig. 4D). While the pathology examination of cardiac tissues showed similar lymphocellular infiltration in both groups without significant differences between treated animals and controls, a trend toward a decreased FoxP3/CD3 ratio of graft-infiltrating T cells was found in RMT3-23–treated CD28-deficient recipients compared with that in controls (Fig. 4B). In addition, consistent with our findings in CD4−/− B6 recipients of BALB/c allografts, TIM-3 blockade did not affect long-term allograft survival in CD4−/− CD28−/− double-deficient recipients (MST > 100 d; n = 6/6; NS), confirming again the essential role of CD4+ T cells for TIM-3–mediated immunoregulation. Next, we tested the effects of neutralizing IL-6 by anti–IL-6 mAb in CD28−/− B6 recipients of BALB/c allografts, in which targeting TIM-3 by RMT3-23 significantly accelerates allograft rejection. Whereas concurrent treatment with lower dose of anti–IL-6 (0.1 mg i.p. on days 0, 1, 2, 3, 7, 10, 14) did not neutralize the graft-abrogating effects of RMT3-23 (MST 10 versus 11 d; n = 6/6; NS), higher doses of IL-6 neutralizing Ab (0.2 mg i.p. on days 0, 1, 2, 3; 0.1 mg i.p. on days 7, 10, 14) reversed the effects of RMT3-23 on allograft survival (MST 16.5 versus 11 d [RMT3-23 alone]; n = 6/6; p = 0.0008; 16.5 versus 15.5 d [control]; n = 6/6; NS) (Fig. 4A). Administration of anti–IL-6 alone did not influence allograft survival (MST 15 [anti-IL6] versus 15.5 d [controls]; n = 6/5; NS).
FIGURE 4.

TIM-3 blockade in CD28-deficient recipients of a fully MHC-mismatched vascularized cardiac allograft. A, Survival of BALB/c allografts in CD28−/− B6 recipients is abrogated by RMT3-23 (n = 6/6; p = 0.002). Anti–IL-6 mAb can neutralize the effects of RMT3-23 in a dose-dependent manner (no effect with lower doses of anti–IL-6 mAb [n = 6]; complete reversal of accelerated allograft rejection after TIM-3 blockade with higher doses of anti–IL-6 mAb [n = 6]). B, Frozen section of cardiac allografts (day 10 posttransplantation) were stained for FoxP3 and CD3 by immunohistochemistry. FoxP3/CD3 ratio of graft-infiltrating T cells is decreased in RMT3-23–treated animals compared with that in controls (n = 6/6). C, Splenocytes from RMT3-23–treated animals (day 10 posttransplantation) show decreased percentages of CD4+CD25+FoxP3+ Tregs and a trend toward higher CD4+ CD44highCD62Llow effector T cells. D, Splenocytes from RMT3-23–treated animals show increased frequencies of IFN-γ–producing, IL-6–producing, IL-17–producing, and granzyme B-producing alloreactive splenocytes as assessed by ELISPOT (data are shown as spot numbers per a total number of 500.000 responder cells on day 10 posttransplantation). Data are representative of three or more independent experiments using at least n = 3 mice per group. All ELISPOT measurements were done in triplicate. *p < 0.05; **p < 0.01; ***p < 0.001.
We also investigated the influence of RMT3-23 on allograft survival in CD80−/−CD86−/− B6 recipients that usually indefinitely accept BALB/c heart allografts (35, 36). Compared with untreated control animals that showed long-term allograft survival, TIM-3 blockade precipitated allograft rejection (Fig. 5A). Graft histopathology showed unaffected myocardial tissue in control animals, whereas treated animals displayed severe necrosis, enhanced cellular myocarditis, and acute vasculitis (Fig. 5B).
FIGURE 5.
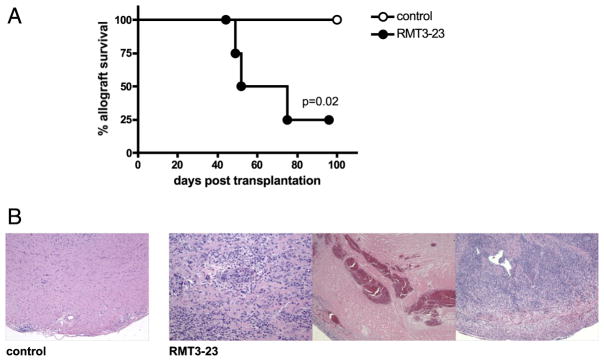
Targeting TIM-3 molecule in B7-1/2–deficient recipients of a fully MHC-mismatched vascularized cardiac allograft. A, Survival of BALB/c allografts in B7-1/2–deficient (CD80−/−CD86−/−) recipients is abrogated by RMT3-23 (n = 6/6). B, BALB/c allografts in control CD80−/−CD86−/− recipients remain unaffected, whereas grafts in RMT3-23–treated animals show strong lymphocellular infiltration, vasculopathy, and necrotic areas. H&E, original magnification ×40–100.
Blockade of TIM-3 increases the polyclonal T cell alloimmune responses in vivo
To quantify polyclonal alloreactive T cell expansion in vivo, we used a graft-versus-host alloimmune response model, which permitted investigation of the effect of TIM-3 blockade on polyclonal expansion of CD4+ and CD8+ T cells. We adoptively transferred CFSE-labeled splenocytes from naive WT (Fig. 6A) or CD28−/− (Fig. 6B) B6 animals into sublethally irradiated fully MHC-mismatched DBA/2 host mice that received a blocking anti–TIM-3 mAb or control Ig (37, 38). TIM-3 blockade resulted in significantly increased proliferation of both CD4+ and CD8+ T cells in WT and CD28−/− animals, confirming its ability to expand alloreactive effector T cells.
FIGURE 6.

TIM-3 blockade increases the polyclonal T cell alloresponse. A, Proliferation of CFSE-labeled CD4+ and CD8+ T cells from WT B6 animals in irradiated, fully MHC-mismatched DBA/2 hosts. RMT3-23 increases proliferation of both CD4+ and CD8+ T cells, as assessed by CFSE dilution. n = 5/5; *p < 0.05. B, Proliferation of CFSE-labeled CD4+ and CD8+ T cells from CD28-deficient B6 animals in irradiated, fully MHC-mismatched DBA/2 hosts. RMT3-23 increases proliferation of both CD4+CD28− and CD8+CD28− T cells, as assessed by CFSE dilution. n = 5/5. *p < 0.05.
To assess to what extent TIM-3 blockade influences apoptosis of CFSE-labeled cells, we analyzed the percentage of apoptotic (7-AAD− annexin-V+) CD4+ and CD8+ T cells. We found slight yet statistically significant decrease of apoptosis of CD4+ but not CD8+ T cells after TIM-3 blockade (Supplemental Fig. 1). Finally, in contrast with CD4+ and CD8+ T cells from WT animals, neither IL-6−/− CD4+ T cells (proliferation 38.6 ± 1.27% versus 39.9 ± 1.12%; n = 5/5; NS) nor IL-6−/− CD8+ T cells (proliferation 31.3 ± 0.72% versus 30.7 ± 0.86%; n = 5/5; NS) showed increased proliferation after TIM-3 blockade in this model.
Blockade of TIM-3 abrogates long-term allograft survival in recipients of MHC class II-mismatched but not MHC class I-mismatched allografts
Allografts mismatched at only MHC class II alleles (bm12 into B6) survive long-term, although they do develop chronic allograft vasculopathy (CAV). This model is unique, not only in that it is characterized by primary activation of alloreactive effector CD4+ T cells leading to CAV, but also in that it is dependent on Tregs for long-term allograft survival. We and others have demonstrated that depletion of Tregs by anti-CD25 mAb can precipitate acute cardiac rejection in this model (MST = 16 versus > 56 d; n = 6/6; p < 0.001). Targeting TIM-3 resulted in accelerated graft rejection of the majority of bm12 hearts (Fig. 7A). Whereas pathology examination of the samples showed a moderate degree of CAV in control animals, most treated animals rejected around day 30 and showed severe lymphocellular infiltration as observed in acute rejection (Fig. 7B). In addition, CAV was much more pronounced in treated animals that maintained beating (though weakened) allografts at 6 wk after transplantation (Fig. 7B). Overall, allografts from RMT3-23–treated animals showed higher degrees of cellular infiltration, CAV, and allograft fibrosis (Fig. 7B). Interestingly, RMT3-23–treated IL-6−/− recipients showed a similar survival to that of untreated control animals (MST > 56 d; n = 6/6; NS). Whereas RMT3-23–mediated rejection in WT B6 recipients of bm12 allografts was accompanied by increased frequencies of both alloreactive IL-17– and IFN-γ–producing splenocytes, RMT3-23 did not increase the frequencies of alloreactive IL-17–or IFN-γ–producing splenocytes in IL-6−/− recipients of bm12 hearts (Fig. 7C).
FIGURE 7.

Blockade TIM-3 molecule in a MHC class II-mismatched model of CAV. A, Survival of bm12 allografts in B6 recipients is abrogated by RMT3-23 (n = 6/6). B, Whereas control animals show only moderate CAV (top photo), RMT3-23–treated animals show either accelerated CAV (middle photo) or more severe lymphocellular infiltration (bottom photo). H&E, original magnification ×40–100. Administration of RMT3-23 results overall in accelerated CAV, as shown by enhanced cellular infiltrates, vasculopathy, and increased fibrosis. C, RMT3-23 increases the frequencies of both IFN-γ– and IL-17–producing alloreactive splenocytes in WT but not in IL-6−/− B6 recipients of bm12 cardiac allografts (ELISPOT; spot numbers per a total number of 500.000 responder splenocytes at day 30 posttransplantation). Data are representative of three or more independent experiments using at least n = 3 mice per group. All ELISPOT measurements were done in triplicate. **p < 0.01; ***p < 0.001.
In contrast with MHC class II-mismatched transplants, MHC class I-mismatched hearts (bm1 into B6) are thought to develop minor CAV, mainly due to activation of CD8+ T cells, and are known to be independent of Tregs (MST > 56 d before and after CD25 depletion; n = 6/6). In keeping with our previous data indicating the essential role of CD4+ T cells for immunoregulatory functions of TIM-3, TIM-3 blockade affected neither the allograft survival nor the histopathological findings in the bm1 cardiac samples examined (data not shown).
Blockade of TIM-3 increases the frequency of allospecific CD4+ effector T cells but not graft-reactive natural Tregs and prohibits induction of adaptive Tregs
Because of the impact of RMT3-23 mAb in B6 recipients of bm12 allografts, the effects of TIM-3 blockade on the fate of allospecific CD4+ T cells were further studied using ABM-TCR transgenic (ABM Tg) mice (32, 33). CD4+ T cells (2 × 106) from ABM Tg mice (B6 Thy1.2) with defined specificity against bm12 were adoptively transferred into B6 Thy1.1 mice who were simultaneously transplanted with a skin graft from a bm12 donor mouse. Animals were then treated with either RMT3-23 or control IgG (500 μg i.p. on day 0; 250 μg i.p. on days 2, 4, and 6). Animals were sacrificed on day 7, and splenocytes and lymphocytes from the draining lymph nodes were analyzed for absolute numbers of total allospecific Thy1.2+ cells (to assess the influence of RMT3-23 on the proliferation/expansion of these cells) and allospecific effector cells using flow cytometry. Seven days after adoptive T cell transfer and skin transplantation, there was significant expansion of allospecific Thy1.2+ cells isolated from the spleens and draining lymph nodes of RMT3-23–treated animals compared with that in controls (Fig. 8A). Similarly, targeting TIM-3 resulted in the expansion of effector CD4+ T cells in spleens and draining lymph nodes (Fig. 8B). Evaluation of apoptotic (7-AAD− annexin-V+) ABM Tg cells showed a minor, yet statistically significant, decrease in apoptosis after TIM-3 blockade, suggesting that apoptosis alone does not account for increased expansion of T cells observed after TIM-3 blockade (Fig. 8C). RMT3-23 also increased the frequencies of allospecific IFN-γ+ and IL-17+ ABM Tg cells and decreased the overall percentage of alloreactive FoxP3+ Tregs (Fig. 8D).
FIGURE 8.

Effects of TIM-3 blockade on allospecific effector T cells. A, Seven days after transplantation of skin grafts from bm12 donors and adoptive transfer of 2 × 106 bm12-specific CD4+ Thy1.2+ T cells from ABM Tg animals, RMT3-23–treated Thy1.1+ B6 mice showed increased total numbers of allospecific Thy1.2+ T cells in both draining lymph nodes and spleens. B, In the same model, targeting TIM-3 results in expansion of allospecific Thy1.2+ effector T cells in spleen and lymph nodes of Thy1.1+ B6 recipients. C, Allospecific Thy1.2+ T cells in RMT3-23–treated animals show decreased apoptosis compared with that in untreated controls. D, In RMT3-23–treated animals, allospecific Thy1.2+ T cells show increased expression of IFN-γ+ and IL-17+ and decreased expression of FoxP3 compared with that in untreated controls. Data are representative of three or more independent experiments using at least n = 3 mice per group. *p < 0.05.
To evaluate concurrently the expansion of effector CD4+ T cells and induction of adaptive Tregs by RMT3-23, the experiment above was repeated by using CD4+GFP− cells (non-Tregs) from ABM Tg.FoxP3-GFP animals that were generated by mating ABM Tg mice with B6 FoxP3-GFP reporter mice. As the in vivo induction of CD4+GFP+ cells from GFP− cells was negligible in rejecting control animals in this model, we were unable to evaluate the effects of targeting TIM-3 on the induction of adaptive Tregs. To circumvent this problem, we used a strategy to enhance the induction of adaptive Tregs by treating the Thy1.1+ B6 recipients of sorted Thy1.2+ FoxP3-GFP− cells and bm12 skin grafts with mATG (39). Using this model, concurrent treatment with RMT3-23 and mATG significantly inhibited the ATG-mediated de novo induction of iTregs, which was observed in the control animals treated only with mATG (Fig. 9D). Treatment with RMT3-23 significantly increased the number of allospecific Thy1.2+CD4+ CD44highCD62Llow effector T cells (Fig. 9A), resulting overall in a significant decline in Treg/effector T cell balance (Fig. 9B). Lastly, we evaluated the effect of targeting TIM-3 on expansion of natural Tregs by adoptive transfer of CD4+GFP+ cells derived from ABM Tg.FoxP3-GFP mice into syngeneic B6 mice that then received a skin graft from a bm12 donor and were treated either with RMT3-23 or control IgG. Enumeration of Thy1.2+GFP+ Tregs revealed no effects of TIM-3 blockade in treated animals compared with that in controls (Fig. 9C). Taken together, these data suggest that TIM-3 may suppress expansion of effector T cells and induction of adaptive Tregs without affecting the frequency of nTregs.
FIGURE 9.
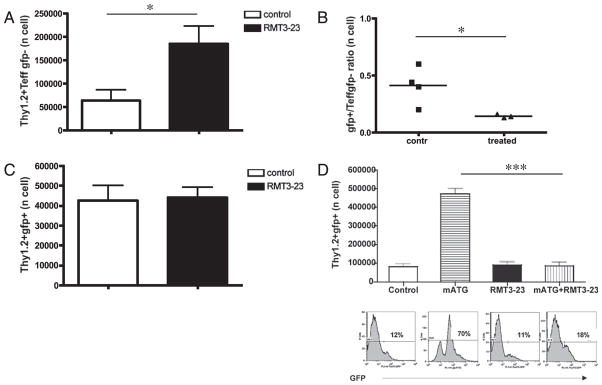
Effects of TIM-3 blockade on expansion of allospecific nTregs and induction of adaptive Tregs. A, Seven days after transplantation of skin grafts from bm12 donors and adoptive transfer of 2 × 106 bm12-specific CD4+ Thy1.2+GFP− T cells from ABM Tg FoxP3-GFP mice, RMT3-23–treated Thy1.1+ B6 mice showed increased total numbers of Thy1.2+ GFP− CD44highCD62Llow allospecific effector CD4+ T cells in spleens. B, In the same model and experiment described earlier, RMT3-23 decreases the Treg/effector T cell ratio in Thy1.1+ B6 recipients. C, Seven days after transplantation of skin grafts from bm12 donors and adoptive transfer of 2 × 106 sorted Thy1.2+GFP+ nTregs originating from ABM Tg FoxP3-GFP mice, TIM-3 blockade does not influence the expansion of nTregs. D, Bar graphs show absolute numbers of Thy1.2+ FoxP3-GFP+ cells in spleens of Thy1.1+ B6 recipients 7 d after bm12 skin transplantation, adoptive transfer of Thy1.2+ FoxP3-GFP− cells, and treatment with either control Ig, mATG, RMT3-23 alone, or RMT3-23 in combination with mATG. Representative histograms of Thy1.2+ FoxP3-GFP+ cells from recipients in each treatment group are shown below the respective bar graphs. While mATG significantly converts Thy1.2+ FoxP3-GFP− cells into Thy1.2+ FoxP3-GFP+ Tregs, this effect is abrogated when RMT3-23 is added. There is no difference in the percentage of Thy1.2+ FoxP3-GFP+ Tregs between Ig-treated controls, RMT3-23–treated and mATG/RMT 3-23-treated animals. Data are representative of three or more independent experiments using at least n = 3 mice per group. *p < 0.05; ***p < 0.001.
Effects of blockade of TIM-3 on the suppressive capacity of natural Tregs
The effect of RMT3-23 on the actual suppressive functions of nTregs was tested in an in vitro suppression IFN-γ ELISPOT assay in which nTregs isolated from naive B6 mice were added to responder cells from B6 mice previously sensitized with BALB/c grafts for 7 d and restimulated with irradiated donor-type BALB/c stimulators. The suppressive effects of nTregs on IFN-γ–producing alloreactive T cells were similar regardless of the presence or absence of RMT3-23 (Fig. 10A). Analysis of supernatants in the same cultures by LUMINEX revealed similar effects of nTregs in lowering the concentration of IFN-γ regardless of the presence of RMT3-23 (Fig. 10B). Consistent with our mechanistic data in RMT3-23–treated animals in vivo, targeting TIM-3 increased IL-6 production in cocultures. Intriguingly, we found a significant increase in IL-17 production when nTregs were added to the assay in the presence of RMT3-23.
FIGURE 10.

Role of TIM-3 in the suppressive capacity of nTregs. A, Splenocytes from sensitized B6 mice were restimulated with irradiated BALB/c splenocytes and incubated with nTregs from naive B6 mice. The in vitro suppressive power of nTregs in the presence or absence of RMT3-23 was assessed by enumeration of allospecific IFN-γ–producing cells (ELISPOT: 0.125 × 106 responder cells from sensitized B6 mice; 0.5 × 106 irradiated donor-type stimulator cells from BALB/c mice; responder/regulator ratio = 1:1). Data are shown as percentage suppression (decrease in spot number). B, Supernatants from this suppression assay were assessed by LUMINEX multicytokine assay. nTreg-mediated suppression of IFN-γ production is not affected by RMT3-23, but RMT3-23 increases IL-6 production independent of the presence or absence of nTregs. Moreover, RMT3-23 strongly increases IL-17 production in the presence of nTregs. Data are representative of three independent experiments using at least n = 3 mice per group. All measurements were done in triplicate. *p < 0.05; **p < 0.01; ***p < 0.001.
To demonstrate which cells (effector cells versus Tregs) are responsible for the increased levels of IL-17 in the presence of Tregs after addition of RMT3-23, we sensitized Thy1.1+ B6 mice with BALB/c, restimulated them with irradiated donor-type APCs from a naive BALB/c, and then added Tregs derived from naive WT (Thy1.2+) B6 in a 1:1 ratio, both in the presence and absence of RMT3-23. After 48 h of culture, cells were collected, and an IL-17 secretion assay was performed. Co-staining of the cells with FACS mAbs against Thy1.1 and Thy1.2 permitted differentiation between responder cells (Thy1.1+) and Tregs (Thy1.2+). We found that Thy1.1+ responder cells are the main source of IL-17 (percentage IL-17+ cells: 2.72 ± 0.29% [Thy1.1+ responder cells] versus 0.36 ± 0.08% [Thy1.2+ Tregs]; n = 2/2; p = 0.02). To confirm this observation, we then used responder cells derived from sensitized B6 IL-17–deficient B6 instead of WT B6 mice and repeated the same in vitro suppression assay as described. In this model, only the WT Tregs, but not the responder cells, could potentially produce IL-17. Again, we found no IL-17–producing cells among Tregs (0.04 ± 0.02%; n = 2). More importantly, IL-17 was not detectable in the supernatants of these cultures, as assessed by LUMINEX assay.
All in all, these data suggest that prorejection effects of targeting TIM-3 may not be due to preventing suppression of alloreactive Th1 cells by nTregs but rather due to enhanced inflammatory cytokines and alloreactivity, as effector T cells inhibited by Tregs begin producing IL-17.
Targeting TIM-3 opposes TGF-β–mediated iTreg differentiation and promotes Th1/Th17 polarization in vitro
To investigate the mechanisms by which RMT3-23 can affect CD4+ T cell differentiation under more controlled conditions, CD4+ CD25− T cells from naive B6 mice were stimulated in vitro under Th1/Th17 and iTreg differentiation conditions with immobilized anti-CD3 and soluble anti-CD28. RMT3-23 strongly inhibits TGF-β–mediated generation of FoxP3+ Tregs and enhances the generation of Th1 and Th17 cells in Th1/Th17-promoting conditions (Fig. 11). Taken together with our previous data in transplant recipients and Tg models treated with RMT3-23, these results suggest that in vitro, blocking the TIM-3 molecule induces IL-6 production, which antagonizes iTreg differentiation and promotes IL-17 production by naive CD4+ T cells.
FIGURE 11.

Role of TIM-3 in differentiation of naive CD4+ T cells into Th1/Th17/iTregs. CD4+CD25− T cells were isolated from B6 splenocytes using appropriate CD4 and CD25 MACS microbeads (purity of CD4+ cells 90–93%). Enriched CD4+CD25− T cells were incubated for 96 h using culture conditions favoring either Th1, Th17, or Treg differentiation. RMT3-23 enhances the in vitro differentiation of Th1 and Th17 cells and impairs the conversion of naive CD4+ cells into Tregs. Data are representative of four independent experiments using at least n = 6 mice per experiment. *p < 0.05; **p < 0.01.
Discussion
The establishment of effective regulation of alloreactive T cell-mediated inflammation is a major goal of tolerance-inducing strategies in transplantation. Acute allograft rejection has been traditionally correlated with Th1 and more recently with Th17 differentiation in some models (6, 7, 9), whereas transplantation tolerance is frequently associated with induction of regulation (4). Thus, CD4+ T cell differentiation to Th1/Th2/Th17 and iTregs plays a central role in transplantation rejection and induction of tolerance (4). TIM molecules represent a unique family of genes and affect many aspects of immunology, including T cell activation, cell survival and death, as well as the capacity of APCs to clear apoptotic cells (17). TIM-3 was originally identified as a surface molecule expressed on Th1 cells (18), and several studies in various immune-mediated diseases suggest that it may be an inhibitory molecule that terminates Th1 immunity after binding to its ligand galectin-9 (22). The goal of our study was to explore the role and mechanisms of action of TIM-3 in physiologic rejection by interruption of its interaction with endogenous galectin-9 using a vascularized cardiac transplant model. To our knowledge, the current report establishes for the first time that TIM-3 is a critical pathway in down- modulating inflammatory responses during acute rejection that involves the Th1/Th17/iTreg branches of adaptive immunity. Rather than only regulating the differential survival of various Th cell subsets by death/apoptosis, TIM-3 can also direct the differentiation of CD4+ T cells by regulating IL-6 production.
Blockade of TIM-3 molecule did not affect graft survival in B6 recipients of fully MHC-mismatched BALB/c allografts that are usually promptly rejected, but accelerated the rejection in CD28-deficient and B7-1/2–double-deficient B6 recipients that usually have longer allograft survival due to the lack of CD28:B7 co-stimulation. Ex vivo mechanistic studies of mice treated with blocking mAb demonstrated a markedly enhanced alloimmune response to donor Ag. One significant observation was the finding that the enhanced alloresponse was not confined to an increase in frequency of IFN-γ–producing allospecific splenocytes, as one would have expected based on previous reports of expression of TIM-3 molecule on predominant Th1 cells (23). In fact, we also found a significant rise in alloreactive IL-6, granzyme B, and IL-17 production. Furthermore, it is noteworthy to point out that we detected a rise in the frequency of IL-17–producing graft-infiltrating lymphocytes in WT recipients after TIM-3 blockade. These data are in agreement with studies demonstrating a pathogenic role for IL-17 in rejection (6, 7). The aggressive proinflammatory responses leading to severe cellular and Ab-mediated rejection after blockade of TIM-3:galectin-9 pathway are very similar to what we have recently reported in T-bet−/− mice in a chronic model of cardiac allograft rejection (7). Intriguingly, in that study, CD4+ Th17 cells were indeed shown to be primarily responsible for severe vascular inflammation and vasculopathy. Finally, the percentage of CD4+ICOS+CXCR5+ T follicular helper cells did not differ between control and anti–TIM-3–treated transplant recipients in any model or at any time point examined (data not shown). Taken together with results presented previously, our data support induction of Th17, but not T follicular helper, T cells after targeting TIM-3 in our models.
Our data demonstrate that TIM-3 protein is not expressed by naive CD4+ and CD8+ T cells but is upregulated during acute allograft rejection. These results are consistent with previous data, albeit in different immune-mediated models, demonstrating increase in TIM-3 expression as T cells differentiate into Th1/Th17 cells (18, 20, 40). Consistent with recent studies, we also found significant expression of TIM-3 on DCs in naive mice, raising the possibility of a role of the innate immune system (17). In fact, several other studies suggest that blocking TIM-3 could alter the inflammatory response by affecting the innate immune system (25, 41), and another study supports a role for TIM-3 in directly activating innate immunity (42). IL-6 is a pleiotropic cytokine with a wide range of biological activities in immune regulation (43). It is produced by various types of lymphoid cells such as T cells, B cells, monocytes, and DCs. Therefore, we then investigated which immune cells are actually involved in alloreactive IL-6 production after TIM-3 blockade. We establish that T cells but not APCs are the main source of IL-6 production in rejecting mice, regardless of treatment with RMT3-23 or control Ig. More intriguingly, the overall rise in alloreactive IL-6 by TIM-3 blockade was due to increased production by CD4+ T cells. Importantly, experiments with IL-6 neutralization or using IL-6–deficient hosts establish this cytokine as a key player in acceleration of physiological allograft rejection after targeting TIM-3.
CD4−/− and CD4−/−CD28−/− recipients accept fully MHC-mismatched heart allografts indefinitely. However, we and others have previously demonstrated that this is not due to inherent inability of CD8+ T cells to reject cardiac allografts, as blockade of B7:CTLA-4 and programmed death 1:programmed death ligand-1 pathways resulted in acute cardiac rejection (36). In contrast, RMT3-23 did not precipitate acute rejection in CD4−/− and CD4−/− CD28−/− recipients of BALB/c hearts. Thus, the presence of host CD4+ T cells seems to be essential for immunoregulatory functions of TIM-3. Taken together, these data confirm that TIM-3 exhibits its regulatory functions by affecting the differentiation of CD4+ effector cells into Th1/Th17/iTreg cells and possibly modulating their ability to provide help to CD8+ and B cells. Accordingly, we observed an increase in granzyme B-producing splenocytes and increased alloantibody production in RMT3-23–treated allograft recipients. In agreement with these data, targeting the TIM-3 molecule had no effects on allograft survival in an MHC class I-mismatched bm1 into B6 allograft model but precipitated acute rejection in an MHC class II-mismatched bm12 into B6 model, which is characterized primarily by CD4+ T cell alloactivation, and in which the long-term allograft survival is dependent on Tregs.
To dissect further the mechanisms of actions of TIM-3 blockade, we made use of several other in vitro and in vivo models: Using the graft-versus-host model, we first confirmed expansion of polyclonal CD4+ and CD8+ alloreactive T cells after blockade of TIM-3, regardless of the presence of the CD28 molecule. These data were then confirmed in an adoptive transfer model (32, 33) in which ABM Tg CD4+ T cells with specificity to bm12 were injected into syngeneic WT mice receiving a bm12 skin transplant and either treated with isotype IgG or RMT3-23. By tracking these T cells, we also found significant expansion of allospecific T cells with an effector and Th1/Th17 cytokine phenotype. Similar results in experiments using CD4+ FoxP3-GFP− T cells sorted from ABM Tg.FoxP3-GFP mice confirmed the expansion of effector T cells after targeting TIM-3. These data are in contrast with findings by Sánchez-Fueyo et al. (20) in NOD mice, in which TIM-3 blockade accelerated diabetes without increasing proliferation of islet-specific Th1 cells. The authors hypothesized that targeting TIM-3 enhanced the capacity of effector T cells to mediate tissue injury. Our data showing enhanced inflammation (IL-6/IL-17) after TIM-3 blockade could support the latter hypothesis. Lastly, in vitro differentiation experiments confirmed enhanced Th1/Th17 polarization in the presence of RMT3-23. In fact, TIM-3 has recently been shown to be expressed by Th17 cells, and TIM-3 interaction with galectin-9 can inhibit the differentiation of naive T cells to Th17 cells in vitro (40, 44). Another important observation was that blockade of TIM-3 resulted in a significant decline in Treg/effector T cell ratio both in spleens and in target allografts of transplant recipients and in experiments using ABM Tg mice. The importance of the ratio of Tregs to effector T cells in target organs is being increasingly recognized both in the tumor and allograft settings (45). The decline of the ratio both in the target organ and peripheral lymphoid tissues argues against inability of Tregs to migrate as a result of TIM-3 blockade. Although the expansion of effector T cells by blocking TIM-3, as clearly established by our data, may by itself account for a decreased ratio, RMT3-23 may also inhibit proliferation of nTregs and/or prevent the conversion of naive alloreactive T cells into iTregs. Sánchez-Fueyo et al. (20) previously demonstrated a possible role of TIM-3 pathway in the augmentation of overall Ag-specific potency of Tregs isolated from mice tolerized to allografts by costimulatory blockade. However, they were not able to distinguish whether allospecific Tregs generated in the presence of intact TIM-3 have enhanced immunosuppressive function or whether TIM-3 may be necessary for expansion of alloantigen-specific Tregs. To address these questions further, we adoptively transferred purified GFP+ nTregs or GFP− T cells derived from ABM Tg.GFP-FoxP3 mice into syngeneic WT recipients of bm12 skins and tracked the expansion of Ag-specific nTregs or induction of Tregs (enhanced by mATG) in vivo with or without treatment with RMT3-23. Targeting TIM-3 did not inhibit expansion of allospecific nTregs in this model. However, RMT3-23 clearly antagonizes iTreg differentiation from naive CD4+ T cells both in vivo and in in vitro polarization experiments. Lastly, we tested the suppressive capacity of nTregs in the presence of mAb against TIM-3. Notably, although targeting TIM-3 did not interfere with the suppression of Th1 alloreactive T cells, it significantly increased IL-17 production by effector T cells in the corresponding culture supernatants. These data are in keeping with a previous report in TIM-3–deficient mice demonstrating that the absence of TIM-3 does not abolish the ability of alloantigen-inexperienced nTregs to suppress Th1 responses (20). However, these findings are consistent with reports that IL-6 can induce differentiation of helper T cells to become Th17 in a TGF-β–dependent manner (13, 14, 46).
In sum, our data show that TIM-3 signaling to CD4+ T cells emerges as a key pathway for development of adaptive Tregs and inhibition of Th1/Th17 differentiation, thus controlling the balance between tolerance and immunity. Modulation of IL-6 production by CD4+ T cells, but not innate immune cells, seems to be a key event; in fact, inflammatory cytokines, particularly IL-6, are able to determine the quality of the cellular alloresponses. Nevertheless, we cannot exclude other possible mechanisms by which targeting TIM-3, as expressed by different subsets of immune cells, could modulate their functions.
The results of this study have clinical implications. First, TIM-3 is a unique molecule in that TIM-3–regulated pathways are important in the differentiation of CD4+ helper T cells into Th1/Th17/Tregs, likely by controlling production of inflammatory cytokines such as IL-6 by T cells and cells of innate immunity. In particular, there is a growing body of evidence about the key role of IL-6 in transplant rejection (9) and inhibition of allograft acceptance (47). Second, enhancing TIM-3:galectin-9 pathway using a stable form of galectin-9 to control inflammatory reactions is feasible and currently under investigation as a promising tool to treat various immune-mediated diseases (48). In sum, harnessing physiologic mechanisms that regulate alloimmunity could lead to development of novel strategies to induce durable and reproducible transplantation tolerance.
Acknowledgments
This work was supported by National Institutes of Health Grants RO1 AI51559 and AI70820. N.N. is a recipient of an American Heart Association Scientist Development Grant and National Institutes of Health Grant 1KO8AI064335-01A2. O.B. is funded by Research Fellowship grants from the German Research Foundation (Deutsche Forschungsgemeinschaft) and the American Society of Transplantation.
Abbreviations used in this paper
- CAV
chronic allograft vasculopathy
- DC
dendritic cell
- DSA
donor-specific Ab
- DST
donor-specific transfusion
- iTreg
induced regulatory CD4+ T cell
- mATG
murine antithymocyte globulin
- MHC-II
MHC class II
- MST
median survival time
- nTreg
natural regulatory T cell
- TIM
T cell Ig domain and mucin domain
- Tg
transgenic
- Treg
regulatory T cell
- WT
wild-type
Footnotes
Disclosures
The authors have no financial conflicts of interest.
The online version of this article contains supplemental material.
References
- 1.Alegre ML, Najafian N. Costimulatory molecules as targets for the induction of transplantation tolerance. Curr Mol Med. 2006;6:843–857. doi: 10.2174/156652406779010812. [DOI] [PubMed] [Google Scholar]
- 2.Li XC, Rothstein DM, Sayegh MH. Costimulatory pathways in transplantation: challenges and new developments. Immunol Rev. 2009;229:271–293. doi: 10.1111/j.1600-065X.2009.00781.x. [DOI] [PubMed] [Google Scholar]
- 3.Boenisch O, Sayegh MH, Najafian N. Negative T-cell costimulatory pathways: their role in regulating alloimmune responses. Curr Opin Organ Transplant. 2008;13:373–378. doi: 10.1097/MOT.0b013e328306117f. [DOI] [PubMed] [Google Scholar]
- 4.Strom TB, Koulmanda M. Recently discovered T cell subsets cannot keep their commitments. J Am Soc Nephrol. 2009;20:1677–1680. doi: 10.1681/ASN.2008101027. [DOI] [PubMed] [Google Scholar]
- 5.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 6.Yuan X, Ansari MJ, D’Addio F, Paez-Cortez J, Schmitt I, Donnarumma M, Boenisch O, Zhao X, Popoola J, Clarkson MR, et al. Targeting Tim-1 to overcome resistance to transplantation tolerance mediated by CD8 T17 cells. Proc Natl Acad Sci USA. 2009;106:10734–10739. doi: 10.1073/pnas.0812538106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan X, Paez-Cortez J, Schmitt-Knosalla I, D’Addio F, Mfarrej B, Donnarumma M, Habicht A, Clarkson MR, Iacomini J, Glimcher LH, et al. A novel role of CD4 Th17 cells in mediating cardiac allograft rejection and vasculopathy. J Exp Med. 2008;205:3133–3144. doi: 10.1084/jem.20081937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burrell BE, Csencsits K, Lu G, Grabauskiene S, Bishop DK. CD8+ Th17 mediate costimulation blockade-resistant allograft rejection in T-bet-deficient mice. J Immunol. 2008;181:3906–3914. doi: 10.4049/jimmunol.181.6.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Ahmed E, Wang T, Wang Y, Ochando J, Chong AS, Alegre ML. TLR signals promote IL-6/IL-17-dependent transplant rejection. J Immunol. 2009;182:6217–6225. doi: 10.4049/jimmunol.0803842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+ CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 11.Pasare C, Medzhitov R. Toll-dependent control mechanisms of CD4 T cell activation. Immunity. 2004;21:733–741. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 13.Zheng SG, Wang J, Horwitz DA. Cutting edge: Foxp3+CD4+ CD25+ regulatory T cells induced by IL-2 and TGF-beta are resistant to Th17 conversion by IL-6. J Immunol. 2008;180:7112–7116. doi: 10.4049/jimmunol.180.11.7112. [DOI] [PubMed] [Google Scholar]
- 14.Dominitzki S, Fantini MC, Neufert C, Nikolaev A, Galle PR, Scheller J, Monteleone G, Rose-John S, Neurath MF, Becker C. Cutting edge: trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. J Immunol. 2007;179:2041–2045. doi: 10.4049/jimmunol.179.4.2041. [DOI] [PubMed] [Google Scholar]
- 15.Xiao S, Najafian N, Reddy J, Albin M, Zhu C, Jensen E, Imitola J, Korn T, Anderson AC, Zhang Z, et al. Differential engagement of Tim-1 during activation can positively or negatively costimulate T cell expansion and effector function. J Exp Med. 2007;204:1691–1702. doi: 10.1084/jem.20062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ueno T, Habicht A, Clarkson MR, Albin MJ, Yamaura K, Boenisch O, Popoola J, Wang Y, Yagita H, Akiba H, et al. The emerging role of T cell Ig mucin 1 in alloimmune responses in an experimental mouse transplant model. J Clin Invest. 2008;118:742–751. doi: 10.1172/JCI32451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodriguez-Manzanet R, DeKruyff R, Kuchroo VK, Umetsu DT. The costimulatory role of TIM molecules. Immunol Rev. 2009;229:259–270. doi: 10.1111/j.1600-065X.2009.00772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–541. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- 19.Sabatos CA, Chakravarti S, Cha E, Schubart A, Sánchez-Fueyo A, Zheng XX, Coyle AJ, Strom TB, Freeman GJ, Kuchroo VK. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003;4:1102–1110. doi: 10.1038/ni988. [DOI] [PubMed] [Google Scholar]
- 20.Sánchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, Manlongat N, Bender O, Kamradt T, Kuchroo VK, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. 2003;4:1093–1101. doi: 10.1038/ni987. [DOI] [PubMed] [Google Scholar]
- 21.Oikawa T, Kamimura Y, Akiba H, Yagita H, Okumura K, Takahashi H, Zeniya M, Tajiri H, Azuma M. Preferential involvement of Tim-3 in the regulation of hepatic CD8+ T cells in murine acute graft-versus-host disease. J Immunol. 2006;177:4281–4287. doi: 10.4049/jimmunol.177.7.4281. [DOI] [PubMed] [Google Scholar]
- 22.Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 23.Hafler DA, Kuchroo V. TIMs: central regulators of immune responses. J Exp Med. 2008;205:2699–2701. doi: 10.1084/jem.20082429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frisancho-Kiss S, Nyland JF, Davis SE, Barrett MA, Gatewood SJ, Njoku DB, Cihakova D, Silbergeld EK, Rose NR, Fairweather D. Cutting edge: T cell Ig mucin-3 reduces inflammatory heart disease by increasing CTLA-4 during innate immunity. J Immunol. 2006;176:6411–6415. doi: 10.4049/jimmunol.176.11.6411. [DOI] [PubMed] [Google Scholar]
- 25.Zhao J, Lei Z, Liu Y, Li B, Zhang L, Fang H, Song C, Wang X, Zhang GM, Feng ZH, Huang B. Human pregnancy up-regulates Tim-3 in innate immune cells for systemic immunity. J Immunol. 2009;182:6618–6624. doi: 10.4049/jimmunol.0803876. [DOI] [PubMed] [Google Scholar]
- 26.Nakae S, Iikura M, Suto H, Akiba H, Umetsu DT, Dekruyff RH, Saito H, Galli SJ. TIM-1 and TIM-3 enhancement of Th2 cytokine production by mast cells. Blood. 2007;110:2565–2568. doi: 10.1182/blood-2006-11-058800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang F, Wan L, Zhang C, Zheng X, Li J, Chen ZK. Tim-3-galectin-9 pathway involves the suppression induced by CD4+CD25+ regulatory T cells. Immunobiology. 2009;214:342–349. doi: 10.1016/j.imbio.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 28.He W, Fang Z, Wang F, Wu K, Xu Y, Zhou H, Du D, Gao Y, Zhang WN, Niki T, et al. Galectin-9 significantly prolongs the survival of fully mismatched cardiac allografts in mice. Transplantation. 2009;88:782–790. doi: 10.1097/TP.0b013e3181b47f25. [DOI] [PubMed] [Google Scholar]
- 29.Wang F, He W, Zhou H, Yuan J, Wu K, Xu L, Chen ZK. The Tim-3 ligand galectin-9 negatively regulates CD8+ alloreactive T cell and prolongs survival of skin graft. Cell Immunol. 2007;250:68–74. doi: 10.1016/j.cellimm.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Wang F, He W, Yuan J, Wu K, Zhou H, Zhang W, Chen ZK. Activation of Tim-3-galectin-9 pathway improves survival of fully allogeneic skin grafts. Transpl Immunol. 2008;19:12–19. doi: 10.1016/j.trim.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 31.Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, Yagita H, Okumura K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009;113:3821–3830. doi: 10.1182/blood-2008-10-185884. [DOI] [PubMed] [Google Scholar]
- 32.Sandner SE, Clarkson MR, Salama AD, Sanchez-Fueyo A, Yagita H, Turka LA, Sayegh MH. Mechanisms of tolerance induced by donor-specific transfusion and ICOS-B7h blockade in a model of CD4+ T-cell-mediated allograft rejection. Am J Transplant. 2005;5:31–39. doi: 10.1111/j.1600-6143.2004.00640.x. [DOI] [PubMed] [Google Scholar]
- 33.Sandner SE, Salama AD, Houser SL, Palmer E, Turka LA, Sayegh MH. New TCR transgenic model for tracking allospecific CD4 T-cell activation and tolerance in vivo. Am J Transplant. 2003;3:1242–1250. doi: 10.1046/j.1600-6143.2003.00220.x. [DOI] [PubMed] [Google Scholar]
- 34.Naka T, Nishimoto N, Kishimoto T. The paradigm of IL-6: from basic science to medicine. Arthritis Res. 2002;4(Suppl 3):S233–S242. doi: 10.1186/ar565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harada H, Salama AD, Sho M, Izawa A, Sandner SE, Ito T, Akiba H, Yagita H, Sharpe AH, Freeman GJ, Sayegh MH. The role of the ICOS-B7h T cell costimulatory pathway in transplantation immunity. J Clin Invest. 2003;112:234–243. doi: 10.1172/JCI17008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ito T, Ueno T, Clarkson MR, Yuan X, Jurewicz MM, Yagita H, Azuma M, Sharpe AH, Auchincloss H, Jr, Sayegh MH, Najafian N. Analysis of the role of negative T cell costimulatory pathways in CD4 and CD8 T cell-mediated alloimmune responses in vivo. J Immunol. 2005;174:6648–6656. doi: 10.4049/jimmunol.174.11.6648. [DOI] [PubMed] [Google Scholar]
- 37.Habicht A, Kewalaramani R, Vu MD, Demirci G, Blazar BR, Sayegh MH, Li XC. Striking dichotomy of PD-L1 and PD-L2 pathways in regulating alloreactive CD4(+) and CD8(+) T cells in vivo. Am J Transplant. 2007;7:2683–2692. doi: 10.1111/j.1600-6143.2007.01999.x. [DOI] [PubMed] [Google Scholar]
- 38.Vu MD, Amanullah F, Li Y, Demirci G, Sayegh MH, Li XC. Different costimulatory and growth factor requirements for CD4+ and CD8+ T cell-mediated rejection. J Immunol. 2004;173:214–221. doi: 10.4049/jimmunol.173.1.214. [DOI] [PubMed] [Google Scholar]
- 39.D’Addio F, Yuan X, Habicht A, Williams J, Ruzek M, Iacomini J, Turka LA, Sayegh MH, Najafian N, Ansari MJ. A novel clinically relevant approach to tip the balance toward regulation in stringent transplantation model. Transplantation. 2010;90:260–269. doi: 10.1097/tp.0b013e3181e64217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakae S, Iwakura Y, Suto H, Galli SJ. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J Leukoc Biol. 2007;81:1258–1268. doi: 10.1189/jlb.1006610. [DOI] [PubMed] [Google Scholar]
- 41.Frisancho-Kiss S, Davis SE, Nyland JF, Frisancho JA, Cihakova D, Barrett MA, Rose NR, Fairweather D. Cutting edge: cross-regulation by TLR4 and T cell Ig mucin-3 determines sex differences in inflammatory heart disease. J Immunol. 2007;178:6710–6714. doi: 10.4049/jimmunol.178.11.6710. [DOI] [PubMed] [Google Scholar]
- 42.Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, Chandwaskar R, Karman J, Su EW, Hirashima M, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science. 2007;318:1141–1143. doi: 10.1126/science.1148536. [DOI] [PubMed] [Google Scholar]
- 43.Nishimoto N, Kishimoto T. Interleukin 6: from bench to bedside. Nat Clin Pract Rheumatol. 2006;2:619–626. doi: 10.1038/ncprheum0338. [DOI] [PubMed] [Google Scholar]
- 44.Seki M, Oomizu S, Sakata KM, Sakata A, Arikawa T, Watanabe K, Ito K, Takeshita K, Niki T, Saita N, et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol. 2008;127:78–88. doi: 10.1016/j.clim.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 45.Chen LC, Delgado JC, Jensen PE, Chen X. Direct expansion of human allospecific FoxP3+CD4+ regulatory T cells with allogeneic B cells for therapeutic application. J Immunol. 2009;183:4094–4102. doi: 10.4049/jimmunol.0901081. [DOI] [PubMed] [Google Scholar]
- 46.Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–6729. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- 47.Shen H, Goldstein DR. IL-6 and TNF-alpha synergistically inhibit allograft acceptance. J Am Soc Nephrol. 2009;20:1032–1040. doi: 10.1681/ASN.2008070778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niwa H, Satoh T, Matsushima Y, Hosoya K, Saeki K, Niki T, Hirashima M, Yokozeki H. Stable form of galectin-9, a Tim-3 ligand, inhibits contact hypersensitivity and psoriatic reactions: a potent therapeutic tool for Th1- and/or Th17-mediated skin inflammation. Clin Immunol. 2009;132:184–194. doi: 10.1016/j.clim.2009.04.012. [DOI] [PubMed] [Google Scholar]