

Abstract
Proteins targeted for degradation by the mycobacterial proteasome are covalently modified with prokaryotic ubiquitin-like protein (Pup) in a process termed “pupylation.” Despite its name, Pup is only ubiquitin-like in function and not sequence or structure. Furthermore, the enzymology of pupylation appears to be distinct from protein modification by ubiquitin (Ub) and other ubiquitin-like proteins (Ubls). Nonetheless, we have adapted methods established in the Ub field for the production of reagents to isolate, identify, and analyze pupylated proteins in mycobacteria. These methods can be modified to study specific pupylated proteins in various Pup-bearing bacteria or to identify posttranslational modifiers in other prokaryotes.
Keywords: Prokaryotes, Bacteria, Posttranslational modifiers, Tandem affinity purification (TAP), Proteomics
1. Introduction
Proteasomes are found in all eukaryotes and archaea, and in some bacterial species (reviewed in ref. 1). Despite the similarity of these protein degradation machines among the domains of life, it was not understood for some time how proteins were targeted for degradation in prokaryotes due to the absence of Ub in these organisms. Recently, the first prokaryotic protein-to-protein post-translational modifier (PTM), Pup, was identified, and shown to target proteins to the proteasome of Mycobacterium tuberculosis (Mtb), a deadly pathogen (2). A parallel study also found Pup in Mycobacterium smegmatis (Msm), a nonpathogenic relative of Mtb (3). Modifiers similar to eukaryotic Ubls were eventually found in archaea (4), suggesting small protein PTMs are universally important for proteasome-dependent degradation.
Like ubiquitin, Pup is a small polypeptide (64 amino acids) that forms a covalent bond with a substrate to doom it for degradation (2, 3). The enzymology of pupylation, however, is completely different from ubiquitylation. To begin with, although Ub uses a C-terminal glycine to attach to substrates, Pup uses a C-terminal glutamate. Furthermore, in contrast to the Ub system, which typically requires several enzymatic steps for substrate conjugation, pupylation requires only one or two enzymes. In mycobacteria, Pup is synthesized with a C-terminal glutamine that must be converted to glutamate by Dop (deamidase of Pup) in order to prepare it for substrate ligation (5). Notably, not all Pup-bearing bacteria require this deamidation step; numerous Actinomycetes encode Pup that naturally terminates in glutamate. Next, PafA (proteasome accessory factor A) ligates the C-terminal glutamate of Pup to a lysine in a target protein, resulting in an isopeptide bond between the γ-carboxylate of Pup’s glutamate and the ε-amino group of the substrate’s lysine (2, 3, 5–7). Pup can then bind to the Mycobacterium proteasomal ATPase, Mpa, in order to deliver the substrate into the proteasome core for destruction (8–12).
In the eukaryotic Ub-proteasome system, Ub can be removed from substrates by deubiquitinases (DUBs) (reviewed in ref. 13). In mycobacteria, it appears that Dop can remove Pup from substrates, thus acting like a “depupylase” or “DPUP” (14, 15). Interestingly, depupylation is facilitated by Mpa in vitro (15) and in vivo (14), suggesting depupylation is coupled to the degradation of some substrates. Taken together, the bacterial Pup-proteasome system is functionally, if not enzymatically, similar to the eukaryotic Ub-proteasome pathway.
Since the discovery of the Pup-proteasome system, a priority was to identify targets of pupylation (the “pupylome”). The proteasome is essential for Mtb to kill mice (reviewed in ref. 16). Therefore, identifying the pupylome and potential proteasome substrates could shed light on the role of this system in Mtb virulence as well as on bacterial physiology. Hundreds of proteins were identified as potential pupylation targets in Mtb, and the Pup attachment site was identified for 55 of these proteins (17). Unlike the presence of poly-Ub chains in the eukaryotic system, no poly-Pup chains were identified in Mtb, and only three proteins had more than one lysine that could be targeted for pupylation. Several proteins appeared to be degradation substrates under routine culture conditions; however, many of the proteins identified in the Mtb pupylome did not appear to be degraded under these conditions. This suggested that pupylation has functions in addition to targeting proteins to the proteasome, although nonproteasomal roles for Pup have yet to be identified. In addition to the Mtb study, two reports analyzed the pupylomes of Msm (18, 19). In all cases, Pup was attached to substrate lysines via its C-terminal glutamate. To date, no motif that predicts whether or not a protein can be pupylated has been identified.
Our work has provided important methodologies and insights for additional studies on the posttranslational regulation of many proteins in Mtb. Here, we discuss the techniques used to isolate the pupylome from Mtb or Msm and to purify specific pupylated proteins of interest. These methods should be easily applicable to other systems with similarly available molecular biology reagents.
2. Materials
2.1. Media
Luria-Bertani (LB-Miller; Difco) broth or agar with appropriate antibiotics for propagation of Escherichia coli.
Middlebrook 7H9 broth (Difco) supplemented with 0.2% glycerol and 0.05% Tween-80 for Msm; further supplemented with 0.5% bovine serum albumin (BSA), 0.2% dextrose, and 0.085% NaCl (“ADN”) for Mtb.
7H11 agar for propagation of Msm; supplement agar with Middlebrook Enrichment (oleic acid, albumin, dextrose, catalase) (Difco) for Mtb.
2.2. Plasmids
2.3. Bacterial Strains
M. smegmatis mc2155 (Msm) (23).
M. tuberculosis H37Rv 25618 (Mtb) (American Type Culture Collection). All work using Mtb must be carried out in a Biosafety Level 3 laboratory. Msm, which is not infectious and not considered a pathogen, can be handled in a standard laboratory.
E. coli DH5α (Gibco, BRL) for routine cloning and transformations.
2.4. Protein Analysis
0.2 μm Nitrocellulose (Whatman PROTRAN BA83).
Nickel-NTA agarose (Ni-NTA, Qiagen).
Strep-Tactin Superflow (Qiagen).
Anti-Penta-His (Qiagen).
c-Myc-tagged protein MILD PURIFICATION GEL (MBL Intl).
Lysis, wash, and elution buffers for Ni-NTA are described in The QiaExpressionist manual (Qiagen).
PBS, Tween-80 (PBST): PBS (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.4), 0.05% v/v Tween-80.
Denaturing lysis buffer: 100 mM NaH2PO4, 10 mM Tris–HCl, 8 M urea, pH 8.
Denaturing wash buffer: As denaturing lysis buffer B, but pH 6.3.
Native wash buffer: 50 mM NaH2PO4, 30 mM NaCl, 20 mM imidazole, pH 8.
Native elution buffer: 50 mM NaH2PO4, 30 mM NaCl, 250 mM imidazole, pH 8.
Strep wash buffer: 50 mM NaH2PO4, 300 mM NaCl, pH 8.0.
Strep elution buffer: 50 mM NaH2PO4, 300 mM NaCl, 2.5 mM desthiobiotin, pH 8.
TBST: 25 mM Tris–HCl, pH 7.4, 125 mM NaCl, 0.05% v/v Tween-20.
Native lysis buffer: 50 mM Tris–HCl, pH 8, 300 mM NaCl, 10 mM imidazole.
Native wash buffer B: 50 mM Tris–HCl, pH 8, 300 mM NaCl, 25 mM imidazole.
Native elution buffer B: 50 mM Tris–HCl, pH 8, 300 mM NaCl, 250 mM imidazole.
Bead beating tubes, zirconia silica beads, Mini Bead Beater (BioSpec Products).
Vesphene, Staphene (Steris).
Biosafe Coomassie Blue (Bio-Rad).
Blocking buffer: 1–3% nonfat dry milk in TBST.
Enhanced chemiluminescence reagent (ThermoScientific Super Signal West Pico or Femto).
Econo-Pac 10DG desalting columns (Bio-Rad).
Microcon centrifugal filter devices (Millipore).
3. Methods
3.1. Culture Conditions and Transformations
Grow Mtb cultures without shaking in 25, 75, or 150-cm2 vented flasks (Corning) in humidified incubators with or without 5% CO2.
For Mtb and Msm, hygromycin is used as needed at 50 μg/mL; for E. coli, hygromycin is used at 150 μg/mL.
Transform E. coli (24) and mycobacteria (25) using standard techniques. Mycobacteria transformants arise after 2–3 weeks (Mtb) or 3–4 days (Msm).
For every transformation, three transformant colonies are inoculated into three wells of a 96-well flat bottom plates, each containing 200 μL 7H9 broth with antibiotics.
After 1 week (Mtb) or 3 days (Msm) at 37°C, add these starter cultures to 5 mL 7H9 in vented flasks with selection for further growth.
After 1 (Msm) to 7 (Mtb) days, freeze cultures in 7H9 with glycerol (final 25%) in 1 mL aliquots, which can be used as starter cultures for future studies.
3.2. Purification of the Pupylome from Mtb
Grow 0.5 L Mtb or Msm (see Note 1) containing pMN-His6-Strep-Pup to an optical density (OD580) of ~1.0. Mycobacteria become clumpy at higher densities making quantification of cell numbers less accurate, thus care must be taken not to overgrow the cultures.
Harvest bacteria in a centrifuge at 3,310 × g for 5–8 min. Wash in an equal volume of PBST.
Resuspend pellets in 14 mL of denaturing lysis buffer (see Note 2) and transfer 1 mL aliquots of resuspended cells into bead beating tubes, each with 250 μL of zirconia silica beads (see Note 3).
Bead beat samples in a BioSpec Mini Bead Beater, three times, 1 min each time and chill tubes on ice for 1 min between beatings (see Note 4).
Microfuge samples for 1 min at top speed to pellet insoluble debris.
Combine and filter sterilize supernatants from like samples through a 0.2 μm nylon syringe filter. Wipe tubes with Vesphene or spray tubes with Staphene to disinfect the outside of the tubes. Samples can now be removed from the BSL3 suite.
Incubate clarified lysate with 1.5 mL of Ni-NTA agarose for 2 h at 4°C with agitation.
Load agarose into a polypropylene column and wash with denaturing wash buffer.
Wash with one column volume plus 10 mL of native wash buffer.
Elute with 3.5 mL native elution buffer.
Incubate eluate with 1 mL of Strep-Tactin Superflow resin for 2 h at 4°C with agitation.
Wash column three times with 5 mL of Strep wash buffer. Elute protein in six 0.5 mL fractions with Strep elution buffer.
Separate proteins on 12% SDS-PAGE gels and visualize with Biosafe Coomassie Blue (Fig. 1a) (see Note 5). For the identification of proteins, in-gel proteolysis and LC-MS/MS analysis is used as described elsewhere (17).
Fig. 1.
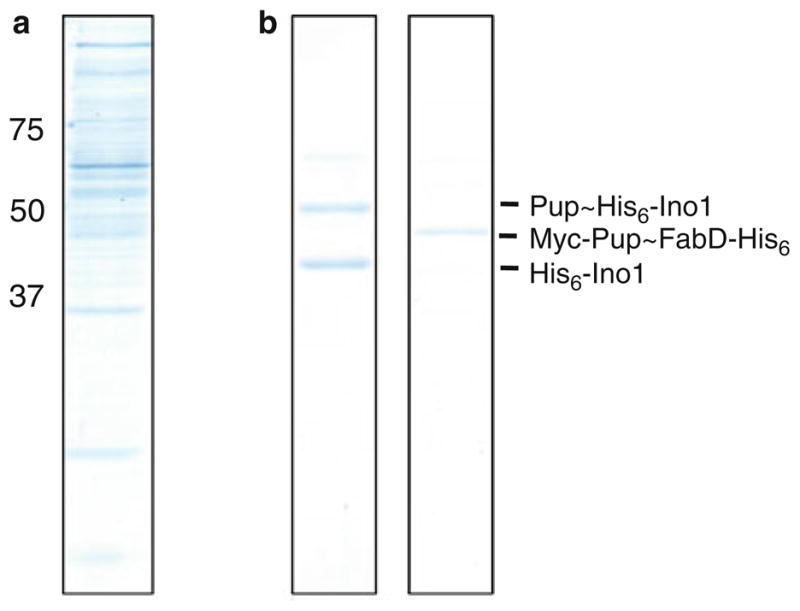
Purification of pupylated proteins. (a) Total Msm pupylome. (b) Pupylated Ino1 (left) or FabD (right ). his6-strep-pup (a) or myc-pup with fabD-his6 (b) was expressed and purified from Msm, as described in Subheading 3.
3.3. Validation of Pupylation
To test if proteins are pupylated in mycobacteria, the gene encoding the protein of interest is expressed with a his6 tag from pSYMP in Msm or Mtb (17) (see Note 6). Culture and transform bacteria as described in Subheading 3.1.
Inoculate 25 mL 7H9/hygromycin in a 75-cm2 vented flask with 1 mL frozen stock of Mtb harboring the pSYMP construct. Incubate at 37°C with or without shaking to the desired growth phase (OD580 ~ 0.5–2 of a 1 mL volume). For Mtb, typically 15–20 optical density units are harvested.
Pellet and wash bacteria with PBST as described above.
Resuspend in 3 mL of denaturing lysis buffer B. Lyse by bead beating as described above.
Process bacteria as described in the QiaExpressionist manual for purification under denaturing conditions. Incubate the clarified lysate with 30 μL Ni-NTA agarose per 1.5 mL lysate for 2 h at 4°C on a rotator.
Pellet the agarose for 1–2 min at 4°C in a microfuge. Remove supernatant and gently resuspend the agarose in 250 μL native wash buffer. Repeat three more times. Elute in 60 μL native elution buffer.
Add protein sample buffer to eluates and analyze by immunoblotting with antibodies to His6. If samples are from Mtb, eluates should be boiled for 5–10 min in sample buffer prior to removal from the BSL3.
Run appropriate gel percentage based on the estimated size of protein (we typically use 10–12% gels) (see Note 7).
We use a semidry method for transferring proteins to nitrocellulose membranes.
Equilibrate gels in transfer buffer (26) for at least 30 min.
Follow the transfer protocol as described by the manufacturer. We typically use 15 V for 15 min for a 12% gel.
After transfer, dry the membrane (see Note 8). Confirm protein transfer by Ponceau S staining of the membrane. Destain in water or TBST. Incubate the membrane in blocking buffer (or 3% BSA in TBS for anti-His5 immunoblotting) for 30 min at room temperature or overnight at 4°C.
Discard blocking solution and incubate membrane in primary antibody diluted in 1–3% nonfat dry milk or BSA (dilution depends on the antibody) at room temperature with shaking overnight at 4°C.
Decant antibody and rinse membrane four times 10 min each in TBST or TBS. Incubate in secondary antibody, 30–60 min.
Decant and rinse four times in TBST or TBS, 10 min each. Detect proteins with enhanced chemiluminescence reagent.
3.4. Purification of a Specific Pupylated Substrate from Mycobacteria
Co-express myc-pup and fabD-his6 in Msm in pOLYG or another mycobacterial plasmid with a strong promoter (see Note 9).
Grow 0.5 L Msm harboring this plasmid in 7H9 broth to an OD580 of ~1 and harvest (see Note 10).
Resuspend cells in 15 mL native lysis buffer and lyse by sonication three times, 30 s each time (1 s pulse on/off). Ice 10 min between sonication pulses.
Apply clarified lysate to 1.5 mL Ni-NTA agarose (preequilibrated in native lysis buffer) by gravity flow and wash with 20 mL native lysis buffer followed by at least 30 mL native wash buffer B.
Elute with 3 mL native elution buffer B.
Buffer exchange sample into 4 mL PBS with Econo-Pac 10DG desalting columns and apply to preequilibrated (with PBS) c-Myc-tagged protein MILD PURIFICATION GEL (0.5 mL) by gravity flow.
Re-apply flow through to MILD PURIFICATION GEL by gravity flow.
Wash the column with PBS (at least ten column volumes) and elute with two 0.5 mL aliquots of 0.17 M glycine, pH 2.3. Quickly neutralize eluate with 20 μL 1 M Tris–HCl, pH 8.
Buffer exchange during concentration using Microcon centrifugal filter devices.
Pupylated substrate is ready for use in enzymatic assays (Fig. 1b).
Acknowledgments
We thank Andrew Darwin and Tony Huang for critical review of this chapter. This work was supported by the NIH (1R01HL092774) and the Irma T. Hirschl Charitable Trust. K. Heran Darwin, Ph.D. holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.
Footnotes
Msm can be lysed by sonication (in place of bead beating/filtering). Similar culture volumes can be used.
Under native conditions, the pupylome co-purifies with Dop. In the presence of ATP (e.g., in cell lysates), Dop depupylates much of the pupylome, reducing the amount and number of pupylated proteins. Thus, purification under denaturing conditions minimizes the loss of pupylated substrates.
Due to the nature of the mycobacterial cell wall, glass beads should not be used as they are insufficient for cell lysis.
The use of a vortex mixer will not ensure consistent and reproducible cell lysis and should not be used as an alternative to a bead beater.
We recommend the use of reagents and containers that have never been used for other protein work to minimize contamination of samples to be analyzed by mass spectrometry.
pSYMP allows for the heterologous expression and translation of putative substrates and so far has not resulted in toxicity to bacteria.
Mycobacterial Pup has a predicted molecular size of 6.9 kDa, however, Pup migrates to approximately 14 kDa using standard SDS-PAGE protocols as well as size exclusion analysis (2, 11). The addition of Pup to target proteins may not always result in a +6.9 kDa or a +14 kDa shift in molecular weight based on SDS-PAGE. As examples: Ino1 (inositol 1-phosphate synthetase) is ~40 kDa, Pup ~ Ino1 migrates to ~50 kDa; FabD (malonyl coA-acyl carrier protein transacylase) is ~31 kDa, Pup~FabD migrates to ~48 kDa.
Drying the membrane prior to Ponceau S staining or blocking helps the detection of certain proteins, in particular, Pup. A hair dryer or fan can be used to facilitate drying.
Certain pupylated proteins, such as Pup ~ Ino1, co-purify with their unpupylated counterparts, despite the use of different tags on Pup and substrate. Ino1 forms tetramers (27) thus we predict that not every monomer of this oligomeric structure is pupylated. This may be true of other multimeric complexes. We routinely use FabD because it is a monomeric protein.
We have found that certain proteins are more pupylated during certain growth phases. It may be necessary to optimize the harvest time in order to increase the yield of pupylated protein.
References
- 1.Cerda-Mairaz F, Darwin KH. The Mycobacterium tuberculosis proteasome: more than just a barrel-shaped protease. Microbes Infect. 2009;11:1150–1155. doi: 10.1016/j.micinf.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearce MJ, Mintseris J, Ferreyra J, et al. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science. 2008;322:1104–1107. doi: 10.1126/science.1163885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burns KE, Liu WT, Boshoff HI, et al. Proteasomal protein degradation in Mycobacteria is dependent upon a prokaryotic ubiquitin-like protein. J Biol Chem. 2009;284:3069–3075. doi: 10.1074/jbc.M808032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Humbard MA, Miranda HV, Lim JM, et al. Ubiquitin-like small archaeal modifier proteins (SAMPs) in Haloferax volcanii. Nature. 2010;463:54–60. doi: 10.1038/nature08659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Striebel F, Imkamp F, Sutter M, et al. Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat Struct Mol Biol. 2009;16:647–651. doi: 10.1038/nsmb.1597. [DOI] [PubMed] [Google Scholar]
- 6.Sutter M, Damberger FF, Imkamp F, et al. Prokaryotic ubiquitin-like protein (Pup) is coupled to substrates via the side chain of its C-terminal glutamate. J Am Chem Soc. 2010;132:5610–5612. doi: 10.1021/ja910546x. [DOI] [PubMed] [Google Scholar]
- 7.Guth E, Thommen M, Weber-Ban E. Mycobacterial ubiquitin-like protein ligase PafA follows a two-step reaction pathway with a phosphorylated Pup intermediate. J Biol Chem. 2011;286:4412–4419. doi: 10.1074/jbc.M110.189282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang T, Darwin KH, Li H. Binding-induced folding of prokaryotic ubiquitin-like protein on the Mycobacterium proteasomal ATPase targets substrates for degradation. Nat Struct Mol Biol. 2010;17:1352–1357. doi: 10.1038/nsmb.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sutter M, Striebel F, Damberger FF, et al. A distinct structural region of the prokaryotic ubiquitin-like protein (Pup) is recognized by the N-terminal domain of the proteasomal ATPase Mpa. FEBS Lett. 2009;583:3151–3157. doi: 10.1016/j.febslet.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 10.Liao S, Shang Q, Zhang X, et al. Pup, a prokaryotic ubiquitin-like protein, is an intrinsically disordered protein. Biochem J. 2009;422:207–215. doi: 10.1042/BJ20090738. [DOI] [PubMed] [Google Scholar]
- 11.Chen X, Solomon WC, Kang Y, et al. Prokaryotic ubiquitin-like protein pup is intrinsically disordered. J Mol Biol. 2009;392:208–217. doi: 10.1016/j.jmb.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Striebel F, Hunkeler M, Summer H, Weber-Ban E. The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup’s N-terminus. EMBO J. 2010;29:1262–1271. doi: 10.1038/emboj.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burns KE, Cerda-Maira FA, Wang T, et al. “Depupylation” of prokaryotic ubiquitin-like protein from mycobacterial proteasome substrates. Mol Cell. 2010;39:821–827. doi: 10.1016/j.molcel.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imkamp F, Striebel F, Sutter M, et al. Dop functions as a depupylase in the prokaryotic ubiquitin-like modification pathway. EMBO Rep. 2010;11:791–797. doi: 10.1038/embor.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darwin KH. Prokaryotic Ubiquitin-Like Protein, Proteasomes, and Pathogenesis. Nat Rev Microbiol. 2009;7:485–491. doi: 10.1038/nrmicro2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Festa RA, McAllister F, Pearce MJ, et al. Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis. PLoS One. 2010;5:e8589. doi: 10.1371/journal.pone.0008589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watrous J, Burns K, Liu WT, et al. Expansion of the mycobacterial “PUPylome”. Mol Biosyst. 2010;6:376–385. doi: 10.1039/b916104j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poulsen C, Akhter Y, Jeon AH, et al. Proteome-wide identification of mycobacterial pupylation targets. Mol Syst Biol. 2010;6:386. doi: 10.1038/msb.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehrt S, Guo XV, Hickey CM, et al. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 2005;33:e21. doi: 10.1093/nar/gni013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scholz O, Thiel A, Hillen W, Niederweis M. Quantitative analysis of gene expression with an improved green fluorescent protein. p6. Eur J Biochem. 2000;267:1565–1570. doi: 10.1046/j.1432-1327.2000.01170.x. [DOI] [PubMed] [Google Scholar]
- 22.Garbe TR, Barathi J, Barnini S, et al. Transformation of mycobacterial species using hygromycin resistance as selectable marker. Microbiology. 1994;140:133–138. doi: 10.1099/13500872-140-1-133. [DOI] [PubMed] [Google Scholar]
- 23.Snapper SB, Melton RE, Mustafa S, et al. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol. 1990;4:1911–1919. doi: 10.1111/j.1365-2958.1990.tb02040.x. [DOI] [PubMed] [Google Scholar]
- 24.Sambrook J, Maniatis T, Fritsch E. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1989. [Google Scholar]
- 25.Hatfull GF, Jacobs WR., Jr . Molecular Genetics of Mycobacteria. ASM Press; Washington, DC: 2000. [Google Scholar]
- 26.Ausubel F, Brent R, Kingston R, et al. Short Protocols in Molecular Biology. Vol. 1. Wiley; 2002. [Google Scholar]
- 27.Norman RA, McAlister MS, Murray-Rust J, et al. Crystal structure of inositol 1-phosphate synthase from Mycobacterium tuberculosis, a key enzyme in phosphatidylinositol synthesis. Structure. 2002;10:393–402. doi: 10.1016/s0969-2126(02)00718-9. [DOI] [PubMed] [Google Scholar]