

Abstract
Vascular endothelial growth factor receptor-3 is a receptor tyrosine kinase that is overexpressed in some human carcinomas, but its role in tumorigenesis has not been fully elucidated. We examined VEGFR-3 expression in normal, nonneoplastic and early stage malignant breast tissues and have shown that VEGFR-3 upregulation in breast cancer preceded tumor cell invasion, suggesting that VEGFR-3 may function as a survival signal. We characterized the biological effects of VEGFR-3 over-expression in human breast cancer cells based on two approaches: gain of function by overexpressing VEGFR-3 in MCF-7 breast cancer cells and loss of function by RNAi-mediated silencing of VEGFR-3 in MCF-7-VEGFR-3 and BT474 cells.
VEGFR-3 overexpression increased cellular proliferation by 40% when MCF7-VEGFR-3 cells were compared to parental MCF7 cells, and proliferation was reduced by more than 40% when endogenous VEGFR-3 was downregulated in BT474 cells. VEGFR-3 overexpression promoted a three-fold increase in motility and invasion and both motility and invasion were inhibited by downregulation of VEGFR-3. Furthermore, VEGFR-3 overexpression promoted cellular survival under stress conditions induced by staurosporine treatment and led to anchorage-independent growth.
VEGFR-3 overexpression dramatically increased tumor formation in both hormone-dependent and independent xenograft models. With estrogen stimulation, MCF7-VEGFR-3 xenografts were ten times larger than control xenografts. Finally, downregulation of VEGFR-3 expression in both xenograft model cell lines led to a significant reduction of tumor growth. For the first time, we have demonstrated that VEGFR-3 overexpression promotes breast cancer cell proliferation, motility, survival, anchorage-independent growth and tumorogenicity in the absence of ligand expression.
Keywords: vascular endothelial growth factor receptor-3, VEGFR-3, Flt-4, receptor tyrosine kinases, breast cancer, tumorigenicity, carcinogenesis
Introduction
The vascular endothelial growth factor receptors (VEGFRs) are a subfamily of receptor tyrosine kinases that play key roles in angiogenesis and lymphangiogenesis. VEGFR-1 (Flt-1) and VEGFR-2 (Flk-1/KDR) are primarily located on vascular endothelial cells and when activated by their cognate ligands play a major role in tumor angiogenesis.1 VEGFR-3 (Flt-4) is primarily located on lymphatic endothelial cells and, along with its ligands VEGF-C and VEGF-D, represents the most extensively studied lymph- angiogenic signaling system in cancer.2–4
Activation of the VEGF-C/VEGF-D/VEGFR-3 axis through ligand overexpression induces intratumoral lymphangiogenesis, peritumoral lymphatic hyperplasia, and/or increased lymph node metastasis in animal models for carcinoma of the breast,5,6 stomach,7 rectum,8 lung9,10 and skin.11 These model systems demonstrate the extensive paracrine effects of tumor ligand secretion on lymphatic endothelia, including increased lymphatic endothelial cell size, proliferation and vessel permeability leading to lymphatic metastasis of primary tumor xenografts.12,13 In humans, upregulation of the VEGF-C/VEGF-D/VEGFR-3 axis through ligand overexpression promotes tumor invasion and increases lymph node metastases in gastric14 and colorectal adenocarcinoma15 and is an independent indicator of poor prognosis in endometrial,16 ovarian17 and esophageal squamous cell carcinoma.18 Despite this abundance of correlative animal xenograft and clinical data regarding ligand overexpression, very little is known about the actual biological effects of VEGFR-3 overexpression on primary tumors.
VEGFR-3 is overexpressed in the primary tumor cells of a subset of patients with gastric adenocarcinoma,19 colorectal adenocarcinoma,15,20 endometrial carcinoma16 and ovarian carcinoma.17 The biological effects of VEGFR-3 overexpression on cancer progression vary with tumor type. Shida et al. reported that VEGFR-3 overexpression did not affect tumor invasion or lymphatic metastasis in gastric adenocarcinoma.19 Yokoyama et al. have shown VEGFR-3 overexpression to be a significant promoter of lymphatic metastasis in ovarian and endometrial carcinoma.16,17 However, these results are complicated by concomitant tumor overexpression of the ligand VEGF-D. The impact of VEGFR-3 overexpression in colorectal adenocarcinoma remains controversial.21,22 While some authors have shown that expression of VEGFR-3 in solid tumors is restricted to blood and lymphatic vessels4 the evidence for expression of VEGFR-3 in tumor cells has continued to accumulate.23,24 We have previously demonstrated VEGFR-3 overexpression in the primary tumor cells of patients with invasive ductal breast adenocarcinoma.25 To further characterize the role of VEGFR-3 in breast tumorigenesis, we first determined the levels of VEGFR-3 in normal and benign human breast tissues as well as atypical ductal hyperplasia and pre-invasive ductal carcinoma-in-situ (DCIS). We then focused on defining the biological effects of VEGFR-3 overexpression in human breast cancer cells in vitro and in vivo. To our knowledge, this is the first report to address the significance of VEGFR-3 overexpression in the absence of ligand overexpression. We have shown that VEGFR-3 upregulation in breast cancer precedes tumor cell invasion or metastasis, suggesting that VEGFR-3 may function as a survival signal and be an early event in breast tumorigenesis. VEGFR-3 overexpression significantly increases the proliferation, motility, viability and tumorigenicity of human breast cancer cells.
Results
Analyses of VEGFR-3 expression in human breast cancer specimens
We used immunohistochemical techniques to assess VEGFR-3 expression in 76 specimens of normal/fibrocystic breast tissue (FCD), atypical ductal hyperplasia (ADH) and ductal carcinoma in situ (DCIS). Formalin-fixed, paraffin-embedded (FFPE) tissue sections were stained with anti-VEGFR-3 monoclonal antibody MAB3757 directed at the extracellular domain of the VEGFR-3 molecule.26 The specificity and immunoreactivity of this antibody in paraffin-embedded samples have been extensively characterized.4,27,28 The intensity of the VEGFR-3 expression in each group ranged from 1 to 3 and there was variability in the percentage of tumor cells demonstrating immunoreactivity against VEGFR-3 ranging from 0 to 90% (Fig. 1). VEGFR-3 expression was scored as either high (2 or 3 intensity and ≥50% positive cells) or low (Table 1). VEGFR-3 expression was high in 7 (31%) of the 22 DCIS tumors, and 7 (50%) of the 14 ADH samples. At the same time, only 1 (2.5%) of the 40 normal/FCD tissues demonstrated high VEGFR-3 expression (p < 0.0001, Table 1). This confirmed that VEGFR-3 was expressed in human breast tumor cells and suggested that VEGFR-3 overexpression preceded tumor cell invasion or metastasis, suggesting that VEGFR-3 may function as a survival signal and be an early event in breast tumorigenesis.
Figure 1.
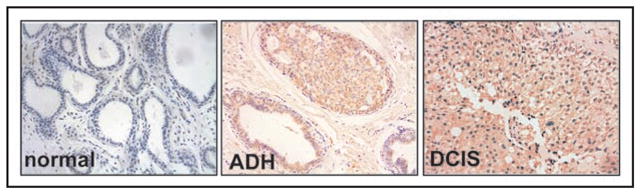
Analyses of VEGFR-3 expression in human breast cancer specimens. Human breast samples staining with VEGFR-3 9D9F9 antibody. Left panel normal tissue, center hyperplasia ADH measured as 2+, right—DCIS staining measured as 2+. Diaminobenzidine (DAB) was used as the chromogen, and the slides were counter-stained with hematoxylin.
Table 1.
Immunohistochemical analysis of VEGFR-3 expression
| Specimen | N Total | N High* | % samples |
|---|---|---|---|
| Normal + FCD | 40 | 1 | 2.5 |
| ADH | 14 | 7 | 50 |
| DCIS | 22 | 7 | 31 |
DCIS, ADH, FCD and normal tissue sections categorized by staining intensity (0, none; 1, borderline; 2, weak; 3, moderate; 4, strong) and percentage of cell staining positive for VEGFR-3.
Tissue sections dichotomized as High (2+ or 3+ intensity and ≥50% positive cells) or low. N Total = N Low + N High.
Establishment of model systems to examine biological effects of VEGFR-3 overexpression in breast cancer
To characterize the biological effects of VEGFR-3 overexpression in human breast cancer cells, we employed two approaches: gain of function by overexpressing VEGFR-3 and loss of function by RNAi-mediated silencing of VEGFR-3. We established model systems using BT474 cells that had high endogenous VEGFR-3 expression and MCF-7 cells that did not express VEGFR-3 by RT-PCR analysis. As our first model, we stably transfected MCF7 cells with a VEGFR-3 expression plasmid, creating the modified cell line called MCF7-VEGFR-3. As a control for this model, we stably transfected the same MCF7 cells with an empty expression vector, pcDNA3.1B called MCF7-pcDNA3. This allowed us to compare the biological effects of VEGFR-3 overexpression in these 2 breast cancer cell lines. In parallel, we attenuated VEGFR-3 expression in the MCF-7-VEGFR-3 and BT474 cells using both transiently-expressed and stably-expressed RNAi.
We demonstrated VEGFR-3 mRNA transcripts in BT474 and MCF7-VEGFR-3 cell lines by RT-PCR (Fig. 2A). Consistent with the transcript level, western blot analysis revealed that both BT474 and MCF7-VEGFR-3 abundantly expressed VEGFR-3, which was highly phosphorylated (Fig. 2B). In contrast, the expression of VEGFR-3 was detected in very low amounts in parental MCF7 and control MCF7-pcDNA3 cells (Fig. 2B). In our second model system, VEGFR-3 expression was downregulated in both cell lines in transient transfection with a receptor-specific pool of siRNAs. We showed both dose-dependent and time-dependent reduction of VEGFR-3 expression in transiently-transfected BT474 and MCF7-VEGFR-3 cells (Fig. 2C). We used lentiviral shRNA constructs (shRNA1 and shRNA2) to create stable clones in both of these cell lines. In the MCF7 model, both shRNA clones showed reduced levels of VEGFR-3 expression (clones shRNA1 and -2, Fig. 3A). However, in the BT474 cells, only shRNA1 was efficient in reducing VEGFR-3 expression. Thus, we used the shRNA2 BT474 clone as an internal control (Fig. 3B and C).
Figure 2.

Establishment of the Model Systems. MCF7 breast cancer cells were stably transfected with expression plasmid for VEGFR-3 or an empty vector pcDNA3 and selected for antibiotic resistance. (A) VEGFR-3 transcription. RT-PCR was performed with VEGFR-3-specific primers to demonstrate transcription of the VEGFR-3 gene in stably transfected MCF7 cells and BT474 cells. (B) VEGFR-3 Expression in Breast Cancer Cell Lines. western blot analysis. BT474 and MCF7-VEGFR-3 cells express highly phosphorylated VEGFR-3. (C) VEGFR-3 knockdown with siRNA. western blot analysis. A dose-dependent downregulation of VEGFR-3 in BT474 cells (left), 48 hours. Time-dependent downregulation of VEGFR-3 expression with 10 nM siRNA (smart pool) in BT474 (central) and MCF7-VEGFR-3 cells (right).
Figure 3.

VEGFR-3 knockdown with shRNA. (A) VEGFR-3 expression in shRNA stable clones of MCF7-VEGFR-3. (B) VEGFR-3 expression in shRNA stable clones of BT474. shRNA2 and shRNA2′ are first and second GFP selection of cells infected with VEGFR-3 shRNA2 lentivirus. (C) Immunoprecipitation assay of VEGFR-3 protein in BT474 VEGFR-3 shRNA knock-down stable clones. Santa Cruz antibody sc321 were used for immunoprecipitation and as a control normal rabbit IgG (CellSignaling) used as a non-specific antibody control.
Since VEGFR-3 overexpression is frequently associated with overproduction of its cognate ligands17 we analyzed production of the ligands VEGF-C and VEGF-D by ELISA of conditioned media from each cell line in this study. The amount of each ligand in the conditioned media was below the minimum detectable dose (13.3 pg/ml) for each cell line (data not shown). This data regarding the absence of ligand expression correlates well with data of Akahane et al.29 who demonstrated VEGF-D transcripts but the absence of expression or secretion of VEGF-D in MCF7 cells, and Mattila et al.30 who demonstrated lack of VEGF-C mRNA transcripts or protein expression in this cell line. We concluded that neither overexpression of VEGFR-3 in MCF7 cells nor down-regulation of VEGFR-3 in BT474 cells led to the activation of an autocrine loop through increased ligand expression. Thus, these model systems allowed us to examine the effects of VEGFR-3 overexpression in the absence of ligand secretion.
VEGFR-3 increases breast cancer cell viability and proliferation
Our first experiment evaluated the impact of VEGFR-3 overexpression on the baseline characteristics of MCF7 and BT474 cells. MCF7-VEGFR-3 cells demonstrated a 36% increase in cell viability compared to control MCF7-pcDNA3 cells after 24 hours of growth (Fig. 4A). Similarly, downregulation of endogenous expression of VEGFR-3 with VEGFR-3-specific siRNA in BT474 cells resulted in a 42% reduction in cell viability when compared with control siRNA-treated cells (Fig. 4B). Approximately the same decrease of viability was measured in cells where VEGFR-3 expression was stably downregulated with VEGFR-3 shRNA constructs (Fig. 4C and D).
Figure 4.

VEGFR-3 increases viability of breast cancer cells. (A) MTS assay (24 h) of MCF7-VEGFR-3 and MCF7-pcDNA3 cells. Viability was normalized relative to MCF7-pcDNA. (B) BT474 cells viability measured 24 h after pretreatment for 48 h with 10 nM VEGFR-3-specific and Control siRNAs. Viability normalized relative to untreated BT474 cells. (C) Viability of MCF7-VEGFR-3 cells in comparison to clones with VEGFR-3 shRNA knockdown, normalized relative to parental clone. (D) Viability of BT474 cells in comparison to clones with VEGFR-3 shRNA knockdown, normalized relative to parental clone.
Since MTT assays are broad indicators of cellular activity, we specifically confirmed the impact of VEGFR-3 overexpression on cellular proliferation using a BrdU incorporation assay. In our VEGFR-3 overexpression model, there was a 40% increase in DNA synthesis after 24 hours, comparing MCF7-VEGFR-3 cells to MCF7-pcDNA3 cells (Fig. 5A). After stably decreasing VEGFR-3 expression in these cells, proliferation decreased 35% and 41% in cells expressing VEGFR-3 shRNA1 and 2, respectively (Fig. 5B). In our model of receptor downregulation, transient treatment of BT474 with VEGFR-3 specific siRNA resulted in a 32% reduction in DNA synthesis when compared to control siRNA treated cells (Fig. 5C). A similar result was obtained in BT474 cells stably expressing shRNA for VEGFR-3, where proliferation of these cells was reduced by 30% in shRNA1, but not in our internal control shRNA2 (Fig. 5D). Taken together, these experiments demonstrate that VEGFR-3 is involved in the regulation of cell viability and its overexpression significantly increases proliferation in human breast cancer cells.
Figure 5.

VEGFR-3 increases proliferation of breast cancer cells. BrdU incorporation was measured after 24 hours in fluorogenic enzyme immunoassay. (A) MCF7-VEGFR-3 proliferation compared to MCF7-pcDNA3 proliferation. (B) Proliferation of MCF7-VEGFR3 shRNA clones. (C) BT474 cells treated with VEGFR-3-specific siRNA and Control siRNA (10 nM) for 48 h and analyzed after 24 h of BrdU incorporation (*p = 0.0019). (D) Proliferation of BT474 in comparison with BT474 shRNA clones. **p < 0.001, RFU—relative fluorescent units.
Overexpression of VEGFR-3 increases breast cancer cell motility and invasion
Next, we characterized the effect of VEGFR-3 overexpression on cellular motility and invasion. In our VEGFR-3 overexpression model, increased receptor expression in MCF7-VEGFR-3 cells resulted in a 3.5-fold increase in motility as compared to MCF7-pcDNA3 cells (Fig. 6A). Furthermore, transient downregulation of VEGFR-3 in MCF7-VEGFR-3 cells with siRNA, resulted in a three-fold reduction in motility when compared to untreated and control siRNA-treated MCF7-VEGFR-3 cells (Fig. 6B). Next, we used our model of endogenous VEGFR-3 downregulation in BT474 cells. Transient treatment of BT474 cells with VEGFR-3 specific siRNA resulted in a 2.5-fold decrease in motility when compared to untreated and control siRNA-treated BT474 cells (Fig. 6C). Similar results were seen in our stable clones of BT474 (shRNA1) (Fig. 6D) and MCF7-VEGFR-3 (shRNA1 and -2) with attenuated VEGFR-3 (Fig. 6E).
Figure 6.

VEGFR-3 increases motility of breast cancer cells. (A) Motility of MCF7-VEGFR-3 cells relative to MCF7-pcDNA3 cells (folds increase, p < 0.05). (B) MCF7-VEGFR-3 cells treated with 10 nM VEGFR-3-specific siRNA for 48 h and motility compared to parental and Control siRNA treated MCF7-VEGFR-3 (fold decrease, **p < 0.001). (C) BT474 cells treated with 10 nM VEGFR-3-specific siRNA for 48 h and motility compared to parental and Control siRNA treated BT474 (fold decrease, *p < 0.05). (D) Motility of BT474 shRNA stable clones. (E) Motility of MCF7-VEGFR-3 shRNA stable clones.
Overexpression of VEGFR-3 in MCF7 cells also increased the invasive potential of these cells, resulting in a four-fold increase in the percentage of MCF7-VEGFR-3 cells capable of invading through a matrigel when compared to pcDNA3-transfected control cells (Fig. 7A). At the same time, downregulation of VEGFR-3 lead to a 2- to 2.8-fold decrease of invasiveness of these cells (Fig. 7A). In a similar fashion, VEGFR-3 downregulation in BT474 cells was seen with shRNA1 but not our internal shRNA2 control caused decrease of invasion (Fig. 7B). These results demonstrated that VEGFR-3 overexpression plays a significant role in promoting breast cancer cell motility and invasion.
Figure 7.
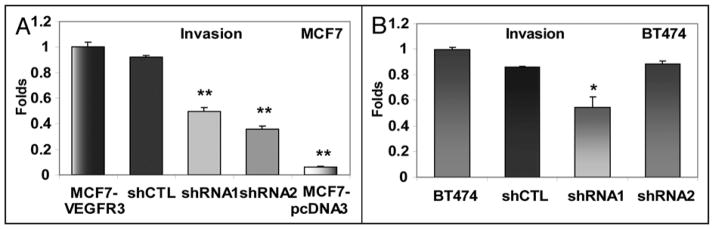
VEGFR-3 downregulation decreases invasion of breast cancer cells. (A) Invasion of MCF7-VEGFR-3 shRNA stable clones. (B) Invasion of BT474 shRNA stable clones (Fold decrease, *p < 0.05; **<0.001).
VEGFR-3 promotes survival in breast cancer cells under stress conditions
Overexpression of many receptor tyrosine kinases often leads to resistance to apoptosis and promotes cell survival in stress conditions.31 To evaluate the effect of VEGFR-3 over-expression on cellular survival under stress conditions, we treated MCF7-VEGFR-3 and MCF7-pcDNA3 cells with the broad-range kinase inhibitor, staurosporine (STS). MTT assays revealed that after staurosporine treatment approximately 23% more MCF7-VEGFR-3 cells were viable when compared to MCF7-pcDNA3 cells (Fig. 8A). Furthermore, TUNEL assays confirmed that approximately 50% of MCF7-pcDNA3 cells underwent apoptosis after 24 hours of treatment with staurosporine, compared to only 27% of the MCF7-VEGFR-3 cells (Fig. 8B). As expected, treatment with staurosporine dephosphorylated VEGFR-3 in MCF7-VEGFR-3 cells (Fig. 8C) and lead to the cleavage of PARP. Additionally, biochemical analysis revealed that the basal level of PARP expression in MCF7-VEGFR-3 cells was approximately three times greater than the basal level of PARP in MCF7-pcDNA3 (Fig. 8C). We estimated the ratio of cleaved to uncleaved PARP in staurosporine treated cell lines and found that MCF7-VEGFR-3 cells treated with STS have less cleaved PARP (1:0.59 ratio) than the MCF7-pcDNA3 cells (ratio of 1:1.2). This difference in PARP cleavage indicates that overexpression of VEGFR-3 promotes breast cancer cell survival under stress conditions. This resistance to apoptosis seen with VEGFR-3 overexpression was associated with increased phosphorylation (activation) of AKT and ERK 1/2 (Fig. 8C).
Figure 8.

VEGFR-3 promotes resistance to apoptosis. (A) Viability of MCF7-VEGFR-3 and MCF7-pcDNA3 cells treated with 100 nM staurosporine for 24 hours, measured by MTS assay, data normalized relative to untreated cells. (B) Apoptosis of MCF7-VEGFR-3 and MCF7-pcDNA3 cells treated with 100 nM staurosporine for 24 hours measured in TUNEL assay. (C) Western Blot analysis demonstrates resistance to apoptosis in MCF7 cells overexpressing VEGFR-3. Control MCF7-pcDNA3 cells (Lanes 1 and 2) compared to MCF7-VEGFR-3 cells (lanes 3 and 4) after 24 h treatment with 100 nM staurosporine. PARP to cleaved PARP ratio was calculated after densitometry. **p < 0.001.
Next we compared the response to staurosporine treatment in breast cancer cell lines with reduced level of VEGFR-3 expression. In both of our cell line models of VEGFR-3 downregulation with stable shRNA, viability was reduced and accompanied by increase of apoptosis (Fig. 9). Together, these results suggest that VEGFR-3 is involved in survival signaling in breast cancer cells.
Figure 9.

VEGFR-3 knockdown decreases resistance to apoptosis in breast cancer cells. (A) Viability and (B) TUNEL assays of MCF7-VEGFR-3 shRNA stable clones treated for 24 hours with increased doses of Staurosporine. (C) Viability and (D) TUNEL assays of BT474 breast cancer cells shRNA stable clones treated for 24 hours with increased doses of Staurosporine. *p < 0.05; **p < 0.001 Viability data normalized as treated/untreated.
VEGFR-3 promotes anchorage-independent growth
Thus far, our data have shown that VEGFR-3 overexpression in human breast cancer cells increased proliferation, motility and survival in vitro. Next, we sought to evaluate the effect of VEGFR-3 overexpression on breast cancer cell tumorigenicity. We used a colony-formation assay to test the effect of VEGFR-3 overexpression on anchorage-independent growth. After 14 days of incubation in soft agar, significantly more colonies were formed by MCF7-VEGFR-3 cells when compared to MCF7-pcDNA3 cells (356 ± 42 versus 122 ± 13; p < 0.001, Fig. 10A, Table 2). Not only did VEGFR-3 overexpression lead to increased colony formation, but also the size of the largest colonies formed by MCF7-VEGFR-3 was significantly larger than those formed by MCF7-pcDNA3 with a mean diameter 5.5 times greater (Fig. 10A). When the level of VEGFR-3 was reduced by stably expressed VEGFR-3 shRNA in MCF7-VEGFR-3 cells (Fig. 10B) and BT474 cells (Fig. 10C), their ability to form and grow colonies in suspension was dramatically reduced. In both shRNA1 and shRNA2 MCF7-VEGFR-3 cells, colony formation was reduced three-fold compared to controls, and the average size of the colonies was reduced at approximately the same level (Table 2). These in vitro data indicated that VEGFR-3 over-expression impacted tumorigenicity of MCF7 and BT474 cells in vitro.
Figure 10.

VEGFR-3 promotes anchorage-independent growth of breast cancer cells. Representative photos and statistical plots of soft agar assay. (A) Effect of VEGFR-3 overexpression on colony formation in soft agar. Phase micrographs are representative examples of largest colonies for each cell type. (B) Effect of VEGFR-3 knockdown with shRNA in MCF7-VEGFR-3 cell line on colony formation in soft agar. (C) Effect of VEGFR-3 knockdown with shRNA in BT474 cell line on colony formation in soft agar. *p < 0.05; **p < 0.001.
Table 2.
VEGFR-3 promotes anchorage-independent growth of breast cancer cells
| Cell line | Total colonies | Mean diameter | Cell line | Total colonies | Mean diameter |
|---|---|---|---|---|---|
| MCF7-VEGFR-3 | 356 ± 41.9 | 31 ± 4.1 | BT474 | 325 ± 46 | 22.3 ± 9 |
| shRNA1 | 133 ± 10.5 | 10.4 ± 5.4 | shRNA1 | 33 ± 25 | 7.5 ± 2.7 |
| shRNA2 | 83 ± 15 | 11.1 ± 3.5 | shRNA2 | 203 ± 41 | 17.6 ± 2.2 |
| shCTL | 316 ± 21 | 30 ± 5.8 | shCTL | 238 ± 35 | 21.1 ± 3.7 |
| MCF7-pcDNA3 | 122 ± 12.7 | 5.5 ± 0.97 |
Colony formation assay.
VEGFR-3 promotes growth of tumors in an animal model
Our final assessment of the biological effect of VEGFR-3 overexpression in human breast cancer cells focused on the tumorigenicity of MCF7 cells in a xenotransplant nude mouse model. First, MCF7-pcDNA3 and MCF7-VEGFR-3 cells were subcutaneously implanted into nude mice in the absence of hormonal stimulation. Twenty-one days after implantation only MCF7-VEGFR-3 implants formed tumors (Table 3). These tumors had a mean tumor volume of 2153 ± 395 mm3.
Table 3.
VEGFR-3 increases tumor formation and tumor growth in xenotransplant mouse model
| Cell line | Tumor incidence | Median tumor volume (mm3) | Estrogen pellet |
|---|---|---|---|
| MCF7-pcDNA3 | 0/11 (0%) | N/A* | No |
| MCF7-VEGFR-3 | 14/15 (93%) | 2153 ± 395 | No |
| MCF7-pcDNA3 | 5/5 (100%) | 239 ± 79 | Yes |
| MCF7-VEGFR-3 | 5/5 (100%) | 2589 ± 429** | Yes |
MCF7-pcDNA3 inoculated mice failed to develop tumors.
p < 0.005.
To drive tumor formation of MCF7-pcDNA3 cells xenografts, we implanted nude mice with estrogen pellets, which increased the tumor incidence of MCF7-pcDNA3 xenografts to 100%. Consistent with our tumorigenicity data in the absence of hormonal stimulation, MCF7-VEGFR-3 xenografts grew at a much faster rate compared to the MCF7-pcDNA3 xenografts (Fig. 11A). Three weeks after implantation the mean tumor volume of MCF7-VEGFR-3 xenografts (2589 ± 429 mm3) was ten times greater than the mean tumor volume of MCF7-pcDNA3 xenografts (239 ± 79 mm3) even in the presence of hormonal stimulation (Table 3, p < 0.01). Tumors representative for each group are shown in Figure 11A. Tumor lysates from harvested xenografts were evaluated by western blot analysis demonstrating overexpression of highly phosphorylated VEGFR-3 in MCF7-VEGFR-3 xenografts compared to the absence of VEGFR-3 expression in MCF7-pcDNA3 xenografts (Fig. 11B). From these in vivo experiments we concluded that VEGFR-3 overexpression significantly impacted the tumorigenicity of breast cancer cells in vivo, leading to the increased formation of rapidly growing xenografts.
Figure 11.

VEGFR-3 increases tumor formation and tumor growth in xenotransplant mouse model. (A) Tumor volume over time for MCF7-pcDNA3 and MCF7-VEGFR-3 xenografts with hormonal stimulation (*p < 0.05: **p < 0.001). Photographs of MCF7-VEGFR-3 (top) and MCF7-pcDNA3 (bottom) xenografts, demonstrating the representative sizes of tumors 18 days after implantation. (B) Western blot analysis of selected tumor lysates, demonstrating overexpression of highly phosphorylated VEGFR-3 in lysates derived from MCF7-VEGFR-3 xenografts. There is no VEGFR-3 expression in lysates from MCF7-pcDNA3 xenografts. (C) Immunohistochemical staining of MCF7-pcDNA3 and MCF7-VEGFR-3 xenografts with lymphatic endothelial cell marker LYVE-1, demonstrating increased peritumoral lymphangiogenesis associated with MCF7-VEGFR-3 xenografts (top) and staining with Ki67 demonstrating increase in proliferation of VEGFR-3 expressing cells (bottom). (D and E) RNAi knockdown of VEGFR-3 expression in breast cancer cells reduces tumor growth. (D) Tumor size at 15th day after subcutaneous injection of MCF7-VEGFR-3 cells and MCF7-VEGFR-3 shRNA stable clones. (E) Tumor size at 15th day after subcutaneous injection of BT474 cells and its shRNA stable clones.
Immunohistochemical analyses of MCF7-pcDNA3 and MCF7-VEGFR-3 breast cancer cell xenografts with Ki67 antibody revealed increased proliferation of VEGFR-3 overexpressing tumors (70% vs. 90% positive cells, respectively, Table 4 and Fig. 11C, bottom) which correlated with our in vitro data. Analysis with the LYVE-1 antibody, which specifically detects lymphatic endothelial cells, demonstrated significantly increased peritumoral lymphangiogenesis associated with MCF7-VEGFR-3 xenografts as compared to MCF7-pcDNA3 xenografts (Fig. 11C, top; p < 0.001). Furthermore, when VEGFR-3 shRNA clones of both MCF7-VEGFR-3 and BT474 cells were introduced into mice, we found that tumor growth was significantly reduced. The VEGFR-3 shRNA1 and shRNA2 clones of MCF7-VEGFR-3 cells produced tumors of negligible size, compared to parental control and to shRNA control (Table 5, Fig. 11D). In the BT474 cells, tumor growth of parental, shRNA control and internal shRNA2 control were similar (Fig. 11E). In contrast, BT474 shRNA1 cells with stably-reduced VEGFR-3 produced xenografts that were approximately half the size of the controls (Fig. 11E). These data demonstrated a significant role for VEGFR-3 in tumor lymphangiogenesis as well as tumor proliferation, survival and progression.
Table 4.
IHC analysis of xenograft tumor samples
| Antibody | Cell line injected | MCF7-pcDNA-3 | MCF7-VEGFR-3 | p value |
|---|---|---|---|---|
| Ki-67 | % Positive | 70 ± 0 | 88 ± 4 | 0.0008 |
| LYVE-1 | % Positive | 5 ± 0 | 61 ± 16 | <0.000001 |
Table 5.
RNAi knockdown of VEGFR-3 expression in breast cancer cells reduces tumor growth
| Cell line stable clone | Median tumor volume (mm3) | Cell line stable clone | Median tumor volume (mm3) |
|---|---|---|---|
| MCF7-VEGFR-3 | 1204.5 ± 695.7 | BT474 | 2141.2 ± 428.3 |
| shCTL | 443.25 ± 91.2 | shCTL | 1649.7 ± 901.6 |
| shRNA1 | 34.575 ± 7.8 | shRNA1 | 849.6 ± 241 |
| shRNA2 | 32.225 ± 19.7 | shRNA2* | 1682 ± 967.9 |
BT474 clone shRNA2 express VEGFR-3 at the same level as parental BT474 and shCTL (control) cell lines (Fig. 2E).
Discussion
VEGFR-3 is overexpressed in a variety of human tumors, but its role in tumorigenesis has not been fully elucidated. In this study, we have shown that VEGFR-3 upregulation in breast cancer precedes tumor cell invasion or metastasis, suggesting that VEGFR-3 may function as a survival signal and be an early event in breast tumorigenesis. We demonstrated that VEGFR-3 overexpression had a significant impact on several important biological characteristics of human breast cancer cells. VEGFR-3 overexpression promoted proliferation, motility, survival and, most importantly, tumorigenicity of MCF7 and BT474 cells. The salience of these results is increased by the fact that neither ligand VEGF-C nor VEGF-D was concomitantly overexpressed in these cell lines. To our knowledge, this is the first study to focus solely on VEGFR-3 overexpression and its biological impact on breast cancer.
Despite the large number of studies of VEGFR-3 expression in human tumors there is some controversy on its expression in tumor cells and thus the significance of this receptor protein kinase in tumor development.4,24 To some extent, it may be related to the variety of antibodies and immunohistochemical techniques used to measure expression. In this study, we used a well-characterized monoclonal antibody to analyze expression of VEGFR-3 in 76 different human breast tissues. Our results showed that VEGFR-3 was expressed at minimal levels in normal and nonneoplastic breast tissues, but was expressed in a significant number of preinvasive breast ductal cells, suggesting VEGFR-3 may have a pro-survival function before tumor invasion has occurred.
Since the landmark paper by Skobe et al.5 demonstrated that overexpression of the ligand VEGF-C increased growth and lymphatic metastasis of breast cancer xenografts, the majority of the work on the VEGF-C/VEGF-D/VEGFR-3 axis has focused on these ligands. Many of the subsequent studies do not even address receptor expression or its impact on tumor biology. Our results show that overexpression of VEGFR-3, by itself, was capable of making MCF-7 cells tumorigenic. Thus, receptor overexpression is at least as important as ligand overexpression, in that either phenomenon promoted characteristics associated with cancer progression, including increased proliferation, resistance to apoptosis as well as promotion of tumor growth. For example, our data show that overexpression of highly phosphorylated VEGFR-3 in MCF7 cells resulted in increased cellular proliferation, motility and invasion. Akahane et al. have reported a similar effect for proliferation when MCF7 cells were stably transfected to overexpress the ligand VEGF-D.29 These authors also demonstrated that VEGF-D overexpression in MCF7 cells led to resistance to apoptosis induced by staurosporine, cycloheximide and oxygen deprivation.32 Our data showed that overexpression of VEGFR-3 alone was sufficient to promote resistance to apoptosis. Furthermore, there was a large difference in basal PARP expression between our control stable clone MCF7-pcDNA3 and our VEGFR-3 overexpressing stable clone MCF7-VEGFR-3. The importance of PARP as a survival factor has been recently demonstrated. Mason et al. have shown that inhibition of PARP sensitized breast cancer cells to chemotherapeutic agents.33 Calabrese et al. demonstrated sensitization to radiation and chemotherapy through PARP inhibition in colorectal cancer cells.34 Finally, we found that expression and activation of AKT was increased in our receptor overexpressing MCF7-VEGFR-3 cells, compared to control MCF7-pcDNA3 cells. AKT is a well-known promoter of resistance to apoptosis through its ability to phosphorylate caspase-9,35 preventing initiation of cell death, to phosphorylate MDM2, increasing inhibition of p53,36 and to phosphorylate BAD, preventing mitochondrial cell death.37
In a similar fashion, downregulation of VEGFR-3 by either transient siRNA expression or stable shRNA transfection reversed its tumorigenic effects. This was seen in vitro, where downregulation of VEGFR-3 caused decreased proliferation, motility and invasion as well as increased levels of apoptosis. Importantly, these in vitro data were confirmed in vivo, where silencing of VEGFR-3 in BT474 cells led to reduced tumor formation in a xenograft tumor model. Conversely, MCF7 xenografts that over-expressed VEGFR-3 formed fast-growing tumors, independent of the estrogen stimulation that was required for control MCF7-pcDNA3 xenograft growth. This is consistent with data published by Akahane et al.29,30 and further demonstrates the biological significance imparted by overexpression of VEGFR-3. In addition, when these MCF7 xenografts were analyzed for proliferation and lymphangiogenesis, the xenografts that overexpressed VEGFR-3 showed increased proliferation as well as significantly increased peritumoral lymphangiogenesis, compared to the vector control cells. This increased peritumoral lymphangiogenesis is consistent with the known biological effects of VEGFR-3,38 and might impact the metastatic potential of the tumor cells.
In summary, VEGFR-3 overexpression has a profound impact on breast cancer cell tumorigenesis and survival. Most importantly, VEGFR-3 overexpression increased tumorigenicity and tumor growth of breast cancer cells in the absence of ligand expression or hormonal stimulation. These data indicate that VEGFR-3 might be a useful therapeutic target in the treatment of breast cancer.
Materials and Methods
Tissue specimens
76 formalin-fixed, paraffin-embedded (FFPE) breast tissue sections were obtained from the University of North Carolina Hospitals Lineberger Comprehensive Cancer Center Tissue Procurement and Analysis Facility. Routine morphological analysis of hematoxylin and eosin stained tissue sections were reviewed and the respective lesions confirmed by a pathologist (N.M.).
Immunohistochemistry
Four μm sections were cut from paraffin-embedded blocks, deparaffinized in xylene and rehydrated in an alcohol series. The sections were pretreated in a steamer in 1X Antigen Retrieval Citra (Biogene) for 30 minutes to retrieve tissue antigen and peroxidase activity was blocked with 3% hydrogen peroxide in methanol for 10 minutes. Nonspecific binding sites were blocked with background Sniper (Biocare Medical) for 15 min. Subsequently, the sections were incubated with anti-VEGFR-3 monoclonal antibodies 9D9F9, which recognize the extracellular domain of VEGFR-3 [24] (1:300; MAB3757 Chemicon) for 1 hour 30 min at 25°C in a humidified chamber. Then the slides were washed three times with TBS for 5 min, followed by 30 min incubation at 25°C with Mach2 goat anti-mouse HRP polymer secondary antibody (MHRP520; Biocare Medical). For detection, we used Vectastain Elite ABC kit (Vector Laboratories). Diaminobenzidine (DAB) was used as the chromogen, and the slides were counterstained with hematoxylin. A negative and positive control was included in each staining.
Immunohistochemistry scoring
One board certified pathologist (N.M.) scored each tissue sections for VEGFR-3 expression based on a scoring system that measured intensity (0, none; 1, borderline; 2, weak; 3, moderate; 4, strong), percentage of positive cells (0–100), cellular localization (cytoplasm, nucleus, membrane or combination) and overall distribution (homogeneous or heterogeneous). VEGFR-3 expression was dichotomized as high or low and was scored as high if the tissue sections had a staining intensity of 2 or 3 and ≥50% of the cell staining positive, otherwise, expression was scored as low.
Cell lines
MCF7 cells were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). The BT474 cells are a subclone of the original cell line that does not express the receptor tyrosine kinase Her-2/neu. BT474 were maintained in RPMI-1640 with 10% fetal bovine serum and insulin 250 ug/ml. MCF7 cells were maintained with Modified minimum Eagle’s media with 10% fetal bovine serum, 1X non-essential amino acids (Cellgro, Herndon, VA, USA), 1 mM sodium pyruvate, and 500 μg/ml insulin.
Antibodies and reagents
Chemicon VEGFR-3 9D9F9 (MAB3757).
Santa Cruz Biotechnology, Inc., (Santa Cruz, CA, USA): VEGFR-3 C-20 (sc-321).
Calbiochem (San Diego, CA, USA): p-VEGFR-3 (pc460), Staurosporine (#569396).
Cell Signaling Technology (Danvers, MA, USA): Pro-caspase-8 (#9746), Erk 1,2 (#9102), p-Erk (#4377S), AKT (#9272), p-AKT (9271S), PARP (#9542), rabbit IgG (#2729).
Dharmacon (Lafayette, CO, USA): Flt-4 Smart Pool siRNA (#L-003138-00).
Western blot
Appropriately treated or non-treated cells were allowed to grow until they were 80–85% confluent or until treatment was completed. Cells were twice washed with ice-cold phosphate-buffered saline (PBS), then incubated on ice with 1% NP-40 lysis buffer with inhibitors—150 mM NaCl, 20 mM Tris-Base, pH 7.4, 5 mM EDTA, 1% Nonidet P-40, 50 mM NaF, 10 mM NaVO4, 10 μl/ml phosphatase inhibitor cocktail I and II, according to the manufacturer’s instructions. (Calbiochem, San Diego, CA, USA), 20 μl/ml Complete Protease Inhibitor, according to the manufacturer’s instructions. (Roche Diagnostics, Mannheim, Germany)—for 30 minutes. Plates were scraped and lysate was centrifuged at 14,000 rpm for 30 minutes at 4°C. Protein concentration of cell lysate was measured in duplicate using the colorimetric BCA Protein Assay Kit (Pierce, Rockford, IL, USA) with bovine serum albumin as a standard. The appropriate amount of Laemmli loading buffer was mixed with protein lysate and boiled for 5 minutes at 95°C. Fifty micrograms of total protein were loaded and resolved by SDS-PAGE using 7.5%, 10% or 4–20% Tris-HCl gel (Biorad, Hercules, CA, USA). Resolved bands were transferred onto PVDF membrane for two hours at 80 mV at room temperature. PVDF membrane was then blocked with 5% BSA in tris-buffered saline tween (TBST, 25 mM Tris-HCl, 125 mM NaCl, 0.1% tween) for one hour. The appropriate primary antibody diluted in 5% BSA-TBST (1:1,000) was added for two hours at room temperature. Membrane was then washed three times for five minutes each with TBST. The appropriate secondary antibody conjugated with horseradish peroxidase, diluted in 5% BSA-TBST was added for one hour at room temperature. Membranes were washed three times with TBST, then developed using Western Lightning™ Chemilumnescence Reagent Plus (Perkin Elmer, Boston, MA, USA). Membrane was stripped using Restore™ western blot Stripping Buffer (Pierce, Rockford, IL, USA) for 15 minutes at 37°C, then washed three times in TBST for five minutes each wash and reused.
Transfection
VEGFR-3 plasmid was kindly provided by Hiroyuki Suzuki, Ph.D. University of Tsukuba, Tsukuba, Japan. Stable transfection of MCF7 cells was performed using SuperFect® transfection reagent (Qiagen, Valencia, CA, USA), according to the manufacturer’s protocol. 5 × 105 cells were plated on a 60-mm plate and allowed to attached overnight. 5 μg of each plasmid expression vector was added to serum-free Eagle’s minimum essential media (DMEM, Mediatech, Inc., Herndon, VA, USA) to make the total volume 150 μl. 60 μl of SuperFect® tranfection reagent was added to the plasmid solution and mixed by pipetting. This reaction mixture was incubated for 10 minutes. During incubation MCF7 were washed with 4 ml of 1X phosphate-buffered saline (PBS, Mediatech, Inc.,). One milliliter of MCF7 growth media was added to the reaction mixture. Complete mixture was added to washed MCF7 cells and incubated at normal growth condition (37°C, 5% CO2) for 2.5 hours. Transfected cells were washed with 7 ml of 1X PBS. 10 ml of MCF7 growth media was added to each plate. Cells grown under normal growth conditions for 24 hours. Cells were then washed with 10 ml of 1X PBS and 10 ml of MCF7 growth media was added supplemented with G418 (MP Biomedicals, Inc., Solon, Ohio, USA) at a concentration of 500 μg/ml.
siRNA transfection
The siRNA specific against Flt4 was a pool containing four different sequences purchased from Dharmacon (On-target plus smart pool L-002138-00). MCF7-VEGFR-3 or BT474 cells were transiently transfected with siRNA using Lipofectamine 2000 (Invitrogen). For analyses of VEGFR-3 expression cells were collected 24, 48 and 72 h after transfection. For motility and invasion assay 48 h later 3 × 104 cells were plated onto the insert of Boiden chamber in 1% FBS-containing medium. After 24 h of incubation migrated cells were fixed, stained and counted.
Stable knock-down of VEGFR-3 in breast cancer cells
A lentiviral-delivery system (a gift from Dr. Peter Chumakov at Lerner Research Institute, CCF, Cleveland) was used to introduce the siRNA expression cassette into MCF7-VEGFR-3 or BT474 cells. In brief, DNA oligo pairs for transcription of VEGFR-3 gene specific hairpin RNA were annealed and cloned into BamHI/EcoRI site of pLSL-GFP vector to obtain the pLSL-siRNA-GFP vector. The sequences of Flt4 NM_182925 from NCBI was used to design the oligo pairs, number in the name of primer represent nucleotide position in the ORF: Flt4_1578_f 5′-GAT CCC CGA GTT TGT GGA GGG AAA CTT CCT GTC ATT TCC CTC CAC AAA CTC GGT TTT TG-3′; Flt4_1578_r 5′-AAT TCA AAA ACC GAG TTT GTG GAG GGA AAT GAC AGG AAG TTT CCC TCC ACA AAC TCG GG-3′; Flt4_232_f 5′-GAT CCG AGA CAA GGA CAG CGA GGA CAC TTC CTG TCA TGT CCT CGC TGT CCT TGT CTC TTT TTG-3′; Flt4_232_r 5′AAT TCA AAA AGA GAC AAG GAC AGC GAG GAC ATG ACA GGA AGT GTC CTC GCT GTC CTT GTC TCG-3′.
An oligo pair unrelated to any gene was used to produce control siRNA construct: UNR_f 5′-GAT CCG CCA CAA GTT CAG CGT GTC CTT CCT GTC AGA CAC GCT GAA CTT GTG GCT TTT TG-3′; UNR_r 5′-GAT CCG CCA CAA GTT CAG CGT GTC CTT CCT GTC AGA CAC GCT GAA CTT GTG GCT TTT TG-3′.
The pLSL-siRNA-GFP vectors were co-transfected with lenti-viral packaging plasmids pCMV-VSV-G and pCMV-deltaR8.2 into 293T cells using Calphos mammalian transfection kit (Clontech Laboratories, Inc., CA). The media containing virus were collected 36, 40, 44 and 48 hours post-transfection. After each collection, media was filtered through 0.22 μM syringe filter and was used to infect cells growing in 6-well plate. Forty-eight hours after the last infection, the cells were collected and sorted for GFP expression by Flow Cytometry. Cells with strong GFP expression were expanded and used for all subsequent experiments. Cell lines expressing VEGFR-3 siRNA 1 and 2 are designated as shRNA1 and shRNA2, respectively. The control cell line that expresses the control siRNA is named shCTL.
BrdU incorporation assay
BrdU incorporation was performed using BrdU Cell Proliferation Assay, HTS (Calbiochem, San Diego, CA, USA), according to the manufacturer’s protocol. 2.5 × 103 cells were plated into a 96-well plate and allowed to attach overnight. 100 μl of fresh growth media or growth media with treatment was added to each well followed by 20 μl of BrdU labeling solution. Cells were incubated for 24 hours. Labeling solution was removed and 200 μl of the fixative solution was added for 30 minutes at room temperature. 100 μl of anti-BrdU antibody solution was added to each well and incubated for 1 hour at room temperature. Cells were washed three times with Wash Buffer. 100 μl of peroxidase Goat Anti-Mouse IgG HRP conjugate solution was added and incubated for 30 minutes at room temperature. Cells were washed three times with Wash Buffer. Wells were flooded with dH2O. 100 μl of Fluorogenic Substrate Working Solution is added to each well and incubated at room temperature for 30 minutes. 100 μl of Stop Solution was added to each well. Plates were read by fluorometer at 320 nm/460 nm.
MTT (cell viability) assay
5.0 × 103 (100 μL) were plated in 96-well plates and were allowed to attach overnight. One hundred microliters of fresh media with or without staurosporine was added to each well. Cells were treated for designated amount of time. Each well was incubated with twenty microliters of CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) for 1–2 hours. Colorimetric changes were read at wavelength of 490 nm using Benchmark microplate reader (Biorad, Hercules, CA, USA).
Detection of apoptosis was performed by TUNEL assay with the APO-DIRECT kit (Pharmingen, BD Biosciences, San Diego, CA) according to the manufacturer’s recommendations and analyzed by flow cytometry. Quantitative analysis of apoptosis was performed using FlowJo program (Tree Star, Ashland, OR).
Motility and invasion assay
Migration of human breast cancer cells was assessed in a modified Boyden chamber assay. Briefly, 50,000 cells were seeded into cell culture inserts containing membranes with 8 μm pores that were placed in wells containing cell culture medium with 10% FBS. Twenty four hours later unmigrated cells were removed with cotton swabs, cells were fixed in cold methanol and stained with Diff-Quik solution. Cells were counted using light microscopy at 40X magnification. Subsequently pictures of the migrated cells were taken and analyzed using Image-Pro Plus 6.0 software. Similar results were obtained in three independent experiments.
Invasion assay was performed using BD BioCoat™ Matrigel™ Invasion Chamber (BD Biosciences, Bedford, MA, USA). Cells were grown overnight in serum-reduced growth media with 1% FBS. Matrigel chambers were rehydrated for two hours using 500 μl of serum-free media. After rehydration, 750 μl normal growth media containing 10% FBS was added to the bottom of the well. 5.0 × 104 cells in serum-reduced growth media (500 ul) were added to the Matrigel chamber. Chambers were incubated overnight in a cell culture incubator, at 37°C, 5% CO2 atmosphere. Non-invading cells were removed with a cotton swab. Diff-Quik solution was added to a 24-well plate. Matrigel chambers were sequentially transferred through a fixative, then two staining solutions, followed by two beakers of double distilled water and were allowed to air dry. Membranes were mounted onto slides using immersion oil and covered with a cover slip. Cells were counted using light microscopy at 40X magnification.
RT-PCR
Protocol was performed using Cloned AMV First-Strand cDNA Synthesis kit (Invitrogen, Carlsbad, CA, USA). 10 μM VEGFR-3 primer (proprietary sequence of R&D Systems, RDP-108-025) was combined with 5 μg of RNA and 10 μM dNTP mix. 8 μl of master mix (cDNA synthesis buffer, 0.1 μM DTT, RNaseOUT™, DEPD-treated water, Cloned AMV reverse transcriptase) was added to the PCR tube on ice. Reaction was incubated at 50°C for 1 hour. Reaction was terminated by incubating at 85°C for 5 minutes. cDNA, primers, dNTP, Herculase are mixed. PCR was performed in the following manner: 10 cycles of 95°C for 30 seconds, 55°C for 30 seconds, 72°C for 70 seconds; 30 cycles of 95°C for 30 seconds, 55°C for 30 seconds, 72°C for 80 seconds.
Colony formation assay
1.5 ml 2X MCF7 or BT474 growth media was combined with 1.5 ml of 1% bact-agar solution, plated onto 60 mm Petri dish and allowed to solidify, creating the bottom layer of 0.5% bact-agar-1x growth media. 1 × 104 cells in 1.5 ml 2X growth media was combined with 1.5 ml of 0.7% agarose solution and plated on top of the bottom layer. After 14 days, plates were stained with 3 ml of 0.005% crystal violet for 3 hours. Plates were divided into quadrants. Colonies were counted in the two quadrants with the highest colony densities.
Animal model
Subconfluent MCF7-pcDNA3 and MCF7-VEGFR-3 cells were collected by centrifugation after treatment with EDTA-trypsin. Cells were washed twice in 1X PBS, diluted in sterile 1X PBS to a concentration of 1 × 106 cells per 100 μl. In accordance with the University of Florida IACUC approved protocol E557, 2 × 106 cells (200 μl) were subcutaneously injected into the right flank of the 6-week old athymic Nude-Foxn 1nu mice. Tumor volume was measured thrice weekly using the formula length × width2 × 0.5. After three weeks animals were sacrificed. Animals were housed in facilities approved by the American Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the U.S. Department of Agriculture.
Animal tissue samples and immunohistochemistry assays
Formalin-fixed, paraffin-embedded tumor blocks were obtained from MCF7-pcDNA3 and MCF7-VEGFR-3 xenografts. LYVE-1 staining was done using polyclonal antibody, LYVE-1 (AbCam, ab14917, Cambridge, MA, USA). Slides were incubated for 60 minutes with anti-LYVE-1 antibody (1:400) after deparaffinization, heat-induced epitope recovery, Citra retrieval (BioGenex, San Ramon, CA), and blocking with Sniper (Biocare Medical, Concord, CA). The slides were washed with TBS and incubated with goat-anti-rabbit Mach2 horseradish peroxidase polymer (Biocare) for 30 minutes. The chromogenic reaction was done with 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA). A single board-certified pathologist (N.M.) scored xenograft tissue sections for LYVE-1 immunoreactivity, based on a scoring system that measured intensity (0, none; 1, borderline; 2, weak; 3, moderate; 4, strong), percent positive cells (0–100). Ki67 staining was done as earlier described. Five-micrometer paraffin sections were microwaved for 7 minutes in 0.01 mol/L citrate buffer (pH 6.0) after deparaffinization and hydration. The sections were then washed with Tris-buffered saline. After Sniper blocking, sections were incubated with mouse anti-human Ki67 antibody (diluted 1:500; Dako M7240) for 1 hour at room temperature and later with Mach2 Ms HRP Polymer for 30 minutes at room temperature. The staining reaction was developed using Vectastain elite ABC kit (Vector Laboratories) and diaminobenzidine tetra-hydrochloride substrate (Vector Laboratories). Counterstain was done with Hem/Blue (Biocare). The proliferative activity was determined by counting the percentage of Ki67-immunoreactive cells. The most immunoreactive areas were selected, and a minimum of 500 cells was assessed.
Statistical analysis
Data presented are the means and 95% confidence intervals of the three or more experiments. For in vitro and in vivo experiments comparison between groups were made using a two-tailed two-sample Student’s t test. Differences for which p value was less than 0.05 were considered statistically significant.
Acknowledgments
This work is supported by NIH grant CA-65910 (W.G.C.). We would like to thank members of the Flow Cytometry Core Laboratory at the University of Florida for help with confocal microscopy and FACS techniques. We would also like to thank Fred Kweh for his help with the mouse experiments and Steven Hochwald for constructive discussion of experiments.
References
- 1.Otrock ZK, Makarem JA, Shamseddine AI. Vascular endothelial growth factor family of ligands and receptors: review. Blood Cells Mol Dis. 2007;38:258–68. doi: 10.1016/j.bcmd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Laakkonen P, Waltari M, Holopainen T, Takahashi T, Pytowski B, Steiner P, et al. Vascular endothelial growth factor receptor 3 is involved in tumor angiogenesis and growth. Cancer Res. 2007;67:593–9. doi: 10.1158/0008-5472.CAN-06-3567. [DOI] [PubMed] [Google Scholar]
- 3.Achen MG, Mann GB, Stacker SA. Targeting lymphangiogenesis to prevent tumour metastasis. Br J Cancer. 2006;94:1355–60. doi: 10.1038/sj.bjc.6603120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petrova TV, Bono P, Holnthoner W, Chesnes J, Pytowski B, Sihto H, et al. VEGFR-3 expression is restricted to blood and lymphatic vessels in solid tumors. Cancer Cell. 2008;13:554. doi: 10.1016/j.ccr.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 5.Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7:192–8. doi: 10.1038/84643. [DOI] [PubMed] [Google Scholar]
- 6.Karpanen T, Egeblad M, Karkkainen MJ, Kubo H, Yla-Herttuala S, Jaattela M, et al. Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer Res. 2001;61:1786–90. [PubMed] [Google Scholar]
- 7.Yanai Y, Furuhata T, Kimura Y, Yamaguchi K, Yasoshima T, Mitaka T, et al. Vascular endothelial growth factor C promotes human gastric carcinoma lymph node metastasis in mice. J Exp Clin Cancer Res. 2001;20:419–28. [PubMed] [Google Scholar]
- 8.Kawakami M, Yanai Y, Hata F, Hirata K. Vascular endothelial growth factor C promotes lymph node metastasis in a rectal cancer orthotopic model. Surg Today. 2005;35:131–8. doi: 10.1007/s00595-004-2896-0. [DOI] [PubMed] [Google Scholar]
- 9.Su JL, Yang PC, Shih JY, Yang CY, Wei LH, Hsieh CY, et al. The VEGF-C/Flt-4 axis promotes invasion and metastasis of cancer cells. Cancer Cell. 2006;9:209–23. doi: 10.1016/j.ccr.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 10.Ishii H, Yazawa T, Sato H, Suzuki T, Ikeda M, Hayashi Y, et al. Enhancement of pleural dissemination and lymph node metastasis of intrathoracic lung cancer cells by vascular endothelial growth factors (VEGFs) Lung Cancer. 2004;45:325–37. doi: 10.1016/j.lungcan.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 11.Lin J, Lalani AS, Harding TC, Gonzalez M, Wu WW, Luan B, et al. Inhibition of lymphogenous metastasis using adeno-associated virus-mediated gene transfer of a soluble VEGFR-3 decoy receptor. Cancer Res. 2005;65:6901–9. doi: 10.1158/0008-5472.CAN-05-0408. [DOI] [PubMed] [Google Scholar]
- 12.Ochi N, Matsuo Y, Sawai H, Yasuda A, Takahashi H, Sato M, et al. Vascular endothelial growth factor-C secreted by pancreatic cancer cell line promotes lymphatic endothelial cell migration in an in vitro model of tumor lymphangiogenesis. Pancreas. 2007;34:444–51. doi: 10.1097/mpa.0b13e31803dd307. [DOI] [PubMed] [Google Scholar]
- 13.He Y, Rajantie I, Pajusola K, Jeltsch M, Holopainen T, Yla-Herttuala S, et al. Vascular endothelial cell growth factor receptor 3-mediated activation of lymphatic endothelium is crucial for tumor cell entry and spread via lymphatic vessels. Cancer Res. 2005;65:4739–46. doi: 10.1158/0008-5472.CAN-04-4576. [DOI] [PubMed] [Google Scholar]
- 14.Kabashima A, Maehara Y, Kakeji Y, Sugimachi K. Overexpression of vascular endothelial growth factor C is related to lymphogenous metastasis in early gastric carcinoma. Oncology. 2001;60:146–50. doi: 10.1159/000055312. [DOI] [PubMed] [Google Scholar]
- 15.Witte D, Thomas A, Ali N, Carlson N, Younes M. Expression of the vascular endothelial growth factor receptor-3 (VEGFR-3) and its ligand VEGF-C in human colorectal adenocarcinoma. Anticancer Res. 2002;22:1463–6. [PubMed] [Google Scholar]
- 16.Yokoyama Y, Charnock-Jones DS, Licence D, Yanaihara A, Hastings JM, Holland CM, et al. Expression of vascular endothelial growth factor (VEGF)-D and its receptor, VEGF receptor 3, as a prognostic factor in endometrial carcinoma. Clin Cancer Res. 2003;9:1361–9. [PubMed] [Google Scholar]
- 17.Yokoyama Y, Charnock-Jones DS, Licence D, Yanaihara A, Hastings JM, Holland CM, et al. Vascular endothelial growth factor-D is an independent prognostic factor in epithelial ovarian carcinoma. Br J Cancer. 2003;88:237–44. doi: 10.1038/sj.bjc.6600701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimura Y, Watanabe M, Ohga T, Saeki H, Kakeji Y, Baba H, et al. Vascular endothelial growth factor C expression correlates with lymphatic involvement and poor prognosis in patients with esophageal squamous cell carcinoma. Oncol Rep. 2003;10:1747–51. doi: 10.3892/or.10.6.1747. [DOI] [PubMed] [Google Scholar]
- 19.Shida A, Fujioka S, Kobayashi K, Ishibashi Y, Nimura H, Mitsumori N, et al. Expression of vascular endothelial growth factor (VEGF)-C and -D in gastric carcinoma. Int J Clin Oncol. 2006;11:38–43. doi: 10.1007/s10147-005-0528-3. [DOI] [PubMed] [Google Scholar]
- 20.Kawakami M, Furuhata T, Kimura Y, Yamaguchi K, Hata F, Sasaki K, et al. Quantification of vascular endothelial growth factor-C and its receptor-3 messenger RNA with real-time quantitative polymerase chain reaction as a predictor of lymph node metastasis in human colorectal cancer. Surgery. 2003;133:300–8. doi: 10.1067/msy.2003.45. [DOI] [PubMed] [Google Scholar]
- 21.Funaki H, Nishimura G, Harada S, Ninomiya I, Terada I, Fushida S, et al. Expression of vascular endothelial growth factor D is associated with lymph node metastasis in human colorectal carcinoma. Oncology. 2003;64:416–22. doi: 10.1159/000070301. [DOI] [PubMed] [Google Scholar]
- 22.Onogawa S, Kitadai Y, Tanaka S, Kuwai T, Kimura S, Chayama K. Expression of VEGF-C and VEGF-D at the invasive edge correlates with lymph node metastasis and prognosis of patients with colorectal carcinoma. Cancer Sci. 2004;95:32–9. doi: 10.1111/j.1349-7006.2004.tb03167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donnem T, Al-Shibli K, Al-Saad S, Delghandi MP, Busund L-T, Bremnes RM. VEGF-A and VEGFR-3 correlate with nodal status in operable non-small cell lung cancer: Inverse correlation between expression in tumor and stromal cells. Lung Cancer. 2009;63:277. doi: 10.1016/j.lungcan.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 24.Su J-L, Chen P-S, Chien M-H, Chen PB, Chen Y-H, Lai C-C, et al. Further evidence for expression and function of the vegf-c/vegfr-3 axis in cancer cells. Cancer Cell. 2008;13:557. doi: 10.1016/j.ccr.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 25.Garces CA, Kurenova EV, Golubovskaya VM, Cance WG. Vascular endothelial growth factor receptor-3 and focal adhesion kinase bind and suppress apoptosis in breast cancer cells. Cancer Res. 2006;66:1446–54. doi: 10.1158/0008-5472.CAN-05-1661. [DOI] [PubMed] [Google Scholar]
- 26.Jussila L, Valtola R, Partanen TA, Salven P, Heikkila P, Matikainen M-T, et al. Lymphatic endothelium and kaposi’s sarcoma spindle cells detected by antibodies against the vascular endothelial growth factor receptor-3. Cancer Res. 1998;58:1599–604. [PubMed] [Google Scholar]
- 27.Bando H, Brokelmann M, Toi M, Alitalo K, Sleeman JP, Sipos B, et al. Immunodetection and quantification of vascular endothelial growth factor receptor-3 in human malignant tumor tissues. Int J Cancer. 2004;111:184–91. doi: 10.1002/ijc.20211. [DOI] [PubMed] [Google Scholar]
- 28.Yonemura Y, Fushida S, Bando E, Kinoshita K, Miwa K, Endo Y, et al. Lymphangiogenesis and the vascular endothelial growth factor receptor (VEGFR)-3 in gastric cancer. Eur J Cancer. 2001;37:918–23. doi: 10.1016/s0959-8049(01)00015-6. [DOI] [PubMed] [Google Scholar]
- 29.Akahane M, Akahane T, Shah A, Okajima E, Thorgeirsson UP. A potential role for vascular endothelial growth factor-D as an autocrine growth factor for human breast carcinoma cells. Anticancer Res. 2005;25:701–7. [PubMed] [Google Scholar]
- 30.Mattila MM, Ruohola JK, Karpanen T, Jackson DG, Alitalo K, Harkonen PL. VEGF-C induced lymphangiogenesis is associated with lymph node metastasis in orthotopic MCF-7 tumors. Int J Cancer. 2002;98:946–51. doi: 10.1002/ijc.10283. [DOI] [PubMed] [Google Scholar]
- 31.Zhou H, Kim YS, Peletier A, McCall W, Earp HS, Sartor CI. Effects of the EGFR/HER2 kinase inhibitor GW572016 on EGFR- and HER2-overexpressing breast cancer cell line proliferation, radiosensitization and resistance. Int J Radiat Oncol Biol Phys. 2004;58:344–52. doi: 10.1016/j.ijrobp.2003.09.046. [DOI] [PubMed] [Google Scholar]
- 32.Akahane M, Akahane T, Matheny SL, Shah A, Okajima E, Thorgeirsson UP. Vascular endothelial growth factor-D is a survival factor for human breast carcinoma cells. Int J Cancer. 2006;118:841–9. doi: 10.1002/ijc.21420. [DOI] [PubMed] [Google Scholar]
- 33.Mason KA, Valdecanas D, Hunter NR, Milas L. INO-1001, a novel inhibitor of poly(ADP-ribose) polymerase, enhances tumor response to doxorubicin. Invest New Drugs. 2007 doi: 10.1007/s10637-007-9072-5. [DOI] [PubMed] [Google Scholar]
- 34.Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004;96:56–67. doi: 10.1093/jnci/djh005. [DOI] [PubMed] [Google Scholar]
- 35.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 36.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA. 2001;98:11598–603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of bad couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 38.Kukk E, Lymboussaki A, Taira S, Kaipainen A, Jeltsch M, Joukov V, et al. VEGF-C receptor binding and pattern of expression with VEGFR-3 suggests a role in lymphatic vascular development. Development. 1996;122:3829–37. doi: 10.1242/dev.122.12.3829. [DOI] [PubMed] [Google Scholar]