

Abstract
The activation of Gα subunits of heterotrimeric G proteins by G protein-coupled receptors (GPCRs) is a critical event underlying a variety of biological responses. Understanding how G proteins are activated will require structural and biochemical analyses of GPCRs complexed to their G protein partners, together with structure-function studies of Gα mutants that shed light on the different steps in the activation pathway. Previously, we reported that the substitution of a glycine for a proline at position 56 within the linker region connecting the helical and GTP-binding domains of a Gα chimera, designated αT*, yields a more readily exchangeable state for guanine nucleotides. Here we show that GDP-GTP exchange on αT*(G56P), in the presence of the light-activated GPCR, rhodopsin (R*), is less sensitive to the β1γ1 subunit complex as compared to wild-type αT*. We solved the x-ray crystal structure for the αT*(G56P) mutant and found that the G56P substitution leads to concerted changes that are transmitted to the conformationally sensitive switch regions, the α4/β6 loop, and the β6 strand. The α4/β6 loop has been proposed to be a GPCR contact site that signals to the TCAT motif and weakens the binding of the guanine ring of GDP, whereas, the switch regions are the contact sites for the β1γ1 complex. Collectively, these biochemical and structural data lead us to suggest that αT*(G56P) may be adopting a conformation that is normally induced within Gα subunits by the combined actions of a GPCR and a Gβγ subunit complex during the G protein activation event.
Transducin is a heterotrimeric guanine nucleotide-binding protein (G protein) that is located in the outer segments of retinal rod cells (reviewed in refs. 1 and 2). In the visual phototransduction cascade, light-activated rhodopsin (R*) catalyzes GDP-GTP exchange on the Gα subunit of transducin (αT). The activated GTP-bound αT subunit converts the signal (i.e. a photon of light) into the final sensory output (the hyperpolarization of rod outer segment membranes and the visual response) by coupling the light-dependent activation of rhodopsin to the stimulation of the effector enzyme, the cGMP phosphodiesterase (PDE). This reduces the concentrations of cGMP in retinal rod cells and results in the closure of cGMP-gated ion channels and membrane hyperpolarization. The GTP hydrolytic activity of αT returns it to the basal GDP-bound state allowing the rebinding of the Gβγ subunit complex (i.e. β1γ1). The rate of GTP hydrolysis can be modulated by the PDE and markedly increased by RGS (Regulators of G protein-signaling) proteins.3
The αT subunit, like other Gα subunits of the heterotrimeric G protein superfamily, has a conserved nucleotide-binding (GTPase) domain similar to that of Ras and other “small G proteins”, and a helical domain connected by flexible linkers that bury the bound guanine nucleotide.4,5 High-resolution structural information has been obtained from x-ray crystallographic structures of various Gα subunits alone,4-9 in complex with their Gβγ partners,10,11 as well as with their effector proteins.12,13 These structures have highlighted the presence of three flexible regions, referred to as switch regions, in the GTPase domain that undergo conformational changes in response to the binding of GTP.
The x-ray crystal structure of the Gs heterotrimer complexed to the β2-adrenergic receptor has been recently achieved.14-16 In this structure, the C-terminal end of the α5-helix of the Gα subunit (referred to as Gαs) makes extensive contacts with the GPCR, confirming biochemical studies using synthetic peptides and mutants of Gα subunits that suggested the binding site for GPCRs on their Gα subunits is located at the C-terminal α-helix of Gα.17-20 The x-ray structure for the β2-adrenergic receptor-Gs complex also confirmed predictions from molecular modeling studies that the binding site on the Gα subunit for the GPCR is separated from the guanine nucleotide-binding (GTPase) domain by ∼30 Å.21
Given this distance, a question of great interest has been how GPCRs are able to transmit changes to the guanine nucleotide-binding sites of their Gα-signaling partners that weaken the binding of GDP and thereby stimulate the rate-limiting step for G protein activation, namely GDP-GTP exchange. In this regard, the x-ray crystal structure of the β2-adrenergic receptor complexed to the heterotrimeric G protein Gs provides an atomic resolution model of the endpoint of G protein activation, subsequent to nucleotide release, and suggests that a significant displacement of the helical-domain from the GTPase domain may be an important contributor to GDP-GTP exchange.14 However, the development of constitutively active Gα mutants that are able to exchange GDP for GTP at a relatively rapid rate in the absence of a GPCR,22-25 together with EPR studies of R*-αT interactions,26,27 have also added to the picture by defining the distinct steps that underlie GPCR-dependent G protein activation. The results obtained thus far point to the involvement of the α5 helix and the β6/α5 loop within Gα subunits in helping GPCRs to stimulate GDP dissociation. In particular, movements in the β6 loop and the β2-β3 turn of Gα subunits have been suggested to be important for GPCR-stimulated GDP-GTP exchange by EPR studies where mutations that caused the β2-β3 strands to move away from the α5 helix led to increases in the basal guanine nucleotide exchange activity of the Gαi1 subunit, whereas mutations within the β6 strand resulted in a decrease in the nucleotide exchange rates.26 Mutational and computational studies have further suggested that a GPCR-induced movement of the α5 helix of Gα subunits can be relayed to the GDP-binding site through the N-terminal α helix and the β2-β3 strands.23,28,29 Moreover, recent EPR studies suggest that engagement of the β6/α5 loop by GPCRs can be translated into significant changes in the juxtaposition of the helical domain relative to the guanine nucleotide-binding (GTPase) domain which help to provide an “escape route” for GDP.27
Still, there are a number of additional questions surrounding the steps that make-up the complete reaction pathway for a G protein activation event that will likely require the combined efforts of solution and crystallographic analyses, as well as biochemical studies of GPCR-G protein complexes and different Gα mutants that will mirror different steps of the pathway. For example, what role does the Gβγ subunit complex play in different stages of G protein activation? It was known for several years that Gβγ increased the affinity of GPCRs for their Gα subunit targets and this was traditionally felt to be the primary role for Gβγ in GPCR-stimulated nucleotide exchange. However, there have been suggestions that the Gβγ subunit complex has a direct involvement in the G protein activation event.30-33 Indeed, mutagenesis studies have pointed to a potentially more direct role for Gβγ in stimulating nucleotide exchange, leading to a model where the Gβγ complex works together with GPCRs to catalyze GDP-GTP exchange by helping to create an “exit” for GDP release by influencing the interactions between the helical and GTPase domains of the Gα subunit.34
Previous work from our laboratory showed that point mutations made within the linker regions that connect the helical and GTPase domains of a chimeric Gα subunit, comprised primarily of the αT subunit and a short segment of Gαi1 (designated as αT*), resulted in an accelerated exchange of GDP for GTP.25 In order to better understand how one such mutant, αT*(G56P), was capable of undergoing constitutive GDP-GTP exchange (i.e. independent of light-activated rhodopsin), and what this might tell us about the G protein activation event, we set out to further characterize its nucleotide exchange activity under various conditions, as well as solve its x-ray crystal structure. Here we show that the αT*(G56P) mutant can be strongly activated in the absence of Gβγ (β1γ1) when assayed together with sufficiently high levels of rhodopsin, thus differing from the case of wild-type αT* (designated αT*(WT)) which exhibits a much greater dependence on β1γ1 for full activation. This suggested that the αT*(G56P) mutant might exist in a conformational state that bears some similarity to that normally induced within the αT*(WT) subunit by Gβγ during the activation event. We were able to solve a 2.9 Å resolution x-ray crystal structure for GDP-bound αT*(G56P) and found that it showed more similarity (particularly in Switch 1) to the activated, GTPγS-bound state of αT*(WT) rather than to the GDP-bound form of αT*(WT). Overall, these findings raise some intriguing possibilities regarding how Gβγ might work together with a GPCR during the G protein activation event, as well as corroborate some earlier suggestions from NMR studies that Gβγ complexes may help to impart “activating conformational changes” within Gα subunits.30
Experimental Procedures
Protein expression and purification
The linker mutation (G56P) described in this study was made in the αT* background as previously reported.25 The recombinant αT*(WT) and αT*(G56P) mutant were expressed in BL21 (DE3) supercompetent cells and purified in the presence of 50 μM GDP as described previously.35 The recombinant proteins were further purified by gel filtration chromatography on a HiLoad Superdex 75 HR26/60 column equilibrated with a buffer containing 20 mM Na-HEPES, pH 7.5, and 10% glycerol. The samples were aliquoted, snap-frozen, and stored at −80°C. The final yield of recombinant αT* proteins ranged from 3 to 5 mg of pure protein/liter of bacterial culture. Retinal αT and the β1γ1 complex were purified from bovine retina as described.25 Urea-washed rod outer segment membranes were prepared as described.36
[35S]GTPγS-binding assays
Rhodopsin, together with the αT* subunits and β1γ1, was incubated in HMDN buffer (20 mM Hepes, pH 7.5, 5 mM MgCl2, 1 mM dithiothreitol, and 100 mM NaCl) for 20 minutes at room temperature and in room light. [35S]GTPγS (final concentration, 5 μM; specific activity, 1 Ci/mmol) was added to initiate the reaction, and the samples were incubated for different time periods. The reaction was quenched by direct application to pre-wetted nitrocellulose filters (Schleicher & Schuell; pore size, 0.45 μm) on a suction manifold. The filters were washed twice with HMN buffer (20 mM Hepes, pH 7.5, 5 mM MgCl2, and 100 mM NaCl), added to scintillation liquid (30% LSC Scintisafe Mixture), and counted in a scintillation counter (LS6500 Multipurpose Scintillation counter).
Crystallization, data collection and structure analysis
The αT*(G56P) mutant complexed with GDP, at 10 mg/ml in 50 mM Na-cacodylate buffer (pH 6.5), was crystallized by the hanging-drop vapor-diffusion method in 1.8-2.1 M ammonium sulfate (NH4)2SO4. Crystals (0.2 mm × 0.5 mm × 0.5 mm) grew in 5-7 days at 20°C and belonged to the space group P43212 (a=b=97.2 Å, c=380.6 Å; α=β=γ=90°). For data collection at 100 K, crystals were transferred to a well solution supplemented with 2.5-3.0 M (NH4)2SO4 for 30-60 seconds, followed by flash freezing in a liquid nitrogen stream immediately before the data collection. Freezing the crystals by any other method or in any other cryoprotectant led to their disintegration. The data sets were collected at beam line A1 at MacCHESS, Cornell University, with an ADSC Quantum-210 CCD detector (four 2048 × 2048-pixel modules). The data sets were indexed and processed using HKL2000.37 The structure was determined by molecular replacement using the coordinates from 1TAG (αT-GDP-Mg2+) as a search model in Molrep from the CCP4 suite (http://www.ccp4.ac.uk). CNS was utilized to obtain simulated annealing-generated models for αT*(G56P)-GDP. Model building was performed in Coot.38 Different parts of the protein and bound nucleotide were fitted into the observed densities followed by refinement using Refmac5 in CCP4 as well as a combination of rigid-body, simulated annealing, energy minimization, and B-factor protocols in CNS.39 The structural superposition was done using the program LSQMAN40 for superimposing the Cα carbon backbone in Coot. The contact and other coordinate analyses were done using the CCP4 suite. All structural images were made with PyMOL (http://www.pymol.org). Structure factors and coordinates are deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) protein database with PDB code 3V00.
Results
The R*-dependent activation of αT*(G56P) is less sensitive to β1γ1
αT*(WT) is capable of only a very slow rate of spontaneous GDP-GTP exchange (Figure 1A, inverted triangles), whereas the αT*(G56P) mutant exhibits a much more rapid rate of intrinsic nucleotide exchange (Figure 1B, inverted triangles), consistent with previous results from our laboratory.25 As expected, the addition of catalytic amounts of R* together with the β1γ1 complex enabled αT*(WT) to achieve high rates of GDP-GTP exchange (Figure 1A, open circles). The presence of relatively high concentrations of R* has been shown to circumvent the requirement for (β1γ1 to increase the affinity of R* for αT.41 Thus, increasing the levels of R* from 20 nM to 200 nM, in the absence of the β1γ1 subunit complex, caused a significant enhancement in the rate of GDP-GTP exchange on αT*(WT) (compare Figures 1A and 1C, closed circles). Still, the addition of β1γ1 to the above assay incubations stimulated even faster rates of GDP-GTP exchange (Figures 1A and 1C, open circles), consistent with the idea that the β1γ1 complex plays a direct role in the R*-dependent activation event.36-40 The αT*(G56P) subunit showed only a slight increase in the rate of GDP-GTP exchange when the concentration of R* was increased from 20 nM to 200 nM in the absence of β1γ1 (Figures 1B and 1D, closed circles). In the presence of catalytic amounts of R* (20 nM), the addition of β1γ1 to αT*(G56P) triggered an increase in the rate of GDP-GTP exchange (Figure 1B, open circles). However, surprisingly, when the assay was performed in the presence of 200 nM R*, the β1γ1 complex conferred only a minor enhancement in the rate of nucleotide exchange on the αT*(G56P) mutant (Figure 1D, open circles). These results seemed to suggest that although the β1γ1 complex might help to enhance the binding of αT*(G56P) to R* at relatively low levels of R*, consistent with the known role of Gβγ complexes in increasing the affinity of GPCRs for their cognate Gα partners, it does not appear to significantly influence the actual GDP-GTP exchange event on αT*(G56P), unlike the case for αT*(WT). This suggested that the αT*(G56P) mutant might resemble a conformation normally induced within αT*(WT) by β1γ1 during the course of R*-stimulated GDP-GTP exchange. Therefore, we set out to determine the three-dimensional x-ray crystal structure of αT*(G56P) as a means of potentially obtaining some insights into the structural changes induced by the β1γ1 complex upon the αT subunit, during the R*-dependent activation process.
Figure 1.

Comparison of the rhodopsin-dependence of nucleotide exchange on αT* and αT*(G56P). 5 μM [35S] GTPγS (specific activity, 1 Ci/mmol) was added to 700 nM α*T (G56P) or wild-type αT*(WT), preincubated with Gβγ (350 nM) and different concentrations of R* (20 nM; A, B), (200 nM; C, D) in 200 μl HMDM buffer. R* (solubilized in 0.01% dodecyl maltoside) was activated in ambient light on ice for 5 min. Aliquots (20 μl) of reaction mixture were removed at the indicated times and added directly to pre-wetted nitrocellulose filters on a suction manifold to quench the reaction. The filters were subsequently washed twice with HM buffer and added to 3 ml of scintillation fluid and counted on a scintillation counter. The extent of binding is plotted as percentage of maximal binding for each of the αT* subunits.
General features of the x-ray structure of the αT*(G56P) mutant
The x-ray crystal structure of the αT*(G56P) mutant was solved to a resolution of 2.9 Å. The data collection and refinement statistics are shown in Table 1. The αT*(G56P) structure exhibits the two major domains characteristic of Gα subunits (Figure 2A): one that is comprised of α-helices, referred to as the helical domain (shown in yellow), and a second domain that exhibits an α/β fold similar to those of GTPases belonging to the Ras superfamily, and thus referred to as the GTPase domain (shown in blue). While it has not been possible to generate suitable diffracting crystals for αT*(WT) alone (i.e. in the absence of β1γ1, see below), we can make comparisons between the x-ray structure for αT*(G56P) and the structure of retinal αT bound to GDP (PDB ID: 1TAG), from here on referred to as αT(WT), which behaves like αT*(WT) with regard to its low intrinsic GDP-GTP exchange activity and its sensitivity to R* and β1γ1.25 Indeed, most of the secondary structural elements exhibited by αT*(G56P) are similar to those of αT(WT). The helical domain of αT(WT) is made up of 6 helices designated αA to αF. In the crystal structure of αT*(G56P), the αB helix is longer by nearly one full turn at the N-terminus beginning at residue 98, instead of residue 95 in the structure of αT(WT). In addition, the amino acid residues comprising the αB helix have poorly defined electron density for the side chains in the structure of αT*(G56P), although the backbone has well-defined electron density. The GTPase domain of αT(WT) is comprised of 6 α-helices, designated α1 to α5 and αG, and a six-stranded β-sheet, with the strands designated as β1 to β6.4,5 In the x-ray structure of αT*(G56P), the α2 helix is completely disrupted, with the β2 strand being longer (i.e. because of residues Ile 180-Phe187) compared to that for αT(WT) (residues Thr183-Phe187).
Table 1. Data Collection and Refinement Statistics.
| space group | P43212 |
| unit cell a, b, c (Å) | 93.2, 93.2, 380.6 |
| Unit cell α, β, γ (deg) | 90.0, 90.0, 90.0 |
| resolution range (Å) | 50.0-2.9a |
| redundancy | 8.6 |
| completeness (%) | 97.4 |
| no. of unique reflections | 37430 |
| Rmerge (%) | 8.0 |
| average I/σ | 22.0 |
| Rcryst | 0.22 |
| Rfree | 0.27 |
| no. of protein atoms | 8381 |
| no. of water molecules | 296 |
| Wilson B value (Å2) | 89.2 |
| average B factor (Å2) | 94.3 |
| Ramachandran Plot | |
| most favored regions (%) | 86.7 |
| additionally allowed (%) | 13.3 |
| RMSD bond lengths (Å) | 0.007 |
| RMSD bond angles (deg) | 1.2 |
Figure 2.
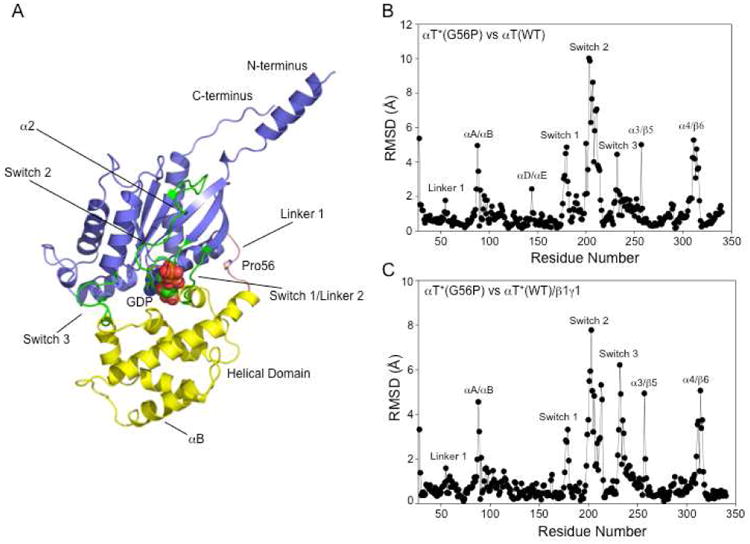
Structural changes in αT*(G56P). (A) The overall structure of αT*(G56P) showing the GTPase (colored purple), helical domains (colored yellow), and GDP (shown as spheres). The switch regions are colored green and the Linker 1 is colored pink. (B) Plot of the root mean square difference (RMSD) between the superimposed Cα atoms of αT*(G56P) and that of the corresponding Cα atoms of αT(WT) (PDB entry 1TAG). (C) Plot of the RMSD between the superimposed Cα atoms of αT*(G56P) and that of the corresponding Cα atoms of αT(WT) in complex with β1γ1 (PDB entry 1GOT).
The RMSD of the Cα atoms of αT*(G56P), when superimposed onto those from the structure of αT(WT), or αT*(WT) bound to the β1γ1 complex, were 1.9 Å and 1.5 Å, respectively. Structural differences were observed in various regions of the αT*(G56P) mutant when compared to αT(WT). These are depicted in a plot of the RMSD of Cα atoms of αT*(G56P) when superimposed against αT(WT) (Figure 2B), or αT*(WT) bound to the β1γ1 complex (Figure 2C). Therefore, while the overall structure of αT*(G56P) is similar to that of other Gα subunits, there are some noteworthy differences, which as discussed below, might shed light on the nature of the conformational changes induced in the αT(WT) subunit by the combined actions of R* and the β1γ1 subunit complex during the G protein activation event.
Arg174 and Lys266 show weaker interactions with GDP in the αT*(G56P)
The x-ray structure of the αT*(G56P) mutant contains a bound GDP molecule. The fact that this mutant exhibits a significantly faster rate of spontaneous GDP-GTP exchange compared to αT(WT) (or αT*(WT)), implies that its interactions with GDP are in some way weakened, particularly given that GDP dissociation is the rate-limiting step for G protein activation. In general, the amino acid residues that interact with GDP in the x-ray structure for αT*(G56P) are similar to those responsible for binding guanine nucleotides in αT(WT) (Figure 3A). However, two interactions appear to be different. One involves Arg174, which in the x-ray structure of αT(WT), is hydrogen-bonded through its side-chain guanidinium groups to the α- and β-oxygens of the phosphate groups of GDP (Figure 3B). What is especially interesting about this difference is that a similar change was observed for the corresponding residue (Arg178) in the x-ray crystal structure of a mutant form of Gαi1, in which the threonine residue at position 329 was mutated to an alanine (i.e. Gαi1(T329A)).42 This Gαi1 mutant exhibited an ∼20-fold increase in its rate of spontaneous GDP-GTP exchange compared to wild-type Gαi1, similar to the increased rate of nucleotide exchange that we see with αT*(G56P). The second interaction with GDP that appears to be altered in the x-ray structure for αT*(G56P), compared to αT(WT), involves Lys266. In αT(WT), this residue helps to stabilize the guanine ring of GDP via both hydrogen bonds and hydrophobic interactions, whereas in the structure for the G56P mutant, its position would suggest that it undergoes weaker interactions with GDP (Figure 3B). These differences between Arg174, Lys266, and GDP likely contribute to the ability of αT*(G56P) to undergo much more rapid GDP-GTP exchange compared to the αT(WT) subunit.
Figure 3.
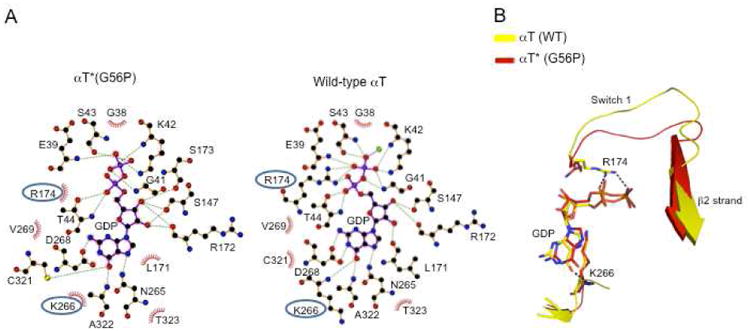
GDP-interacting residues within αT*(G56P). (A) Schematic view of the amino acid residues that interact with GDP in αT*(G56P) (left panel) and αT(WT) (right panel) was made using the program ligplot.43 Hydrogen bonded interactions are shown as dotted lines and hydrophobic interactions are shown using spoked arcs. Residues whose interactions are different between αT(WT) and αT*(G56P) are circled. (B) The Cα atoms of αT*(G56P) were superimposed on αT(WT). The changes in the interactions involving Arg174 and Lys266 in GDP-bound αT(WT) (colored yellow) and αT*(G56P) (colored red), with the bound GDP (shown as sticks), are presented.
The switch regions of GDP-bound αT*(G56P) adopt conformations that are distinct from those for GDP-bound αT(WT)
It is well documented from structural analyses that three regions on the Gα subunits (designated Switch 1-3) undergo conformational changes upon the binding of GTP, when compared to their conformations in the GDP-bound state.5,6 The x-ray crystal structure of the GDP-bound αT*(G56P) subunit shows that each of the three switch regions exhibit differences when compared to their conformations in the x-ray structure for GDP-bound αT(WT) (Figures 2B and 2C). These differences in the switch regions do not match the changes that accompany GDP-GTP exchange and can be distinguished from the conformations of Switch 1-3 seen in the x-ray structure for GTPγS-bound αT(WT).5 The differences in the switch domain conformations exhibited by GDP-bound αT*(G56P) (Figure 4, left panel) appear to be triggered by the loss of the hydrogen bond that is normally formed between the main-chain amide of Gly56 in αT(WT) and the main-chain carbonyl of Lys50 (Figure 4, right panel). This in turn causes a change in the conformation of Linker 1 in the G56P mutant, resulting in the loss of a hydrogen bond between the main-chain amide of Tyr57 and the side-chain carboxyl oxygen of Glu167. The loss of this hydrogen bond then causes a conformational change in the adjacent Linker 2/Switch 1 region, which is transmitted to other regions of the protein. These include the following residues: amino acids 196 to 210 (Switch 2), amino acids 226 to 238 (Switch 3), amino acids 249-264, and amino acids (305-319).
Figure 4.
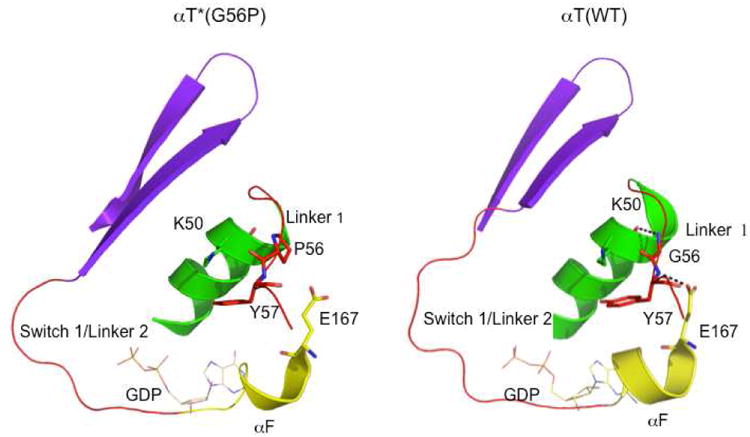
Transmission of conformational changes from Linker 1 in αT*(G56P). The Cα atoms of αT*(G56P) were superimposed on the corresponding Cα atoms of αT(WT) using the LSQMAN program. The G56P mutation results in the loss of a hydrogen-bond between the main-chain amide of Gly56 in Linker 1 and the main-chain carbonyl of Lys50 in the N-terminal helix (colored green). This in turn results in the loss of a hydrogen-bond between the main-chain amide of Tyr57 in Linker 1 and the side-chain carbonyl oxygen of Glu167 in the αF helix. These interactions are shown in the αT*(G56P) structure in the left panel and in the αT(WT) structure in the right panel. The hydrogen-bonding interactions are shown by dashed lines. Also, shown is the Switch 1 region as well as the β2 and β3 strands.
The Linker 2/Switch 1 region, along with Linker 1, has been proposed to serve as a hinge around which the helical domain would need to be rotated relative to the GTPase domain, in order to achieve the accelerated release of GDP during a G protein activation event.5,25 Indeed the conserved glycine residue in Linker 2/Switch 1, Gly179, shows the highest RMSD of all Cα atoms in this region of αT*(G56P) as compared to αT(WT) (Figure 2B). In addition, we have shown that the mutation of Gly179 to proline (αT*(G179P)) results in constitutive GDP-GTP exchange, similar to the αT*(G56P) mutant.25 The differences in Switch 1 exhibited by the GDP-bound αT*(G56P) mutant, as compared to GDP-bound αT(WT), appear to have an influence on Switch 2. One likely reason is due to the loss of a hydrogen-bonding interaction in the G56P mutant that normally occurs between the main-chain oxygen of Thr178 and the side-chain amino group of Lys205 in αT(WT). Several other interactions are also lost when comparing the x-ray structure of GDP-bound αT*(G56P) with that of GDP-bound αT(WT) (Figure 5). These include: (1) A hydrophobic cluster that forms between Val197, Trp207 and Phe211 in αT(WT) but is not evident in the αT*(G56P) structure. (2) A hydrogen bond between the main-chain carbonyl oxygen of Asp196 and the side-chain amino group of Lys206 in the structure of αT(WT) that is missing in the αT*(G56P) mutant. (3) A hydrogen bond between the nitrogen atom of the indole ring of Trp207 and the main-chain carbonyl oxygen of Gly198 in αT(WT) that is absent in the G56P mutant.
Figure 5.

Interactions that are lost in αT*(G56P) compared to αT(WT). The Cα atoms of αT*(G56P) were superimposed on the corresponding Cα atoms of αT(WT) using the LSQMAN program. A hydrophobic cluster (shown as dots) that forms between Trp207, Val197 and F211 in αT(WT) (right panel) is weakened in αT*(G56P) (left panel). Also, shown is the hydrogen bond (dashed lines) between the nitrogen atom of the indole ring of Trp207 and the main-chain carbonyl oxygen of Gly198 in αT(WT) (right panel), that is absent in αT*(G56P) (left panel).
The loss of these interactions in the GDP-bound αT*(G56P) subunit leads to several new interactions between Switch 1 and Switch 2, as well as within the Switch 2 domain (Figure 6A). These are the following: (1) A hydrogen bond between the main-chain amide of Gly199 and the main-chain carbonyl oxygen of Thr178. (2) A hydrogen bond between the main chain amide of Ile181 and the main-chain carbonyl oxygen of Asp196. (3) A hydrogen bond between the main chain carbonyl oxygen of Ile181 and the main-chain amide nitrogen of Asp196. (4) A hydrogen bond between the side-chain nitrogen (NH1) of Arg 201 and the side-chain oxygen (OE2) of Glu 241. Thus, the x-ray structure of the GDP-bound αT*(G56P) subunit shows the establishment of a new network of interactions between Switch 1 and Switch 2 (Figure 6A, left panel). Interestingly, Thr178 and Arg 201 from Switch 1 and Switch 2 are involved in contacting the Gβ subunit in the structure of αT*(WT) complexed with β1γ1 (Figure 6B).11 This, along with our findings that the αT*(G56P) subunit shows a reduced requirement for β1γ1 during R*-mediated GDP-GTP exchange, suggests that the structural differences observed in the Switch 1 and Switch 2 regions of the G56P mutant compared to αT(WT) might resemble changes induced by the β1γ1 complex in the presence of R*. In addition, a sequence alignment of αT against Gα subunits belonging to both the Gi family as well as other G protein families shows that all of the residues that either lose or gain interacting partners in the x-ray structure for αT*(G56P), compared to αT(WT), are conserved. This suggests that structural changes similar to those observed in αT*(G56P) might also be occurring in other Gα subunits.
Figure 6.
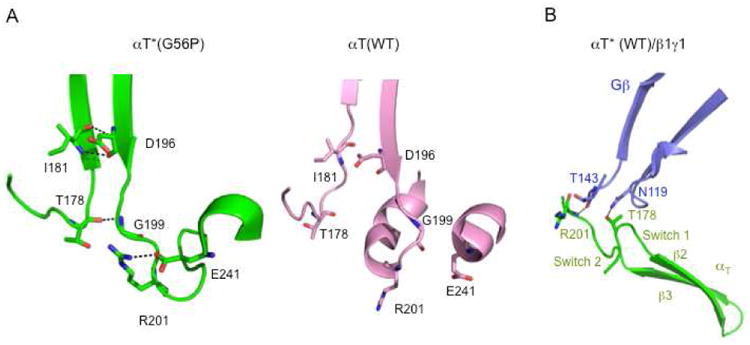
Interactions that are gained in αT*(G56P) compared to αT(WT). The Cα atoms of αT*(G56P) were superimposed on to αT(WT), or αT(WT) complexed to β1γ using the LSQMAN program. (A) Hydrogen bonds are present between the following pairs of residues in αT*(G56P) (left panel, dashed lines): Gly199/Thr178, Ile181/Asp196 and Arg201/Glu241. The same region in αT(WT) (right panel) shows the absence of these hydrogen bonds. (B) Hydrogen bonding between Arg201 and Thr178 in αT(WT) (colored green) to residues in the β1 subunit (colored purple) of the β1γ1 complex.
Discussion
Understanding the steps that comprise the conversion of GDP-bound heterotrimeric G proteins to their GTP-bound Gα subunits and free Gβγ complex ranks among the most fundamentally important issues in signal transduction, as these activation events are pre-requisite to a host of G protein-dependent changes in second messenger levels and cellular responses. In order to develop a complete picture of how G protein activation occurs, it will be necessary to combine the information obtained from structural and biophysical analyses of GPCRs complexed to their G protein-signaling partners, with the studies of various Gα mutants that might mimic individual species that form along the activation reaction pathway. A major breakthrough in this regard has been provided by the x-ray crystal structure of the β-adrenergic receptor in complex with Gs, its cognate G protein, in its nucleotide-free state.14-16 Here we describe efforts to obtain a Gα subunit that contains a single mutation in a linker region connecting the helical and GTPase domains, with the goal being to better understand how perturbing this linker might impact the activation event.
Our initial assumption was that by making substitutions within Linker 1 and/or Linker 2 of the αT* subunit, and in particular by changing conserved glycine residues to proline within these regions (i.e. G56P or G179P substitutions), we would alter the flexibility of the linkers that connect the GTPase and helical domains and cause a change in their relative juxtaposition. The idea being that such substitutions would help to open the “clam shell” that is formed by these two domains, and in doing so, facilitate the release of bound GDP and potentially create novel constitutively active Gα subunits. In fact, both the αT*(G56P) and αT*(G179P) mutants showed an enhanced ability to undergo GDP-GTP exchange in the absence of R*, when compared to the αT(WT) and αT*(WT) subunits.25 However, the x-ray crystal structure that we present here for the αT*(G56P) mutant does not show a significant alteration in the relative juxtaposition of the GTPase domain relative to the helical domain, presumably because the G56P mutant has retained bound GDP which helps to maintain a “closed” conformation rather than an “open” state. This suggests that the x-ray structure for the GDP-bound form of αT*(G56P) might mimic an intermediate state along the receptor-mediated nucleotide exchange pathway which would form prior to the actual release of GDP, and therefore might offer some potentially useful insights into different aspects of the G protein activation event.
One in particular concerns the role that the Gβγ complex may play in working together with an activated GPCR to help drive the activation process. There have been various suggestions that Gβγ plays multiple roles in G protein activation, first by significantly enhancing the affinity of Gα subunits for their GPCRs, but secondly and more directly, by working together with GPCRs to stimulate GDP-GTP exchange.30-33 In one such model, the Gβγ complex is proposed to function as a lever to help separate the β3-α2 loop from Switch 1 and thereby provides an “exit route” for GDP.32 A second potential mechanism for Gβγ involvement, referred to as the “gear-shift” model, suggests that the amino-terminal tail of the Gγ subunit helps to displace the helical domain from the GTPase domain and in this manner, provides an exit for GDP.33 Our characterization of the αT*(G56P) mutant shows that when assaying at relatively high levels of R*, so that the αT* subunit is no longer dependent upon the β1γ1 complex for binding to R*, there is little additional advantage provided by β1γ1 in terms of stimulating GDP-GTP exchange. This is not the case when assaying αT*(WT), as β1γ1 provides a significant stimulation of the GDP-GTP exchange activity of αT*, even at relatively high levels of R*. Thus, it is tempting to consider that the G56P mutant might adopt a conformation that is normally induced within αT(WT) by β1γ1 during the R*-stimulated activation reaction. In particular, the structural differences observed in the Switch 1 and Switch 2 regions of αT*(G56P), as compared to the switch regions of αT(WT), might reflect the types of changes mediated by β1γ1 if it were acting as proposed in the lever model to help stimulate GDP-GTP exchange and G protein-activation. These changes in Switch 1 and Switch 2 would also be consistent with the idea that Gα subunits adopt a “pre-activated” conformation when bound to Gβγ, presumably to help stabilize the GTP-bound state following nucleotide exchange.30
Another potentially interesting possibility that arises from an analysis of the x-ray crystal structure of the αT*(G56P) mutant concerns how the helical domains of Gα subunits might communicate with their carboxyl terminal regions that contain the putative contact sites for GPCRs and represent the starting point for the signal that triggers GDP release and its exchange for GTP. Specifically, it has been suggested that an activated GPCR, by contacting the α4/β6 loop of a Gα subunit, induces a rotation and translation of the α5 helix and concomitant conformational changes in the β6 strand. These conformational changes would then be transmitted to the β6/α5 loop, which makes contact with the guanine ring of GDP via the conserved TCAT motif, thereby influencing the hold on GDP. However, starting with the initial descriptions of the x-ray crystal structures for Gα subunits, and the realization that the helical domain folds over the nucleotide bound to the GTPase domain (i.e. to form a “clam shell”), it has been assumed that the GPCR-mediated activation event would necessitate an opening of the clam shell such that GDP was free to exit from Gα. Indeed, a recent study using site-directed spin labeling and electron-electron resonance spectroscopy suggested that GPCR-catalyzed nucleotide exchange in G proteins requires large-scale changes in the relative orientation of the helical and GTPase domains.27 Also, in the aforementioned structure of the nucleotide-free form of the Gs heterotrimer complexed to the β2-adrenergic receptor, the helical domain is rotated by ∼120° relative to the Ras-like GTPase domain, compared to the x-ray crystal structure of αs complexed to GTPγS.14-16
The lack of the availability of an x-ray structure of Gαs complexed to GDP makes it difficult to map out the conformational changes that occur in the switch regions in the case of the β-adrenergic receptor-induced release of GDP. This also prevented a direct mapping of the conformational changes that occur in the αT*(G56P), compared to those of αT(WT), onto the structure of the complex of the β2-adrenergic receptor and the Gs heterotrimer. The x-ray structure of the αT*(G56P) mutant demonstrates how a series of conformational transitions occur in response to a single substitution within the linker region that connects the helical domain to the GTPase domain. These changes proceed through the Switch 1 and Switch 2 regions and continue through Switch 3 to residues 305-319 comprising the α4/β6 loop and the β6 strand. It is possible that the engagement of GPCRs with the carboxyl-terminal region of Gα subunits could initiate a similar set of changes, but in reverse order relative to what we see with the αT*(G56P) mutant, thereby providing a conformational connection between the GPCR-binding site and the helical domain on Gα subunits. As we learn more about the distinct species that form along the G protein activation pathway, we can expect that the roles played by GPCRs and Gβγ complexes in the actual GDP-GTP exchange event, as well as the potential involvement of Gβγ in helping Gα subunits assume a GTP-bound activated state, will become much better defined. The insights gained from these studies, together with future structure-function analyses of additional Gα mutants that reflect intermediate states in the G protein activation process, should ultimately provide us with a more comprehensive picture for this critically important event in signal transduction.
Acknowledgments
We would like to acknowledge Cindy Westmiller for her expert secretarial assistance.
This work was supported by National Institutes of Health grant R01 GM047458 and by resources provided by MacCHESS (RR001646).
Abbreviations
- Gα
G protein alpha subunit
- αT
G protein alpha subunit of the vertebrate vision system (transducin)
- GDP
Guanosine diphosphate
- GTP
Guanosine triphosphate
- Gβγ
G protein beta gamma subunit complex
- GPCRs
G protein-coupled receptors
- G protein
Guanine nucleotide binding protein
- PDE
cyclic-GMP phosphodiesterase
- EPR
Electron paramagnetic resonance
- HEPES
4-(2-hydroxyethyl)-6-sulfonic acid
- MacCHESS
Macromolecular Crystallography Facility at CHESS (Cornell High Energy Synchrotron Source)
- CCP4
Comprehensive computing suite for protein crystallography
- CNS
Crystallography and NMR (Nuclear Magnetic Resonance) system
- RMSD
Root mean square deviation
- PDB
Protein Data Bank
References
- 1.Burns ME, Arshavsky VY. Beyond counting photons: trials and trends in vertebrate visual transduction. Neuron. 2005;48:387–401. doi: 10.1016/j.neuron.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 2.Chen CK. The vertebrate phototransduction cascade: amplification and termination mechanisms. Rev Physiol Biochem Pharmacol. 2005;154:101–121. doi: 10.1007/s10254-005-0004-0. [DOI] [PubMed] [Google Scholar]
- 3.He W, Cowan CW, Wensel TG. RGS9, a GTPase accelerator for phototransduction. Neuron. 1998;20:95–102. doi: 10.1016/s0896-6273(00)80437-7. [DOI] [PubMed] [Google Scholar]
- 4.Noel JP, Hamm HE, Sigler PB. The 2.2 Å crystal structure of transducin-α complexed with GTPγS. Nature. 1993;366:654–663. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- 5.Lambright DG, Noel JP, Hamm HE, Sigler PB. Structural determinants for activation of the α-subunit of a heterotrimeric G protein. Nature. 1994;369:621–628. doi: 10.1038/369621a0. [DOI] [PubMed] [Google Scholar]
- 6.Mixon MB, Lee E, Coleman DE, Berghuis AM, Gilman AG, Sprang SR. Tertiary and quaternary structural changes in Giα1 induced by GTP hydrolysis. Science. 1995;270:954–960. doi: 10.1126/science.270.5238.954. [DOI] [PubMed] [Google Scholar]
- 7.Sunahara RK, Tesmer JJ, Gilman AG, Sprang SR. Crystal structure of the adenylyl cyclase activator Gsα. Science. 1997;278:1943–1947. doi: 10.1126/science.278.5345.1943. [DOI] [PubMed] [Google Scholar]
- 8.Kreutz B, Yau DM, Nance MR, Tanabe S, Tesmer JJ, Kozasa T. A new approach to producing functional Gα subunits yields the activated and deactivated structures of Gα12/13 proteins. Biochemistry. 2006;45:167–174. doi: 10.1021/bi051729t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slep KC, Kercher MA, Wieland T, Chen CK, Simon MI, Sigler PB. Molecular architecture of Gαo and the structural basis for RGS16-mediated deactivation. Proc Natl Acad Sci USA. 2008;105:6243–6248. doi: 10.1073/pnas.0801569105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, Sprang SR. The structure of the G protein heterotrimer Giα1β1γ2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 11.Lambright DG, Sondek J, Bohm A, Skiba NP, Hamm HE, Sigler PB. The 2.0 A crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- 12.Slep KC, Kercher MA, He W, Cowan CW, Wensel TG, Sigler PB. Structural determinants for regulation of phosphodiesterase by a G protein at 2.0 A. Nature. 2001;409:1071–1077. doi: 10.1038/35059138. [DOI] [PubMed] [Google Scholar]
- 13.Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR. Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsα.GTPγS. Science. 1997;278:1907–1916. doi: 10.1126/science.278.5345.1907. [DOI] [PubMed] [Google Scholar]
- 14.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westfield GH, Rasmussen SG, Su M, Dutta S, DeVree BT, Chung KY, Calinski D, Velez-Ruiz G, Oleskie AN, Pardon E, Chae PS, Liu T, Li S, Woods VL, Jr, Steyaert J, Kobilka BK, Sunahara RK, Skiniotis G. Structural flexibility of the Gαs α-helical domain in the β2-adrenoceptor Gs complex. Proc Natl Acad Sci USA. 2011;108:16086–16091. doi: 10.1073/pnas.1113645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung KY, Rasmussen SG, Liu T, Li S, DeVree BT, Chae PS, Calinski D, Kobilka BK, Woods VL, Jr, Sunahara RK. Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature. 2011;477:611–615. doi: 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamm HE, Deretic D, Arendt A, Hargrave PA, Koenig B, Hofmann KP. Site of G protein binding to rhodopsin mapped with synthetic peptides from the α subunit. Science. 1988;241:832–835. doi: 10.1126/science.3136547. [DOI] [PubMed] [Google Scholar]
- 18.Osawa S, Weiss ER. The effect of carboxyl-terminal mutagenesis of Gtα on rhodopsin and guanine nucleotide binding. J Biol Chem. 1995;270:31052–31058. doi: 10.1074/jbc.270.52.31052. [DOI] [PubMed] [Google Scholar]
- 19.Sullivan KA, Miller RT, Masters SB, Beiderman B, Heideman W, Bourne HR. Identification of receptor contact site involved in receptor-G protein coupling. Nature. 1987;330:758–760. doi: 10.1038/330758a0. [DOI] [PubMed] [Google Scholar]
- 20.West RE, Moss J, Vaughan M, Liu T, Liu TY. Pertussis toxin-catalyzed ADP-ribosylation of transducin Cysteine 347 is the ADP-ribose acceptor site. J Biol Chem. 1985;260:14428–14430. [PubMed] [Google Scholar]
- 21.Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. The G protein-coupled receptor rhodopsin in the native membrane. FEBS Lett. 2004;564:281–288. doi: 10.1016/S0014-5793(04)00194-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marin EP, Krishna AG, Sakmar TP. Rapid activation of transducin by mutations distant from the nucleotide-binding site: evidence for a mechanistic model of receptor-catalyzed nucleotide exchange by G proteins. J Biol Chem. 2001;276:27400–27405. doi: 10.1074/jbc.C100198200. [DOI] [PubMed] [Google Scholar]
- 23.Marin EP, Krishna AG, Sakmar TP. Disruption of the α5 helix of transducin impairs rhodopsin-catalyzed nucleotide exchange. Biochemistry. 2002;41:6988–6994. doi: 10.1021/bi025514k. [DOI] [PubMed] [Google Scholar]
- 24.Posner BA, Mixon MB, Wall MA, Sprang SR, Gilman AG. The A326S mutant of Giα1 as an approximation of the receptor-bound state. J Biol Chem. 1998;273:21752–21758. doi: 10.1074/jbc.273.34.21752. [DOI] [PubMed] [Google Scholar]
- 25.Majumdar S, Ramachandran S, Cerione RA. Perturbing the linker regions of the α-subunit of transducin: a new class of constitutively active GTP-binding proteins. J Biol Chem. 2004;279:40137–40145. doi: 10.1074/jbc.M405420200. [DOI] [PubMed] [Google Scholar]
- 26.Oldham WM, Van Eps N, Preininger AM, Hubbell WL, Hamm HE. Mechanism of the receptor-catalyzed activation of heterotrimeric G proteins. Nat Struct Mol Biol. 2006;13:772–777. doi: 10.1038/nsmb1129. [DOI] [PubMed] [Google Scholar]
- 27.Van Eps N, Preininger AM, Alexander N, Kaya AI, Meier S, Meiler J, Hamm HE, Hubbell WL. Interaction of a G protein with an activated receptor opens the interdomain interface in the α subunit. Proc Natl Acad Sci USA. 2011;108:9420–9424. doi: 10.1073/pnas.1105810108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herrmann R, Heck M, Henklein P, Hofmann KP, Ernst OP. Signal transfer from GPCRs to G proteins: role of the Gα N-terminal region in rhodopsin-transducin coupling. J Biol Chem. 2006;281:30234–30241. doi: 10.1074/jbc.M600797200. [DOI] [PubMed] [Google Scholar]
- 29.Ceruso MA, Periole X, Weinstein H. Molecular dynamics simulations of transducin: interdomain and front to back communication in activation and nucleotide exchange. J Mol Biol. 2004;338:469–481. doi: 10.1016/j.jmb.2004.02.064. [DOI] [PubMed] [Google Scholar]
- 30.Abdulaev NG, Ngo T, Zhang C, Dinh A, Brabazon DM, Ridge KD, Marino JP. Heterotrimeric G-protein α-subunit adopts a “preactivated” conformation when associated with βγ-subunits. J Biol Chem. 2005;280:38071–38080. doi: 10.1074/jbc.M505259200. [DOI] [PubMed] [Google Scholar]
- 31.Oldham WM, Van Eps N, Preininger AM, Hubbell WL, Hamm HE. Mapping allosteric connections from the receptor to the nucleotide-binding pocket of heterotrimeric G proteins. Proc Natl Acad Sci USA. 2007;104:7927–7932. doi: 10.1073/pnas.0702623104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iiri T, Farfel Z, Bourne HR. G-protein diseases furnish a model for the turn-on switch. Nature. 1998;394:35–38. doi: 10.1038/27831. [DOI] [PubMed] [Google Scholar]
- 33.Cherfils J, Chabre M. Activation of G-protein Gα subunits by receptors through Gα-Gβ and Gα-Gγ interactions. Trends Biochem Sci. 2003;28:13–17. doi: 10.1016/s0968-0004(02)00006-3. [DOI] [PubMed] [Google Scholar]
- 34.Rondard P, Iiri T, Srinivasan S, Meng E, Fujita T, Bourne HR. Mutant G protein α subunit activated by Gβγ: a model for receptor activation. Proc Natl Acad Sci USA. 2001;98:6150–6155. doi: 10.1073/pnas.101136198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skiba NP, Bae H, Hamm HE. Mapping of effector binding sites of transducin α-subunit using Gαt/Gαi1 chimeras. J Biol Chem. 1996;271:413–424. doi: 10.1074/jbc.271.1.413. [DOI] [PubMed] [Google Scholar]
- 36.Min KC, Gravina SA, Sakmar TP. Reconstitution of the vertebrate visual cascade using recombinant heterotrimeric transducin purified from Sf9 cells. Protein Expr Purif. 2000;20:514–526. doi: 10.1006/prep.2000.1326. [DOI] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweet RM, editors. Methods Enzymology. 276: Macromolecular Crystallography, part A. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 40.Kleywegt GJ. Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr D Biol Crystallogr. 1996;52:842–857. doi: 10.1107/S0907444995016477. [DOI] [PubMed] [Google Scholar]
- 41.Phillips WJ, Wong SC, Cerione RA. Rhodopsin/transducin interactions. II Influence of the transducin-βγ subunit complex on the coupling of the transducin-α subunit to rhodopsin. J Biol Chem. 1992;267:17040–17046. [PubMed] [Google Scholar]
- 42.Kapoor N, Menon ST, Chauhan R, Sachdev P, Sakmar TP. Structural evidence for a sequential release mechanism for activation of heterotrimeric G proteins. J Mol Biol. 2009;393:882–897. doi: 10.1016/j.jmb.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 43.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1996;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]