

Abstract
Mother-to-child-transmission (MTCT) of human immunodeficiency virus (HIV) remains a significant cause of new HIV infections in many countries. To examine whether fetal immune activation as a consequence of prenatal exposure to parasitic antigens increases the risk of MTCT, cord blood mononuclear cells (CBMCs) from Kenyan and North American newborns were examined for relative susceptibility to HIV infection in vitro. Kenyan CBMCs were 3-fold more likely to be infected with HIV than were North American CBMCs (P = .03). Kenyan CBMCs with recall responses to malaria antigens demonstrated enhanced susceptibility to HIV when compared with Kenyan CBMCs lacking recall responses to malaria (P = .03). CD4+ T cells from malaria-sensitized newborns expressed higher levels of CD25 and human leukocyte antigen DR ex vivo, which is consistent with increased immune activation. CD4+ T cells were the primary reservoir of infection at day 4 after virus exposure. Thus, prenatal exposure and in utero priming to malaria may increase the risk of MTCT.
Malaria and human immunodeficiency virus (HIV) infections are leading causes of death among children and adults, respectively, in Africa [1, 2]. Morbidity attributable to these pathogens is particularly high during pregnancy and among neonates. Pregnant women infected with malaria or HIV have an increased risk of adverse events and outcomes, including anemia, spontaneous abortion, premature delivery, low neonatal birth weight, and increased infant mortality [3–8]. Maternal co-infection with both malaria and HIV may result in worse birth outcomes than either infection alone. Malaria-infected, HIV-positive pregnant women experience an increased prevalence of placental malaria, higher parasitemia, and clinical disease, compared with HIV-negative women [6, 9–14]. Conversely,malaria infection has been shown to increase HIV load and damage the placenta, causing microtransfusions from maternal to fetal circulation, and both of these are important risk factors in the vertical transmission of HIV from mother to child (MTCT) [15–19].
It is unclear whether malaria co-infection of HIV-positive women increases the risk for MTCT. Epidemiologic studies that have addressed this question have produced conflicting results [20]. A study from Uganda found that placental malaria was associated with a ∼3-fold greater risk for HIV vertical transmission [21]. Similarly, a study from Cameroon found a positive association between MTCT and the peak rainy season when malaria transmission was high, suggesting a possible correlation between MTCT and maternal malaria infection, although malaria burden was not directly assessed [22]. By contrast, no association between placental malaria and increased risk of MTCT was found on the Kenyan coast, which had lower levels of infection than did the studies from Uganda and Cameroon [9]. Interestingly, a large study from western Kenya showed that high-density placental parasitemia was associated with an increased risk of MTCT, whereas low-density placental parasitemia was not [23], which suggest that the intensity or frequency of placental malaria may explain differences among studies.
These studies suggest that the relationship between placental malaria and MTCT is complex. To our knowledge, no study, however, has evaluated the potential impact of fetal factors on the risk of MTCT. For example, immune activation is vital for HIV infection; thus, in utero exposure to exogenous antigens and consequent fetal lymphocyte activation may increase susceptibility to HIV infection [24, 25]. Newborns of mothers infected with malaria and other intravascular parasites, such as schistosomiasis and lymphatic filariasis, are often primed to parasite antigens [26–30]. This activation of cord blood mononuclear cells (CBMCs) could increase the risk for HIV infection if exposed to virus in utero or perinatally. In support of this hypothesis, we previously demonstrated that helminth co-infections in HIV-positive women during pregnancy increase the risk for MTCT, especially among newborns who demonstrate in utero sensitization to helminth antigens [31]. Here, we tested the hypothesis that CBMCs from Kenyan newborns of women infected with malaria exhibit greater susceptibility to HIV infection in vitro than do CBMCs collected from newborns of parasite-uninfected Kenyan or North American women. We also show that Kenyan CBMCs that demonstrate in utero priming to malaria blood stage antigens exhibit the greatest susceptibility to HIV infection in vitro.
Materials and Methods
Study population. Samples were obtained from newborns born at the Msambweni District Hospital, Coast Province, Kenya an area where malaria, schistosomiasis, lymphatic filariasis, and intestinal helminthes are endemic [26, 32]. The mothers were assessed for helminth and malaria infection during repeated assays performed on samples collected during antenatal clinical visits. All women were tested for HIV, and the offspring of HIV-positive women were excluded from the current study. Control CBMCs were prepared from healthy North American newborns delivered at University Hospitals of Cleveland (Cleveland, Ohio). Ethical approval was obtained from the Institutional Review Boards of the University Hospitals of Cleveland and the Kenya Medical Research Institute in Nairobi, and informed consent for the study was obtained from study subjects.
Determination of maternal parasite infection. Malaria infection was determined by blood smear and/or polymerase chain reaction performed on samples of maternal peripheral and intervillous blood; urinary schistosomiasis was assessed by the presence of ova in the urine; lymphatic filariasis was assessed by antigen detection in peripheral blood samples; and intestinal helminth infections were assessed by examination of stool samples, as described elsewhere [32].
CBMC sample collection and sensitization determination. CBMC isolation was performed by Ficoll-Hypaque density gradient separation. A subset of freshly collected CBMC sample was immediately cultured with media alone or with the following malaria antigens: recombinant malaria surface protein 1–42 (MSP1-42), alleles corresponding to 3D7 and FVO parasite lines (5 ug/mL each, kindly provided by Sanjay Singh, Carole Long, and David Narum of the Malaria Vaccine Development Unit, National Institutes of Health), pooled MSP1-42 peptides previously determined to be dominant CBMC epitopes in this population [33] (10 ug/mL for each peptide), Plasmodium falciparum ribosomal phosphoprotein peptides (PfP0; 10 ug/mL), and phytohemagglutinin (PHA; 1 ug/mL). Remaining CBMCs were cryopreserved.
Quantification of interleukin (IL)-2, IL-5, interferon γ, IL- 10, and IL-13 was performed on culture supernatants collected at 96 h using multiplex immunoassay (Millipore). Results were expressed in pg/mL by interpolation from standard curves. A positive response was scored when the following 2 criteria were fulfilled: (1) a net value for antigen-stimulated cells that was at least 2-fold greater than that of parallel cultures containing media alone and (2) responses to ⩾2 malaria antigens. If cytokine production was not detectable in the negative control cultures, then >40 pg/mL cytokine was considered to be a positive response.
In vitro HIV infection. Cryopreserved CBMCs were thawed (all samples showed >85% viable cell recovery with preservation of immune function, as indicated by similar recall responses to malaria antigens between fresh and cryopreserved aliquots from the same cord blood sample) and rested overnight in Roswell Park Memorial Institute (RPMI) 1640 plus L-glutamine supplemented with penicillin, streptomycin (100 units/ mL), HEPES (10 mM), and 10% pooled human AB serum (cRPMI+10%). From this point forward, culture media consisted of cRPMI+10% supplemented with 1.0 ng/mL recombinant human IL-2 (rhIL-2; BD Biosciences). Infection with the R5 tropic virus HIVBaL (kindly provided by Dr. Eric Arts of Case Western Reserve/University Hospitals Cleveland Center for AIDS Research) was performed as follows: triplicate wells of 200,000 cells/well (1×106 cell/mL) in a 96-well tissue culture plate were cultured in the following conditions: (1) media alone, (2) immediate HIVBaL exposure with subsequent culture in media alone (virus plus media), (3) immediate HIVBaL exposure with subsequent culture in media containing 5.0 µg/mL PHA (Sigma-Aldrich; virus plus PHA), (4) 3 days of culture in media followed by HIVBaL exposure (media plus virus), (5) 3 days of culture in media containing pooled MSP1-42 peptides at 10 µg/mL each followed by HIVBaL exposure (malaria plus virus), and (6) 3 days of culture in media containing 5.0 mg/ mL PHA followed by HIVBaL exposure (PHA plus virus). Initially, a dose response curve was created to determine the optimal multiplicity of infection (MOI) for infection (see Results). Subsequently, cells were exposed to virus at an MOI of 0.001 for 4 h at 37°C after which cells were centrifuged, washed with fresh media, and resuspended according to the conditions outlined above. Every 3 days, one-half of the culture volume was removed, frozen, and replaced with fresh media containing appropriate supplements.
Reverse transcriptase (RT) assay. The removed culturemedia was stored at −80°C prior to the RT assay, which was performed according to standard protocols [34]. In brief, 10 µL of culture supernatant was incubated with 25 µL of RT mix composed of Tris-HCl, KCl, dithiothretiol (DTT), 32P thymidine- triphosphate (dTTP), Poly r(A) template, poly d(T) primer, nonidet-P40, and α-32PdTTP for 2 h at 37°C. Ten µL of this reaction mixture was spotted onto Whatman filter paper, which was washed successively with 1 × saline-sodium citrate and 85% ethanol and allowed to dry. A known quantity of virus stock was included in all RT assays to verify consistency. Counts per min per microliter culture supernatant (cpm/µL) were quantified using a beta counter.
Flow analysis. One million cells were washed and stained with fluorochrome-labeled antibodies for 30 min in the dark at 4°C. For analysis of HIV infection, the following antibodies were used: CD3-PerCP (BD Biosciences); CD4-APC-Cy7, CD14-PE (eBioscience); and p24-FITC (Beckman Coulter). For analysis of CD4+ T cell activation markers the following antibodies were used: CD3-PerCP, Ki-67-PE (BD Biosciences); CD4-APC-Cy7, CD25-PE-Cy7, HLA-DR-FITC, CCR5-APC (eBioscience). Samples were processed (50,000 events) on a BD LSRII flow cytometer, and data was analyzed with FlowJo software (Tree Star). Gating was determined by fluorescence minus 1 staining, and compensation was calculated using CompBeads (BD Biosciences) and compensation platform in FlowJo. Integrated mean fluorescent indices were calculated as the product of percentage of positive cells and the geometric mean fluorescence intensity of the positive cells.
Statistical analysis. Fisher's exact test was used to make comparisons between groups for frequency of infection. Differences in peak viral replication between groups were determined by either Student's t test or analysis of variance (ANOVA). For expression of activation markers by flow cytometry, integrated mean fluorescent indices were log transformed and compared by ANOVA.
Results
Determination of optimal MOI. To determine the optimal MOI for distinguishing potential differences in susceptibility to HIV in vitro, CBMCs from Kenyan (n = 6) and North American (n = 3) subjects were exposed to varying viral quantities (Figure 1). Maximal viral replication was observed at day 15 for all samples, as indicated by incorporation of α-32P-dTTP (data not shown). At day 15, an MOI of 0.05 infected all samples tested, whereas an MOI of 0.001 infected 4 of 6 Kenyan CBMCs and none of the North American CBMCs. This difference in infection at a lower MOI suggested heightened susceptibility to HIV in Kenyan CBMCs, compared with North American CBMCs.
Figure 1.

Multiplicity of infection (MOI) dose response curve of human immunodeficiency virus BaL (HIVBaL). 32P-dTTP incorporation for individual cord blood mononuclear cell samples at varying MOI. Six samples were from Kenyan subjects, and 3 samples were from North American subjects. Mean 32P-dTTP incorporation was statistically significantly different between Kenyans and North Americans at an MOI of 0.001 (P = .02, by Student's t test).
Kenyan CBMCs are more susceptible to in vitro HIV infection than are North American CBMCs. To further examine this increased susceptibility of CBMCs from Kenyan newborns, compared with CBMCs from North American newborns, to HIV in vitro, additional cryopreserved CBMCs from Kenyan (n = 60) and North American newborns (n = 25) were examined. Samples were considered to be infected for a given culture if the measured cpm/µL was at least 5-fold greater than the no virus condition at 2 or more time points. In the absence of any exogenous stimulation (virus plus media), CBMCs from Kenyan newborns were significantly more likely to be infected with HIV, compared with North American CBMCs (21 of 60 vs 3 of 25 samples; P = .03; Figure 2A). This difference was further enhanced when viral exposure was followed by exogenous stimulation (virus plus PHA) (36 of 60 vs 6 of 25 samples; P = .004). PHA stimulation preceding viral exposure (PHA plus virus) resulted in HIV infection of 59 of 60 (98%) Kenyan and 25 of 25 (100%) North American CBMC samples.
Figure 2.

Cord blood mononuclear cells (CBMCs) from Kenyan newborns demonstrate increased susceptibility to in vitro human immunodeficiency virus (HIV) infection, compared with CBMCs from North American newborns. The frequency of infection (A) and levels of viral replication (B) of in vitro CBMCs in media alone (virus + media), HIV exposure prior to addition of phytohemagglutinin (PHA; virus + PHA), and PHA stimulation proceeding viral exposure (PHA + virus). The frequency of infection (C) and levels of viral replication (D) of in vitro Kenyan CBMCs stratified by prenatal immune sensitization status, as described in the Materials and Methods section, is shown. In B and D, lines indicate mean counts per min per microliter culture supernatant (cpm/µL) of samples meeting the definition of productive HIV infection. P values were determined by Fisher's exact test. Thirty samples were malaria sensitized, 30 samples were malaria nonsensitized, and 25 samples were from North American newborns.
Peak virus replication, as indicated by incorporation of α-32P-dTTP, was observed at day 15 for all samples and all culture conditions except PHA stimulation preceding viral exposure (PHA plus virus) when the peak was consistently observed at day 12. Among samples that were infected, peak viral production tended to be higher in Kenyan CBMC samples than in North American control samples at day 15 for virus plus media and virus plus PHA conditions, although differences were not statistically significant (Figure 2B).
To confirm the reproducibility of results, a second aliquot of Kenyan (n = 10) and North American (n = 10) CBMCs was thawed and infected as before. These replicates exactly match the results from earlier experiments in terms of classification of productive HIV infection, whereas peak levels of viral replication, as determined by RT assay, were also consistent (<20% difference in cpm/µL between replicate experiments; data not shown).
CBMCs of newborns sensitized to malaria antigens have increased HIV susceptibility. There was no clear association of either parity or maternal malaria, schistosomiasis, or geohelminth infection during pregnancy with increased CBMC susceptibility to HIV infection (Table 1). To examine the possibility that in utero immune priming to parasite antigens may increase susceptibility of Kenyan CBMCs to HIV infection in vitro, individual samples were stratified on the basis of prenatal immune priming to malaria blood stage antigens (Figure 2C and 2D). One-half of Kenyan CBMC samples (30 [50%] of 60 samples) demonstrated recall responses to ⩾2 malaria antigens and were defined as “malaria sensitized” (Figure 3). Samples classified as “malaria nonsensitized” generally did not produce any malaria antigen-driven cytokine. A few samples, however, demonstrated an isolated recall response to a single antigen, thereby failing to meet our criteria for immune priming.
Table 1.

Association of Maternal Parasite Infection and Parity with Susceptibility of Cord Blood Mononuclear Cells (CBMCs) from their Offspring to Human Immunodeficiency Virus (HIV) Infection in vitro
Figure 3.
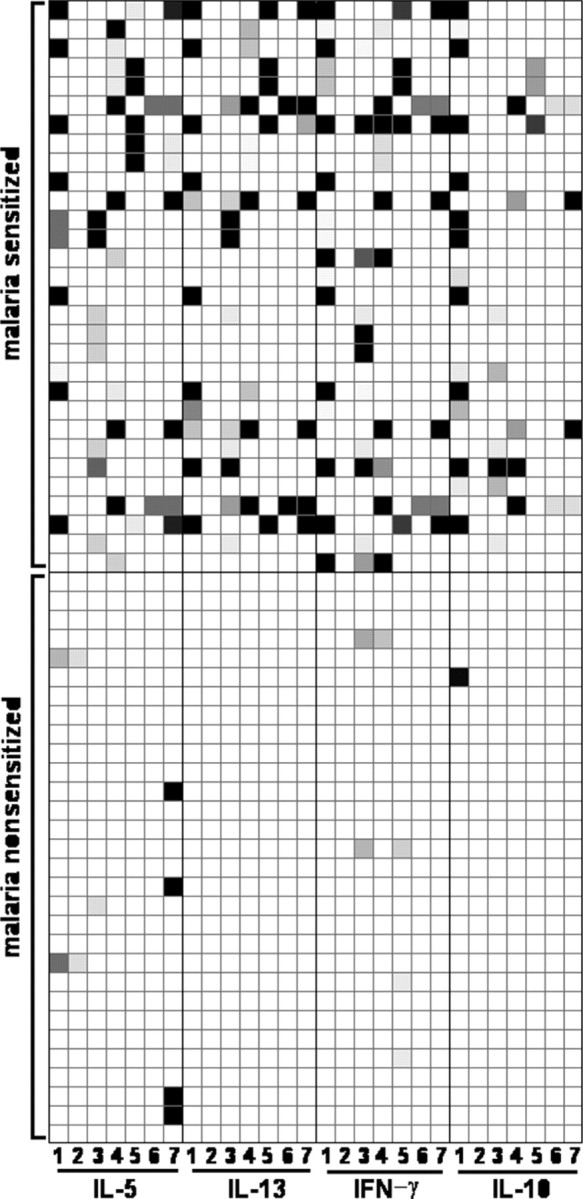
Cytokine production profiles by Kenyan cord blood mononuclear cell (CBMC) samples in response to recall stimulation with malaria antigens. Each row represents an individual CBMC sample, and each column represents a cytokine:antigen combination. The grayscale corresponds to varying levels of cytokine production, with the darkest color indicating the greatest production. Malaria antigens used for the columns were as follows: column 1, recombinant MSP142-3D7; column 2, recombinant MSP142-FVO; column 3, pooled MSP1 peptides 2+3; column 4, PfP0; column 5, MSP142 peptide K49, column 6, MSP142 peptide K30; and column 7, MSP142 peptide M42. Figures depicting recall responses were created using Mayday software [35]. IFN, interferon; IL, interleukin.
Malaria-sensitized CBMCs were significantly more likely to be infected with HIV in vitro in the absence of exogenous stimulation (virus plus media) when compared with either malaria- nonsensitized (15 of 30 vs 6 of 30 samples; P = .03) or to North American CBMC samples (15 of 30 vs 3 of 25 samples; P = .004; Figure 2C). Although malaria-sensitized and malarianonsensitized CBMCs were similarly susceptible when virus exposure was followed by PHA stimulation (20 of 30 vs 16 of 30 samples; P = .43), malaria sensitized CBMC samples remained more likely to be infected, compared with North American CBMC samples (20 of 30 vs 6 of 25; P = .003). Among infected CBMC samples, peak viral replication was similar for malaria-sensitized versus malaria-nonsensitized CBMC samples (Figure 2D).
Stimulation with malaria antigens increases susceptibility of malaria-sensitized CBMCs to HIV infection. We further examined whether recall responses to malaria blood stage antigens increased susceptibility to HIV infection in vitro. Before virus exposure, a subset of CBMC samples from newborns described in Figure 2 (samples with sufficient numbers of frozen CBMCs) were exposed to pooled MSP1-42 peptides previously demonstrated to be dominant T cell epitopes in CBMCs from newborns on the coast of Kenya [33]. In contrast to the addition of virus to CBMCs at the beginning of culture, preculture of CBMCs with media alone for 3 days prior to virus exposure failed to establish a productive infection in most samples from both Kenyan and North American subjects (4 of 20 vs 2 of 20 samples; Figure 4A). As expected, the addition of malaria peptides to North American CBMCs failed to enhance CBMC susceptibility to HIV infection (1 of 20 samples). In contrast, the addition of malaria peptides to CBMC samples classified as malaria sensitized showed a trend toward augmented HIV susceptibility, compared with parallel cultures of malaria-nonsensitized CBMCs (11 of 20 vs 6 of 20 samples; P = .2; Figure 4A). Malaria antigen- and PHA-driven Kenyan CBMC cultures infected with HIV produced similar levels of virus production, regardless of malaria sensitization status (Figure 4B).
Figure 4.

Pretreatment with malaria peptides increases susceptibility to in vitro human immunodeficiency virus (HIV) infection of malaria-sensitized Kenyan cord blood mononuclear cells (CBMCs). A, Frequency of CBMC samples productively infected with HIV in vitro. B, 32P-dTTP incorporation for individual CBMC samples. Lines indicate mean counts per min per microliter culture supernatant (cpm/µL) of samples with productive HIV infection. Twenty samples were used for all conditions except for in vitro CBMCs in media alone (media + virus), for which 10 malaria-sensitized samples and 10 malaria-nonsensitized samples were used. Malaria + virus, 3 days of culture in media containing pooled MSP1-42 peptides at 10 µg/mL each followed by HIVBaL exposure; PHA + virus, phytohemagglutinin stimulation preceeding viral exposure; media + virus, in vitro CBMCs in media alone for 3 days preceding viral exposure.
CD4+ T cells are a primary reservoir for HIV at day 4 in vitro. Prenatal sensitization to malaria antigens has been shown to be predominantly attributable to CD4+ T cell responses [28]. Therefore, we hypothesized that CD4+ T cells were the early target of HIV infection in this in vitro model. Flow cytometric analysis of p24 expression was performed for CD3+CD4+ T cells and CD14+ cells from malaria-sensitized samples known to be susceptible to HIV infection in the absence of exogenous stimulation (n = 5). Expression of p24 was undetectable in all samples at 24 h after virus exposure. However, at 96 h after virus exposure, 1.3% of CD3+ lymphocytes stained positively for p24 (Figure 5). Additional analysis indicated that all CD3+ cells that expressed p24 also expressed CD4 (data not shown). p24 was not detected in CD14+ cells (data not shown). Thus, productive viral infection is primarily detectable in CD3+CD4+ cells at 96 h after virus exposure.
Figure 5.

Detection of p24 in CD3+ T cells of malaria sensitized Kenyan cord blood mononuclear cells (CBMCs) by flow cytometry at 96 h. Gated on viable lymphocytes. CD3 expression is plotted on the y-axis, and p24 expression is plotted on the x-axis. Left panel, no virus exposure (negative control); right panel, in vitro CBMCs in media alone (virus + media). Plots shown are representative of data from 5 samples.
Ex vivo flow staining of activation markers on CD4+ T cells. To determine whether malaria-sensitized newborns had evidence of increased immune activation, compared with malarianonsensitized or North American CBMC samples, ex vivo flow cytometric analysis of various CD4+ T cell activation markers was performed (n = 10 for each group). Little ex vivo proliferation, measured by Ki-67 expression, was observed, with no differences between groups (data not shown). Similarly, no differences were observed in the expression of CCR5 on CD4+ cells (data not shown). Malaria-sensitized CD3+CD4+ T cells demonstrated higher frequency and expression levels of CD25 (integrated mean fluorescent index, 6224) compared with malaria- nonsensitized (2049; P = .04) and North American CBMCs (1688; P = .04; Figure 6). Similarly, the frequency and expression of HLA-DR on CD4+ T cells was highest for malariasensitized CBMCs (integrated mean fluorescent index, 995), compared with malaria-nonsensitized (304;P = .05) andNorthAmerican CBMCs (328; P = .03). Thus malaria-sensitizedCBMCs contained CD4+ T cells that demonstrated increased ex vivo activation, which correlated with increased susceptibility to HIV infection in vitro.
Figure 6.

Expression of CD25 and HLA-DR are increased on CD3+CD4+ cord blood mononuclear cells (CBMCs) from malaria-sensitized Kenyans ex vivo. Integrated mean fluorescent indices for CD25 and HLA-DR gated on viable CD3+CD4+ lymphocytes. Ten samples were examined for each group. Lines indicate geometric mean values. Statistics calculated by analysis of variance.
Discussion
Here, we show that CBMCs from Kenyan newborns in an area where multiple chronic parasitic infections are endemic are more susceptible to infection with an R5-tropic strain of HIV in vitro than are CBMCs from offspring born in North America. This increased susceptibility of Kenyan CBMCs to HIV infection was associated with in utero priming to malaria blood stage antigens and upregulation of CD25 and HLA-DR on CD4+ T cells. Stimulation of malaria-sensitized CBMCs further enhanced their susceptibility to HIV infection in vitro. HIV infected women are often coinfected with malaria and other chronic helminthic infections, which may lead to activation of fetal lymphocytes and, therefore, potentially increase the risk of MTCT. Thus prevention of malaria and chronic helminth infections (such as schistosomiasis, lymphatic filariasis, and geohelminths in pregnant women) may reduce MTCT, especially in conditions in which antiretroviral treatment may be difficult to reliably administer during pregnancy.
The failure to observe an association of maternal malaria or helminth infection with in vitro susceptibility to HIV is not surprising, because not all women expose their fetus to the parasite antigens necessary for in utero priming. The circumstances that might affect in utero sensitization are not clear, but it is likely affected by the duration, timing, and intensity of the parasite infection during gestation. Additional factors may include the integrity of the placenta and the pregnant woman's ability to generate appropriate antibodies to certain parasite antigens, because we have shown that immune complexes may be an important mechanism for transplacental transfer of malaria antigens [36].
Viral dose affected CBMC susceptibility to infection. At higher viral doses, all CBMCs tested became infected in vitro; however, at lower viral doses, more Kenyan samples thanNorth American samples remained susceptible. This increased susceptibility of Kenyan CBMCs occurred in the absence of exogenous stimulation, implicating in utero activation to exogenous antigens, particularly malaria and potentially other intravascular parasites, such as schistosomiasis and lymphatic filariasis, that are common in our population. In the event that the fetus or newborn should be exposed to virus, particularly at lower viral doses, this prenatal priming may increase the likelihood of MTCT. Importantly, when strong polyclonal activation preceded virus exposure in vitro, both Kenyan and North American samples were equally susceptible to infection.
One potential mechanism for fetal activation leading to increased viral susceptibility is by upregulation of CCR5, thereby facilitating viral invasion [37, 38]. Interestingly, such upregulation was not observed in the present study. This suggests that viral entry may not be the limiting step, which is an observation that is supported by similar levels of HIV minus strand strongstop DNA detected by real-time quantitative polymerase chain reaction 24 h after virus exposure among Kenyan and North American CBMCs (unpublished data). Rather, activated cells are likely to have greater pools of nucleotide precursors necessary for viral progression through reverse transcription, and/ or proviral gene transcription and translation is enhanced by host cell transcription factors, such as nuclear factor kB, activation protein 1, and/or nuclear factor of activated T cells [39]. Although p24 was detected only in CD3+CD4+ T cells 4 days after virus exposure, the initial cell type infected in CBMCs remains unclear. Such studies are difficult because of the very low frequency of initially infected cells (unpublished data).
There are several limitations to the study. It is generally accepted that initial HIV infection occurs at mucosal surfaces [40–44], with dissemination to lymph nodes, where CD4+ cells act a reservoir [45–47]. Circulating CBMCs may not reflect those lymphocytes observed either at mucosal surfaces or in lymph nodes. Thus, our in vitro model of HIV infection may not fully represent in vivo conditions. In addition, although maternal infection with helminths during gestation was documented and failed to account for differences in observed in vitro HIV susceptibility, fetal sensitization to helminth antigens was not fully assessed. Because the prevalence of maternal helminth infection was high in the present cohort, and because we have previously shown that helminth-sensitized newborns born of HIV-positive mothers had an increased risk of MTCT in vivo [31], we cannot rule out the likelihood of fetal helminth sensitization contributing to the observed increased HIV susceptibility in vitro for Kenyan CBMCs.
In conclusion, this study demonstrates that CBMCs from Kenyan newborns primed in utero to parasite antigens aremore likely to become infected with HIV. Prevention of malaria and other chronic parasitic infections in HIV-positive pregnant women to block prenatal sensitization could reduce the risk of MTCT.
Acknowledgments
We thank medical superintendent Dr. Dawood Mwaura and nurses Victoria Saidi, Hashora Mwanguku, Zaituni Mwakileo, Fatuma Ngare, Ruth Notina, and Florence Wambua, for assisting with recruitment of women to the study, collection of cord blood samples, and care of the women and their newborns; Elton Mzungu, Kefar Wambua, Charles NgaNga, and Alex Osore, who performed many of the immunologic assays and parasitological examinations; Grace Methenge and Christine Lucas, for data entry and management; Drs. Zahra Toossi and Eric Arts, for assistance with the study concept and critical reading of the manuscript; and the women residing in the Msambweni location who participated in the study.
Footnotes
Potential conflicts of interest: none reported.
Financial support: National Institutes of Health (NIH) (grant AI064687), Merit Review Award from the Veterans Affairs Research Service, Case MSTP (NIH grant T32 GM07250 to K.S.), Case Geographic Medicine and Infectious Diseases Training Grant (NIH grant T32 A107024 to K.S.), and Case/University Hospitals of Cleveland Center for AIDS Research (NIH grant number: AI36219).
Presented in part: American Society for Tropical Medicine and Hygiene 58th Annual Meeting, November 2009, Washington, DC [abstract 368].
References
- 1.Abu-Raddad L, Patnaik P, Kublin J. Dual infection with HIV and malaria fuels the spread of both diseases in sub-Saharan Africa. Science. 2006;314(5805):1603–1606. doi: 10.1126/science.1132338. [DOI] [PubMed] [Google Scholar]
- 2.UNAIDS . Geneva, Switzerland: UNAIDS; 2007. AIDS Epidemic Update: 2007. [Google Scholar]
- 3.Dreyfuss ML, Msamanga GI, Spiegelman D, Hunter DJ, Urassa EJ, Hertzmark E, Fawzi WW. Determinants of low birth weight among HIVi-nfected pregnant women in Tanzania. Am J Clin Nutr. 2001;74(6):814–826. doi: 10.1093/ajcn/74.6.814. [DOI] [PubMed] [Google Scholar]
- 4.Zijenah L, Mbizvo M, Kasule J, et al. Mortality in the first 2 years among infants born to human immunodeficiency virus-infected women in Harare, Zimbabwe. J Infect Dis. 1998;178(1):109–113. doi: 10.1086/515604. [DOI] [PubMed] [Google Scholar]
- 5.Taha TET, Dallabetta GA, Canner JK, et al. The effect of human immunodeficiency virus infection on birthweight, and infant and child mortality in urban Malawi. Int J Epidemiol. 1995;24(5):1022–1029. doi: 10.1093/ije/24.5.1022. [DOI] [PubMed] [Google Scholar]
- 6.Parise ME, Ayisi JG, Nahlen BL, et al. Efficacy of sulfadoxine-pyrimethamine for prevention of placental malaria in an area of Kenya with a high prevalence of malaria and human immunodeficiency virus infection. Am J Trop Med Hyg. 1998;59(5):813–822. doi: 10.4269/ajtmh.1998.59.813. [DOI] [PubMed] [Google Scholar]
- 7.Matteelli A, Caligaris S, Castelli F, Carosi G. The placenta and malaria. Ann Trop Med Parasitol. 1997;91(7):803–810. doi: 10.1080/00034989760563. [DOI] [PubMed] [Google Scholar]
- 8.Bulmer J, Rasheed F, Morrison L, Fancis N, Greenwood B. Placental malaria. II. A semi-quantitative investigation of the pathological features. Histopathology. 1993;22(3):219–225. doi: 10.1111/j.1365-2559.1993.tb00111.x. [DOI] [PubMed] [Google Scholar]
- 9.Inion I, Mwanyumba F, Gaillard P, et al. Placental malaria and perinatal transmission of human immunodeficiency virus type 1. J Infect Dis. 2003;188(11):1675–1678. doi: 10.1086/379737. [DOI] [PubMed] [Google Scholar]
- 10.van Eijk A, Ayisi J, ter Kuile F, et al. HIV increases the risk of malaria in women of all gravidities in Kisumu, Kenya. AIDS. 2003;17(4):595–603. doi: 10.1097/00002030-200303070-00015. [DOI] [PubMed] [Google Scholar]
- 11.Ladner J, Leroy V, Simonon A, et al. HIV infection, malaria, and pregnancy: A prospective cohort study in Kigali, Rwanda. Am J Trop Med Hyg. 2002;66(1):56–60. doi: 10.4269/ajtmh.2002.66.56. [DOI] [PubMed] [Google Scholar]
- 12.ter Kuile F, Parise M, Verhoeff F, et al. The burden of co-infection with human immunodeficiency virus type 1 and malaria in pregnant women in sub-saharan Africa. Am J Trop Med Hyg. 2004;71(2):41–54. [PubMed] [Google Scholar]
- 13.Brahmbhatt H, Sullivan D, Kigozi G, et al. Association of HIV and malaria with mother-to-child transmission, birth outcomes, and child mortality. J Acquir Immune Defic Syndr. 2008;47(4):472–476. doi: 10.1097/QAI.0b013e318162afe0. [DOI] [PubMed] [Google Scholar]
- 14.Ticconi C, Mapfumo M, Dorrucci M, et al. Effect of maternal HIV and malaria infection on pregnancy and perinatal outcome in Zimbabwe. J Acquir Immune Defic Syndr. 2003;34(3):289–294. doi: 10.1097/00126334-200311010-00005. [DOI] [PubMed] [Google Scholar]
- 15.Ayisi JG, van Eijk AM, ter Kuile FO, et al. The effect of dual infection with HIV and malaria on pregnancy outcome in western Kenya. AIDS. 2003;17(4):585–594. doi: 10.1097/00002030-200303070-00014. [DOI] [PubMed] [Google Scholar]
- 16.Mwapasa V, Rogerson S, Molyneux M, et al. The effect of Plasmodium falciparum malaria on peripheral and placental HIV-1 RNA concentrations in pregnant Malawian women. AIDS. 2004;18(7):1051–1059. doi: 10.1097/00002030-200404300-00014. [DOI] [PubMed] [Google Scholar]
- 17.Kublin JG, Patnaik P, Jere CS, et al. Effect of Plasmodium falciparum malaria on concentration of HIV-1-RNA in the blood of adults in rural Malawi: a prospective cohort study. Lancet. 2005;365(9455):233–240. doi: 10.1016/S0140-6736(05)17743-5. [DOI] [PubMed] [Google Scholar]
- 18.Fawzi W, Msamanga G, Renjifo B, et al. Predictors of intrauterine and intrapartum transmission of HIV-1 among Tanzanian women. AIDS. 2001;15(9):1157–1165. doi: 10.1097/00002030-200106150-00011. [DOI] [PubMed] [Google Scholar]
- 19.Mofenson LM, Lambert JS, Stiehm ER, et al. Risk factors for perinatal transmission of human immunodeficiency virus type 1 in women treated with zidovudine. N Engl J Med. 1999;341(6):385–393. doi: 10.1056/NEJM199908053410601. [DOI] [PubMed] [Google Scholar]
- 20.Ned R, Moore J, Chaisavaneeyakorn S, Udhayakumar V. Modulation of immune responses during HIV-malaria co-infection in pregnancy. Trends Parasitol. 2005;21(6):284–291. doi: 10.1016/j.pt.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 21.Brahmbhatt H, Kigozi G, Wabwire-Mangen F, et al. The effects of placental malaria on mother-to-child HIV transmission in Rakai, Uganda. AIDS. 2003;17(17):2539–2541. doi: 10.1097/00002030-200311210-00020. [DOI] [PubMed] [Google Scholar]
- 22.Ayouba A, Nerrienet E, Menu E, et al. Mother-to-child transmission of human immunodeficiency virus type 1 in relation to the season in Yaounde, Cameroon. Am J Trop Med Hyg. 2003;69(4):447–449. [PubMed] [Google Scholar]
- 23.Ayisi J, van Eijk A, Newman R, et al. Maternal malaria and perinatal HIV transmission, western Kenya. Emerg Infect Dis. 2004;10(4):643–652. doi: 10.3201/eid1004.030303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zack J, Arrigo S, Weitsman S, Go A, Haislip A, Chen I. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell. 1990;61(2):213–222. doi: 10.1016/0092-8674(90)90802-l. [DOI] [PubMed] [Google Scholar]
- 25.Zack J, Haislip A, Krogstad P, Chen I. Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. J Virol. 1992;66(3):1717–1725. doi: 10.1128/jvi.66.3.1717-1725.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malhotra I, Ouma J, Wamachi A, et al. In utero exposure to helminth and mycobacterial antigens generates cytokine responses similar to that observed in adults. J Clin Invest. 1997;99(7):1759–1766. doi: 10.1172/JCI119340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ismaili J, van der Sande M, Holland M, et al. Plasmodium falciparum infection of the placenta affects newborn immune responses. Clin Exp Immunol. 2003;133(3):414–421. doi: 10.1046/j.1365-2249.2003.02243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malhotra I, Mungai P, Muchiri E, et al. Distinct Th1- and Th2-Type prenatal cytokine responses to Plasmodium falciparum erythrocyte invasion ligands. Infect Immun. 2005;73(6):3462–3470. doi: 10.1128/IAI.73.6.3462-3470.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xi G, Leke R, Thuita L, et al. Congenital exposure to Plasmodium falciparum antigens: prevalence and antigenic specificity of in utero-produced antimalarial immunoglobulin M antibodies. Infect Immun. 2003;71(3):1242–1246. doi: 10.1128/IAI.71.3.1242-1246.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malhotra I, Mungai P, Muchiri E, Kwiek J, Meshnick S, King C. Umbilical cord-blood infections with Plasmodium falciparum Malaria Are Acquired Antenatally in Kenya. J Infect Dis. 2006;194(2):176–183. doi: 10.1086/505150. [DOI] [PubMed] [Google Scholar]
- 31.Gallagher M, Malhotra I, Mungai P, et al. The effects of maternal helminth and malaria infections on mother-to-child HIV transmission. AIDS. 2005;19(16):1849–1855. doi: 10.1097/01.aids.0000189846.90946.5d. [DOI] [PubMed] [Google Scholar]
- 32.Malhotra I, Dent A, Mungai P, et al. Can prenatal malaria exposure produce an immune tolerant phenotype?: a prospective birth cohort study in Kenya. PLoS Med. 2009;6(7):e1000116. doi: 10.1371/journal.pmed.1000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malhotra I, Wamachi AN, Mungai PL, et al. Fine specificity of neonatal lymphocytes to an abundant malaria blood-stage antigen: epitope mapping of Plasmodium falciparum MSP133. J Immunol. 2008;180(5):3383–3390. doi: 10.4049/jimmunol.180.5.3383. [DOI] [PubMed] [Google Scholar]
- 34.Ostrowski M, Chun T, Cheseboro B, Stanley S, Tremblay M. Current protocols in immunology. New York, NY: John Wiley & Sons; 2007. Unit 12.5: detection assays for HIV proteins. [DOI] [PubMed] [Google Scholar]
- 35.Battke F, Symons S, Nieselt K. Mayday- integrative analytics for expression data. BMC Bioinformatics. 2010;11(1):121. doi: 10.1186/1471-2105-11-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.May K, Grube M, Malhotra I, et al. Antibody-dependent transplacental transfer of malaria blood-stage antigen using a human ex vivo placental perfusion model. PLoS ONE. 2009;4(11):e7986. doi: 10.1371/journal.pone.0007986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu L, Paxton WA, Kassam N, et al. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1 in vitro. J Exp Med. 1997;185(9):1681–1692. doi: 10.1084/jem.185.9.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paxton WA, Liu R, Kang S, et al. Reduced HIV-1 infectability of CD4+lymphocytes from exposed-uninfected individuals: association with low expression of CCR5 and high production of [beta]-chemokines. Virology. 1998;244(1):66–73. doi: 10.1006/viro.1998.9082. [DOI] [PubMed] [Google Scholar]
- 39.Lawn S, Butera S, Folks T. Contribution of immune activation to the pathogenesis and transmission of human immunodeficiency virus type 1 infection. Clin Microbiol Rev. 2001;14(4):753–777. doi: 10.1128/CMR.14.4.753-777.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chuachoowong R, Shaffer N, Siriwasin W, et al. Short-course antenatal zidovudine reduces both cervicovaginal human immunodeficiency virus type 1 RNA levels and risk of perinatal transmission. J Infect Dis. 2000;181(1):99–106. doi: 10.1086/315179. [DOI] [PubMed] [Google Scholar]
- 41.John G, Nduati R, Mbori-Ngacha D, et al. Correlates of mother-to-child human immunodeficiency virus type 1 (HIV-1) transmission: association with maternal plasma HIV-1 RNA load, genital HIV-1DNA shedding, and breast infections. J Infect Dis. 2001;183(2):206–212. doi: 10.1086/317918. [DOI] [PubMed] [Google Scholar]
- 42.Tuomala R, O'riscoll P, Bremer J, et al. Cell-associated genital tract virus and vertical transmission of human immunodeficiency virus type 1 in antiretroviral-experienced women. J Infect Dis. 2003;187(3):375–384. doi: 10.1086/367706. [DOI] [PubMed] [Google Scholar]
- 43.Van de Perre P. Mother-to-child transmission of HIV-1: the ‘all mucosal’hypothesis as a predominant mechanism of transmission. AIDS. 1999;13(9):1133–1138. doi: 10.1097/00002030-199906180-00018. [DOI] [PubMed] [Google Scholar]
- 44.Gaillard P, Verhofstede C, Mwanyumba F, et al. Exposure to HIV-1 during delivery and mother-to-child transmission. AIDS. 2000;14(15):2341–2348. doi: 10.1097/00002030-200010200-00015. [DOI] [PubMed] [Google Scholar]
- 45.Pope M, Haase AT. Transmission, acute HIV-1 infection and the quest for strategies to prevent infection. Nat Med. 2003;9(7):847–852. doi: 10.1038/nm0703-847. [DOI] [PubMed] [Google Scholar]
- 46.Schacker T, Little S, Connick E, et al. Rapid accumulation of human immunodeficiency virus (HIV) in lymphatic tissue reservoirs during acute and early HIV infection: implications for timing of antiretroviral therapy. J Infect Dis. 2000;181(1):354–357. doi: 10.1086/315178. [DOI] [PubMed] [Google Scholar]
- 47.Schacker T, Little S, Connick E, et al. Productive infection of T cells in lymphoid tissues during primary and early human immunodeficiency virus infection. J Infect Dis. 2001;183(4):555–562. doi: 10.1086/318524. [DOI] [PubMed] [Google Scholar]