

Abstract
Hepatic fibrosis, a disease characterized by altered accumulation of extracellular matrix, can cause cirrhosis and liver failure. There is growing interest in the impact of coactivators on hepatic fibrogenesis. Here, we provided genetic evidence that mice lacking steroid receptor coactivator-3 (SRC-3) were protected against carbon tetrachloride (CCl4) induced acute liver necrosis and chronic hepatic fibrosis. After acute CCl4 treatment, SRC-3−/− mice showed attenuated profibrotic response and hepatocyte apoptosis, while hepatocyte proliferation was elevated in SRC-3−/− mice versus SRC-3+/+ mice. Similarly, chronically CCl4-treated SRC-3−/− mice showed significant weakening of inflammatory infiltrates, hepatic stellate cell activation and collagen accumulation in the liver compared to SRC-3+/+ mice. Further investigation revealed that TGFβ1/Smad signaling pathway was impaired in the absence of SRC-3. Moreover, the expression levels of SRC-3, as assessed in human tissue microarray of liver diseases, correlated positively with degrees of fibrosis. These data revealed that SRC-3−/− mice were resistant to CCl4 induced acute and chronic hepatic damage and TGFβ1/Smad signaling was suppressed in the lack of SRC-3. Our results established an essential involvement of SRC-3 in liver fibrogenesis, which might provide new clues to the future treatment of hepatic fibrosis.
Keywords: Chronic hepatic fibrosis, Smad2/3, Steroid receptor coactivator-3, Tissue microarray, Transforming growth factor β1
Hepatic fibrosis is usually resulted from prolonged liver injury caused by chronic hepatitis, alcohol or chemical insults. Cirrhosis, as the end stage of hepatic fibrosis, is a major clinical issue for its high prevalence in the world and its tight relationship with hepatocellular carcinoma incidence. Thus, great research efforts have been aimed on elucidating hepatic fibrogenesis, and eventually, finding a therapeutic way to abrogate its progression. 1, 2
Extensive studies have characterized hepatic fibrosis as an excessive and aberrant deposition of extracellular matrix (ECM) proteins in the liver, the most abundant of which is the collagen family.3 Transforming growth factor β (TGFβ), a profibrotic cytokine with potent collagen synthesis stimulatory effect, plays a fundamental role through its intracellular signal transducers Smads in the pathogenesis of hepatic fibrosis. After TGFβ-induced activation of the TGFβ receptor, Smads are phosphorylated and translocate into the nucleus to transactivate expression of profibrotic target genes, i.e. collagen type I.4
Through decades of research, numerous molecules have been identified to be involved in the TGFβ/Smad-mediated fibrogenesis.5 Among them, a relatively new and thriving branch of work is focused on the role of coactivators in the fibrogenesis process. Ghosh AK’s group implicates that coactivator p300/CBP contributes to fibroblast biology, connective tissue homeostasis and fibrosis.6 TGFβ responses are dramatically amplified with ectopic expression of p300/CBP in fibroblasts, whereas selective p300 depletion results in abrogation of TGFβ-induced Smads signaling and subsequent collagen synthesis. Another transcriptional coactivator P/CAF is reported to be able to potentiate transcriptional activity of heterologous Gal4-Smad2 and Gal4-Smad3 fusion proteins and TGF-β/Smad3-induced transcriptional responses in vitro, which can be further enhanced by coactivators p300 and Smad4.7 In addition, Sylviane et al. demonstrate that coactivator SRC-1, one of the p160 family members, works as a novel Smad3/4 transcriptional partner, facilitating the functional links between Smad3 and p300/CBP.8
As one of the members of the p160 coactivator family, the steroid receptor coactivator-3 (SRC-3) is a broad-specificity transcriptional coregulator which mediates the activating functions of nuclear receptors and other transcription factors.9 Studies on the molecular mechanism of SRC-3 transactivation function reveal that SRC-3 interacts with other coactivators, i.e. p300/CBP and P/CAF10 and forms a coactivator complex with them to facilitate gene transcription.
Since new and emerging researches have emphasized an indispensable role of coactivators during Smads-mediated hepatic fibrogenesis and SRC-3 is a coactivator essential for other cofactors like p300/CBP and P/CAF to exert their functions, we determine to investigate whether SRC-3 is involved in hepatic fibrosis through TGFβ/Smad pathway using a genetic SRC-3 ablation mice model. In this study, Carbon tetrachloride (CCl4)-induced mice model was established to reveal the significance of SRC-3 in acute liver necrosis and chronic hepatic fibrosis. Our results demonstrate that SRC-3−/− mice are protected against liver injury and fibrosis development as compared with SRC-3+/+ mice due to, at least in part, a defect in TGFβ/Smad signaling.
MATERIALS AND METHODS
Experimental Animals
The generation of SRC-3−/− mice was described in ref 9. Male, age matched (8–10 weeks old) SRC-3−/− and wild type mice (both in C57BL/6×129Sv background) were used. Animals were maintained in humidity and temperature controlled rooms, kept on a 12h light/dark cycle with free access to food and water. For acute liver damage study, mice were sacrificed 24, 36, 48 and 72 hours, respectively, after a single intraperitoneal (i.p.) injection of 1 ml/kg body weight CCl4 (1:7 v/v in olive oil). For chronic liver fibrosis study, mice were administered i.p. with 0.5 ml/kg body weight CCl4 (1:7 v/v in olive oil) twice per week for 3 and 6 weeks, separately. Mice were sacrificed 3 days after the last CCl4 administration. After sacrifice, blood samples were obtained and livers were harvested for future analysis. No difference was observed in wild type and SRC-3−/− control mice injected i.p. with olive oil in both studies. All experiments were carried out under accepted ethical guidelines.
RNA Isolation and Quantitative PCR
Aliquots of liver tissue were snapped frozen and kept at −80°C until RNA isolation. Total RNA was extracted from mouse livers using TRIzol Reagent (Invitrogen, CA, USA). One microgram of sample RNA was transcribed to cDNA with the Reverse Transcription Kit (Promega Corp., WI, USA). Quantitative PCR was performed on the ABI 7900 Fast Real-Time PCR System (Applied Biosystem, CA, USA) in 10μl volume, using SYBR Premix EX Taq Kit (Takara, Japan) under the manufacturer’s instructions. The sequences of all used primers are available upon request. PCR array data were calculated by the ΔΔCt method, normalized against house keeping gene β-actin and expressed as mean fold change in SRC-3−/− samples relative to SRC-3+/+.
Histological Analysis and Collagen Content Measurement
Liver tissues were fixed in 4% formalin and embedded in paraffin according to standard procedure. Paraffin-embedded tissues were cut 5 μm thick and stained with hematoxylin and eosin (H&E) for morphological analysis or with Sirius Red (Sigma, MO, USA) for collagen content measurement. For computer quantification of collagen deposition, slides were prepared from the tissues of 3 individual mice of each genotype. Six fields were randomly selected per slide and calculated for collagen accumulation using Leica QWin software package under ×10 objective. All slides were examined under the Nikon Eclipse80i microscope with Dxm1200F digital camera.
Terminal dUTP Nick-End Labeling Assay
Paraffin-embedded liver tissues were assayed for DNA fragmentation using a terminal deoxynucleotidyl transferasemediated dUTP-biotin nick end labeling (TUNEL) reaction according to the manufacturer’s instructions (Roche, IN, USA). Results were examined under a fluorescence Nikon Eclipse80i microscope. To quantitate the TUNEL positive cells, slides were prepared from the tissues of 3 individual mice of each genotype and assayed for TUNEL reaction as described above. Ten fields were then randomly selected per slide at 400× magnification. TUNEL positive cells were counted on each field and averaged to give the TUNEL positive cells per field.
Western blotting
Lysates from liver tissues were separated on SDS-PAGE, transferred to nitrocellulose and blotted with primary antibodies directed against Collagen Type I (Boster, Wuhan, China), anti-smooth muscle α-actin (α-SMA, Sigma), TGFβ1 (Sigma), Smad2, phospho-Smad2, Smad3, phospho-Smad3, Cyclin D1, Bcl2-associated X protein (Bax, all purchased from Cell Signaling, MA, USA) and Glyceraldehtde-3-phosphate dehydrogenase (GAPDH, Cell signaling, as loading control) followed by appropriate secondary antibodies and chemiluminescent detection.
Immunohistochemistry
Sections were incubated with anti-proliferating cell nuclear antigen (anti-PCNA, 1/200, Cell signaling) or α-SMA (1/200, Sigma) for 1 hour. Secondary antibody was biotinylated universal secondary antibody (Vector lab, CA, USA). Negative controls were incubated with no primary antibody. After washing, the epitopes were detected with the Vectstain R.T.U. Elite ABC Reagent (Vector lab) and revealed with liquid DAB Substrate Chromogen System (DakoCytomation, Cambridgeshire, UK) under manufacturer’s instructions. For slides stained with PCNA, sections were then stained with hematoxylin to show PCNA negative hepatocytes. To quantitate the PCNA positive rate, slides were prepared from the tissues of 3 individual mice of each genotype and stained with PCNA and hematoxylin as described above. Ten fields were randomly selected per slide at 400× magnification and counted for PCNA positive cells. PCNA positive cell counts were normalized to total cell counts to give the PCNA positive rate.
Human Tissue Microarray
The human liver tissue microarray is commercially provided by Shanghai Biochip Co., Ltd., Shanghai, China (National Engineering Center for Biochip at Shanghai). Microarray construction was described in ref 11.
To assess the extent of fibrosis in samples of viral hepatitis or hepatic cirrhosis, the slide containing the microarray was dewaxed and rehydrated with standard procedure and stained with Sirius Red to show the fibrotic area. The fibrotic area was then graded into four groups based on the overall intensity and the percentage of stained area of each sample (−, negative; +, low; ++, moderate; and +++, strong) at 100× magnification by experienced, unbiased histology technicians. Variations in grading between the two observers were identified and the cases were individually discussed and a final consensus was made. The Kruskal-Wallis test was used to evaluate the difference in the extent of fibrosis (all four groups) between the human samples of viral hepatitis and hepatic cirrhosis.
For SRC-3 immunoreactivity, the slide containing the microarray was processed according to the standard immunohistological procedure with a primary antibody against human SRC-3. The positive SRC-3 nucleus staining rate of each sample was evaluated quantitatively by experienced, unbiased histology technicians and divided into four groups (−, negative; +, <30% positive rate; ++, 30–60% positive rate; and +++, >60% positive rate). The Kruskal-Wallis test was used to evaluate the difference in the intensity of SRC-3 nucleus staining (all four groups) between the human samples of viral hepatitis and hepatic cirrhosis. Correlation between SRC-3 staining intensity and the grades of Sirius Red staining was evaluated by Spearman’s Rank Correlation.
ELISA Assay and Biochemistry Analysis
Enzyme-linked immunosorbent assay (ELISA) was performed with mouse TGFβ1 immunoassay kit (R&D systems, MN, USA) following manufacturer’s instructions. Each sample was assayed in duplicate. Serum alanine transaminase (ALT) activity was measured by the Department of Clinical Chemistry, Ruijin Hospital, using automated procedures.
Statistics
Animal experiments were performed with between 6–10 animals per experimental group. Results were expressed as the mean ± standard error. Comparisons between groups were performed using t-test (GraphPad Prism version 4.03), the Kruskal-Wallis test (SAS 8.0 statistical software) or the Spearman’s Rank Correlation (SPSS 13.0 statistical software). Differences were considered significant if P <0.05.
RESULTS
Mice Lacking SRC-3 are Protected Against Acute CCl4-induced Liver Necrosis
SRC-3+/+ and SRC-3−/− mice were injected i.p. with a single dose of CCl4 and sacrificed after 24, 36, 48 and 72 hours, respectively. As shown in Figure 1a, similar liver necrosis was induced 24h and 36h after CCl4 injection in SRC-3+/+ and SRC-3−/− mice. However, 48h after CCl4 administration, hepatic damage lesion was significantly decreased in SRC-3−/− mice as compared with SRC-3+/+ mice. Hepatic lesion regression continued and 72h after CCl4 injection the necrotic area decreased to a similar extent in SRC-3+/+ and SRC-3−/− mice. Serum ALT levels paralleled the histopathological findings (Figure 1b). Twenty-four hours after CCl4 injection, serum ALT levels elevated substantially in SRC-3+/+ and SRC-3−/− mice. ALT level had a mild but not significant decrease in SRC-3−/− mice compared to SRC-3+/+ mice 36h after CCl4 challenge. When ALT levels started to decrease in mice from both genotypes 48h after the insult, the difference became apparent as ALT level was significantly lower in SRC-3−/− mice than SRC-3+/+ mice at this time point. Then, at 72h, ALT levels dropped back to the levels seen before CCl4 treatment in both SRC-3+/+ and SRC-3−/− mice. Acute CCl4-induced liver injury can also be reflected by the upregulation of proteins involved in the profibrotic response12. As shown in Figure 1c, CCl4 insult-induced elevation of TGFβ1, Collagen Type I and α-SMA expression were significantly suppressed in SRC-3−/− mice 48h after liver intoxication, which was in consistent with our histological and serological findings. These results indicate that SRC-3−/− mice were protected against acute CCl4-induced liver damage, possibly due to an early onset of tissue recovery.
Figure 1.

SRC-3−/− mice are protected against liver necrosis after acute CCl4 administration. (a) Representative H&E stained liver sections from SRC-3+/+ and SRC-3−/− mice 24, 36, 48 and 72 hours after acute CCl4 administration. Liver necrosis in SRC-3−/− mice was significantly alleviated compared to SRC-3+/+ mice 48 hours after the insult (×100 magnification). (b) ALT serum concentrations in SRC-3+/+ and SRC-3−/− mice 24, 36, 48 and 72 hours after acute CCl4 administration. Note the significant reduction in serum ALT level in SRC-3−/− mice compared to SRC-3+/+ mice 48 hours after the insult. (c) Western blot analysis of TGFβ1, Collagen Type I and α-SMA expression levels in livers from SRC-3+/+ and SRC-3−/− mice 24, 36, 48 and 72 hours after acute CCl4 administration. GAPDH was used as an invariant control. All data are mean ± standard error. * *P<0.01, WT vs KO.
Enhanced Hepatocyte Proliferation and Suppressed Hepatocyte Apoptosis in SRC-3−/− Mice after Acute CCl4 Injection
Since acute liver necrosis and profibrotic response were significantly ameliorated in SRC-3−/− mice 48h after CCl4 injection (Figure 1c and Figure 2a), we focused on the CCl4-treated mice from this time point to study the intrinsic mechanism. The increase in TGFβ1 serum level was significantly blunted in SRC-3−/− mice compared to wild type mice (Figure 2b), which was in consistent with its tissue expression level presented in Figure 1c. As the hallmark of liver injury and a major profibrogenic cytokine, TGFβ1 has been reported to be a potent proliferation inhibitor and apoptosis inducer in hepatocytes.13, 14 Thus, hepatocyte proliferation and apoptosis were examined in SRC-3+/+ and SRC-3−/− mice 48h after CCl4 administration. As shown in Figure 2c and 2d, hepatocyte proliferation was significantly elevated in SRC-3−/− mice compared to SRC-3+/+ as demonstrated by PCNA immunochemistry staining on liver sections. Besides necrosis, hepatocytes may undergo apoptosis in response to CCl4 treatment. TUNEL assay revealed a significant reduction in the number of apoptotic hepatocytes in SRC-3−/− mice compared to SRC-3+/+ mice (Figure 2e, 2f). These data suggested that hepatocyte proliferation was enhanced while apoptosis was reduced in SRC-3−/− mice after acute CCl4 injection, probably due to suppressed TGFβ1 induction in the lack of SRC-3.
Figure 2.

Enhanced hepatocyte proliferation and suppressed hepatocyte apoptosis in SRC-3−/− mice. (a) Representative H&E stained liver sections from SRC-3+/+ and SRC-3−/− mice 48 hours after acute CCl4 administration. Liver necrosis was significantly attenuated in SRC-3−/− mice compared to SRC-3+/+ mice (×200 magnification). (b) Quantification of serum TGFβ1 level by ELISA in SRC-3+/+ and SRC-3−/− mice treated acutely with CCl4. (c) Immunohistochemical analysis of PCNA expression in livers from SRC-3+/+ and SRC-3−/− mice treated acutely with CCl4 (×100 magnification). Slides were stained with hematoxylin to show PCNA negative cells. Images were magnified digitally (below) to show details of the staining. Percentages of PCNA positive cells were calculated and shown in Figure 1d. (e) TUNEL staining was performed on liver sections from SRC-3+/+ and SRC-3−/− mice treated acutely with CCl4. Results were examined using fluorescence microscope (×100 magnification). Images were magnified digitally (below) to show details of the staining. TUNEL positive cells per field were calculated and shown in Figure 1f. All data are mean ± standard error. *P<0.05, **P<0.01 and *** P<0.001, WT vs KO.
SRC-3−/− Mice are Protected Against Chronic Hepatic Fibrosis
The enhanced tissue damage repair and the decreased profibrotic response discovered in SRC-3−/− mice after acute CCl4 administration suggested that the knockout mice might also be protected against chronic hepatic fibrosis. To address this question, we subjected SRC-3+/+ and SRC-3−/− mice under prolonged CCl4 treatment. After 3 and 6 weeks of CCl4 administration, SRC-3+/+ mice had prominently more inflammatory infiltration in the liver compared to similarly treated SRC-3−/− mice as demonstrated by histological analysis (Figure 3a) and CD8 immunofluerence analysis (Figure 3b). Similar to mice treated acutely with CCl4, serum TGFβ1 level was lower in SRC-3−/− mice vs. SRC-3+/+ mice (Figure 3c). Next, we examined α-SMA expression in livers from SRC-3+/+ and SRC-3−/− mice as it is a well accepted marker for hepatic stellate cell (HSC) activation. Stronger intensity of α-SMA immunohistological staining and significantly more α-SMA positive cells were observed in liver sections of SRC-3+/+ mice than that of SRC-3−/− mice (Figure 3d). Consistently, α-SMA mRNA expression level was elevated in the livers of SRC-3+/+ mice compared to SRC-3−/− mice (Figure 3e). Sirius red staining for collagen deposition showed that SRC-3+/+ mice developed extensive fibrosis, disclosing a honeycomb pattern of fibrous septa, bridging the centrilobular veins whereas collagen accumulation was greatly subdued in SRC-3−/− mice (Figure 3f). Digital quantification of fibrotic area confirmed this observation (Figure 3g). All these data indicated that under prolonged CCl4 treatment, SRC-3−/− mice were resistant to chronic hepatic fibrosis as demonstrated by the significant reduction in inflammatory infiltration, TGFβ1 secretion, HSC activation and collagen accumulation.
Figure 3.
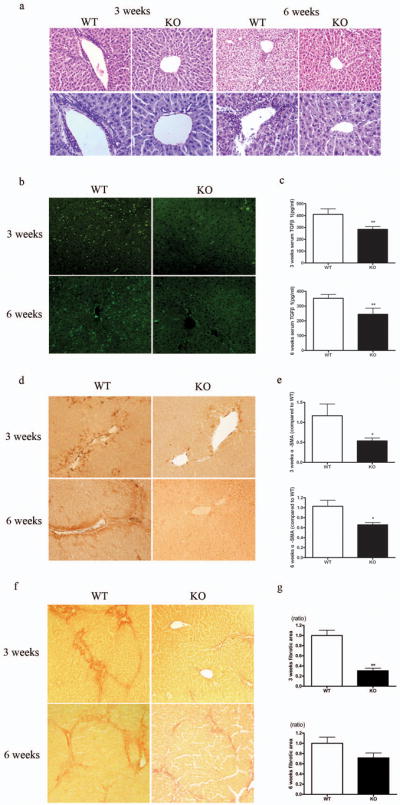
SRC-3−/− mice were protected against chronic hepatic fibrosis. (a) Representative H&E stained liver sections from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 3 and 6 weeks, respectively. Inflammatory infiltration was significantly attenuated in SRC-3−/− mice compared to SRC-3+/+ mice (upper panels, ×100 magnification; lower panels, ×200 magnification). (b) Immunofluerence staining for CD8 positive cells in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 3 and 6 weeks, respectively (×100 magnification). (c) Quantification of serum TGFβ1 level by ELISA in SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 3 and 6 weeks, respectively. (d) Immunohistochemical analysis of α-SMA expression in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 3 and 6 weeks, respectively (×100 magnification). (e) Expression of α-SMA mRNA levels was analyzed in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 3 and 6 weeks, respectively. All data were normalized against house keeping gene β-actin and expressed as mean fold change in SRC-3−/− samples relative to SRC-3+/+. (f) Representative Sirius Red stained liver sections from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 3 and 6 weeks, respectively. Livers from SRC-3−/− mice had less collagen deposition compared to SRC-3+/+ mice (×100 magnification). (g) Digital quantification of Sirius Red staining on liver sections from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 3 and 6 weeks, respectively. Data were expressed as mean fold change in SRC-3−/− samples relative to SRC-3+/+. All data are mean ± standard error. *P<0.05, **P<0.01, WT vs KO.
Altered Fibrotic Gene Expression, Reduced Hepatocyte Apoptosis and Increased Hepatocyte Proliferation in SRC-3−/− Mice
Since hepatic fibrosis was attenuated in SRC-3−/− mice after both 3 and 6 weeks CCl4 treatment, with a more significant difference between SRC-3+/+ and SRC-3−/− mice in the 3-week-treatment group, we used the CCl4-treated mice from this experiment group for further investigation. The mRNA expression levels of the collagen family molecules were significantly reduced in SRC-3−/− mice compared to wild type mice (Figure 4a), which was in accordance with the results from Sirius Red staining analysis (Figure 3f, g). Moreover, expression levels of matrix metalloproteinases (MMPs), a proteolytic enzyme family involved in HSC activation and hepatic fibrosis process,15 were much lower in SRC-3−/− mice than SRC-3+/+ mice (Figure 4b). Similar to mice after acute CCl4 treatment, in SRC-3−/− mice with chronic CCl4 administration, hepatocyte proliferation was enhanced as demonstrated by PCNA immunostaining (Figure 4c, d) and hepatocyte apoptosis was suppressed as revealed by TUNEL assay (Figure 4e, f). Furthermore, similar results on PCNA staining and TUNEL assay were obtained using 6-week-treated mice (Supplementary Figure 1). Collectively, these data confirmed our previous observation that chronic fibrosis was attenuated in SRC-3−/− mice.
Figure 4.
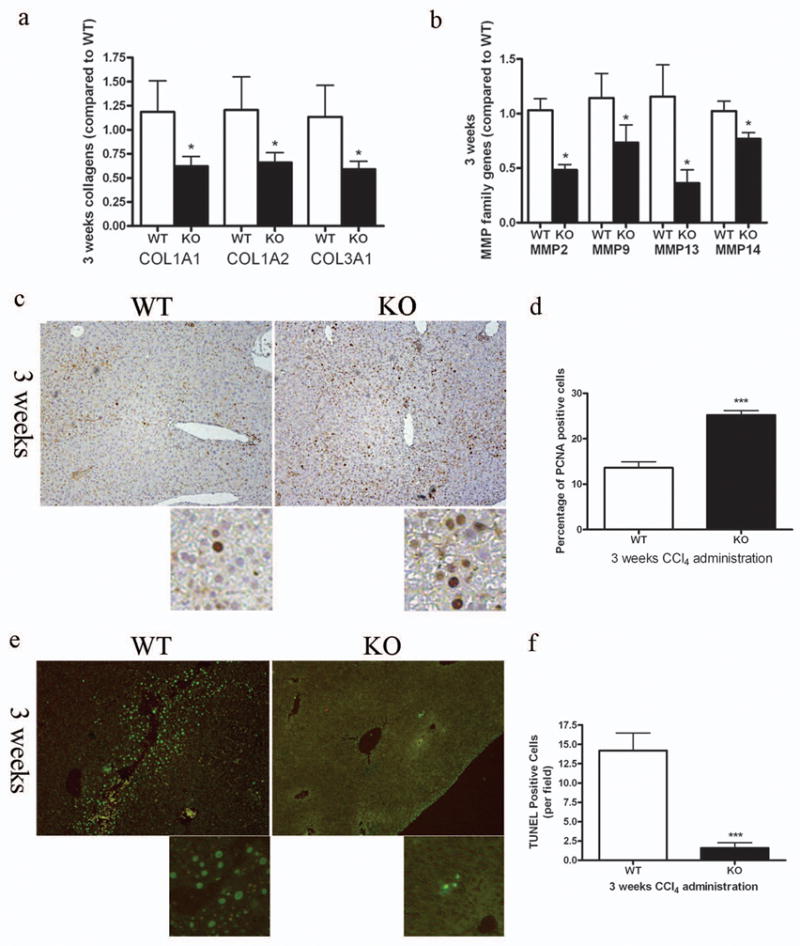
Decreased fibrotic gene expression, enhanced hepatocyte proliferation and suppressed hepatocyte apoptosis in SRC-3−/− mice. (a) Expressions of COL1A1, COL1A2 and Collagen 3α1 (COL3A1) mRNA levels were analyzed in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 3 weeks. All data were normalized against house keeping gene β-actin and expressed as mean fold change in SRC-3−/− samples relative to SRC-3+/+. (b) Expressions of MMP-2, -9, -13 and -14 mRNA levels were analyzed in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 3 weeks. All data were normalized against house keeping gene β-actin and expressed as mean fold change in SRC-3−/− samples relative to SRC-3+/+. (c) Immunohistochemical analysis of PCNA expression in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 3 weeks (×100 magnification). Slides were stained with hematoxylin to show PCNA negative cells. Images were magnified digitally (below) to show details of the staining. Percentages of PCNA positive cells were calculated and shown in Figure 4d. (e) TUNEL staining was performed on liver sections from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 3 weeks. Results were examined using fluorescence microscope (×100 magnification). Images were magnified digitally (below) to show details of the staining. TUNEL positive cells per field were calculated and shown in Figure 4f. All data are mean ± standard error. *P<0.05 and *** P<0.001, WT vs KO.
SRC-3 Ablation Impairs TGFβ1/Smad Signaling
The results we obtained from the SRC-3−/− chronic fibrosis mice models were similar with our observations in the SRC-3−/− acute necrosis mice models, with significantly reduced TGFβ1 serum levels in both models. This motivated us to examine whether the altered TGFβ1/Smad signaling, the key pro-fibrogenesis pathway, in SRC-3−/− mice could be the inner mechanism behind its resistance to CCl4-induced fibrosis. As shown in Figure 5a, TGFβ1 protein expression level was sharply decreased in the liver of SRC-3−/− mice compared to SRC-3+/+ mice. Meanwhile, the expressions of phospho-Smad2 and phospho-Smad3, mediators of the TGFβ1 signaling, were also greatly suppressed in SRC-3−/− mice. No significant difference in Smad2 and Smad3 total protein levels was observed between the two genotypes. Consistently, liver lysates from SRC-3−/− mice showed increased cell cycle-promoting molecule CyclinD1 and decreased pro-apoptotic molecule Bax expression levels, both were reported to be the targets of TGFβ1/Smad pathway (Figure 5b). Similar results were obtained on 6-week-treated mice (Supplementary Figure 2). These data indicated that SRC-3−/− mice were protected against chronic hepatic fibrosis, at least partially, due to impaired TGFβ1/Smad signaling.
Figure 5.
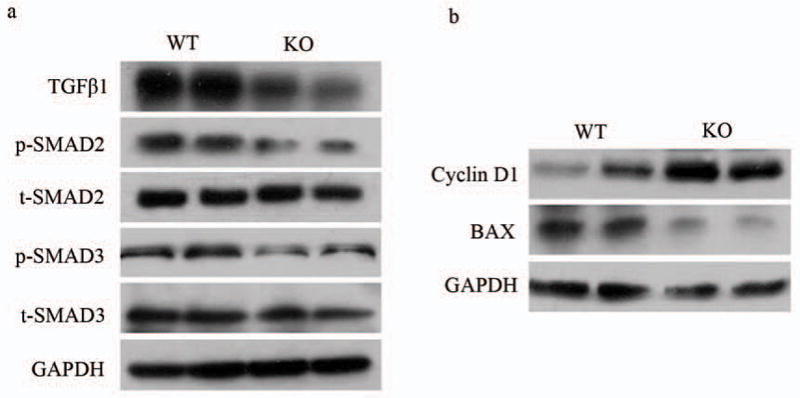
Impaired TGFβ1/Smad Signaling in SRC-3−/− mice. (a) Western blot analysis of TGFβ1, phospho-Smad2 (p-SMAD2), total Smad2 (t-SMAD2), phospho-Smad3 (p-SMAD3) and total Smad3 (t-SMAD3) expression levels in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 3 weeks. GAPDH was used as an invariant control. (b) Western blot analysis of Cyclin D1 and BAX expression levels in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 3 weeks. GAPDH was used as an invariant control.
Positive correlation between SRC-3 and degrees of fibrosis in human hepatic diseases
Various degrees of hepatic fibrosis are frequently observed in livers of patients with chronic hepatitis, and prolonged hepatic fibrosis will finally result in hepatic cirrhosis.16 To study the expression of SRC-3 gene in fibrogenesis progression, we further analyzed SRC-3 immunoreactivity in a human tissue microarray consisting of human hepatic disease samples, including viral hepatitis, hepatic cirrhosis and hepatocellular carcinomas (HCC). As demonstrated by the Kruskal-Wallis test, the degrees of fibrosis in the samples of hepatic cirrhosis were significantly stronger than that in the samples of viral hepatitis (Figure 6a-d and Supplementary Table 1). Positive SRC-3 immunoreactivity showed a diffuse intra-cytoplasmic granular staining and part of nucleus staining in liver sections prepared from patients. Since SRC-3 exerts its function in the nucleus, positive rates of SRC-3 nucleus staining were graded (see “MATERIALS AND METHODS”) and studied. As shown in Table 1, Sixty-eight percent of the hepatitis group stained negative (-) for SRC-3 immunoreactivity and no samples from this group showed strong SRC-3 staining (+++). In contrast, only 23% of the cirrhosis group had negative staining and it had higher percentages of samples with various SRC-3 staining amplitude (+, ++ and +++) than the hepatitis group. The Kruskal-Wallis test demonstrated that the intensity of SRC-3 immunoreactive staining showed a statistically different pattern between the hepatitis and the cirrhosis samples (Figure 6e-h and Table 1). Furthermore, Spearman’s correlation test showed that the intensity of SRC-3 nucleus immunoreactivity and the degrees of fibrosis in each sample were significantly correlated (r=0.462, p=0.008). Compared to other groups, HCC group had the highest percentage of samples with strong SRC-3 staining (+++) (Supplementary Figure 3) and served as a positive control since SRC-3 has been identified as an oncogene in the liver and was reported to be over-amplified in hepatocellular carcinoma17. These data from clinical human samples suggested that SRC-3 expression level was positively correlated with hepatic fibrosis extent in human hepatic diseases.
Figure 6.
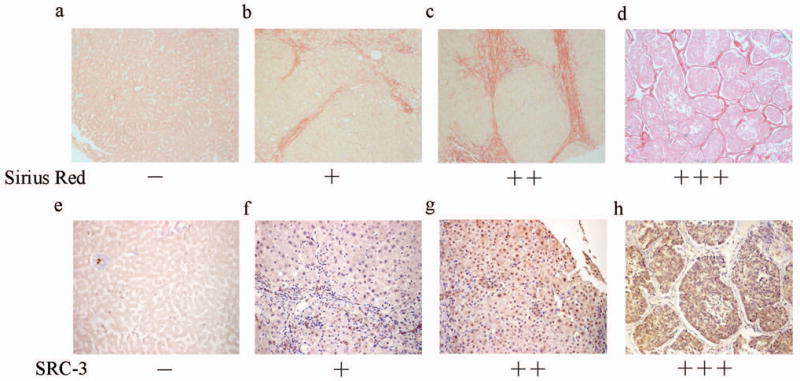
Positive correlation between SRC-3 and fibrosis degrees in human hepatic diseases. (a-d) Fibrosis extents in liver samples from patients with viral hepatitis or hepatic cirrhosis were examined by Sirius Red staining and representative images were shown for grading and grouping. (a) Negative staining of hepatitis. (b)+ of hepatitis. (c) ++ of hepatitis. (d) strong staining (+++) of hepatic cirrhosis (×100 Magnification). (e-h) SRC-3 nuclues immunoreactivity in the same samples shown in Figure 6a-d. They are also representative images for the grading and grouping of SRC-3 immunoreactivity. (e) Negative staining of hepatitis. (f)+ of hepatitis. (g) ++ of hepatitis. (h) strong staining (+++) of hepatic cirrhosis (×200 Magnification).
Table 1.
Immunoreactivity of SRC-3 in human samples of viral hepatitis and hepatic cirrhosis. Immunohistochemistry study of the SRC-3 gene in human viral hepatitis and hepatic cirrhosis indicated that SRC-3 expression correlated positively with fibrosis extent.
| Viral Hepatitis (%) | Hepatic cirrhosis (%)* | |
|---|---|---|
| + + + | 0 | 2 (15) |
| + + | 2 (11) | 2 (15) |
| + | 4 (21) | 6 (47) |
| − | 13 (68) | 3 (23) |
| N | 19 | 13 |
Compared to viral hepatitis group, P=0.012
DISCUSSION
The present study shows that SRC-3 plays an important role in liver injury and fibrogenesis. Mice lacking SRC-3 have attenuated fibrosis following CCl4 treatment, which might be resulted from impaired TGFβ1/Smad profibrotic signaling pathway. Our results indicate that SRC-3 could interfere with the TGFβ1/Smad signaling pathway, which in turn modulates critical pathological events such as HSC activation, collagen deposition and hepatocyte proliferation and apoptosis in the development of hepatic fibrosis.
When treated acutely with CCl4, SRC-3+/+ and SRC-3−/− mice had similar liver damage 24h and 36h after the insult, but the necrosis extent was greatly ameliorated after 48h in SRC-3−/− mice whereas severe hepatic damage in SRC-3+/+ mice remained, as demonstrated by histological analysis and serum ALT levels from both genotypes. This inspired us to examine hepatocyte proliferation12 in SRC-3+/+ and SRC-3−/− mice. The expression level of PCNA, a nuclear protein highly expressed during the DNA synthesis phase of the cell cycle, is tightly correlated with the proliferative state of the cells18. PCNA expression was significantly elevated in SRC-3−/− mice, indicating an increase in hepatocyte proliferation in the lack of SRC-3, which could lead to the enhanced hepatic damage repair and thus the significantly alleviated necrosis in SRC-3−/− mice observed 48h after CCl4 challenge. It should be noted that upon CCl4 treatment, SRC-3+/+ mice demonstrated significant necrotic lesion and dramatically increased serum ALT level. Interestingly, in a previous report, injection of another hepatotoxin, lipopolysaccharide, causes only a mild hepatic injury with a small increase in serum ALT levels in both SRC-3−/− and SRC-3+/+ mice.19 It seems that lipopolysaccharide, as a hepatotoxin, is not involved in the promotion of acute liver damage, which is different from CCl4.
In response to CCl4 treatment, hepatocytes may undergo apoptosis in addition to necrosis12. In our mice models under both acute and chronic CCl4 administration, suppressed hepatocyte apoptosis and reduced pro-apoptotic protein Bax expression were observed in livers of SRC-3−/− mice. Apoptosis of hepatocytes is one of the major promoting factors in the development of liver fibrosis. Mehal’ group demonstrates that DNA from apoptotic hepatocytes acts as an important mediator of HSC differentiation by stopping mobile HSCs around the apoptosing hepatocytes and inducing HSC collagen production, thus promotes liver fibrogenesis.20 We observed suppressed HSC activation, significantly suppressed Collagen I messenger RNA levels and less collagen accumulation in SRC-3−/− mice, which might be contributed in part by reduced hepatocyte apoptosis in the absence of SRC-3.
In liver tissue, the MMP family plays a pivotal role in hepatic fibrosis. The current knowledge obtained from various literatures showed that MMPs might play different roles in fibrogenesis and fibrolysis15. Literature that focused on the fibrogenesis process indicated a fibrosis-promoting role of the MMPs family. For example, MMP-2 mRNA expression increases during experimental fibrogenesis induced by CCl4 injections and remains elevated while its activation is mediated by MMP-14.21–24 MMP-2 and MMP-14 are expressed by HSCs during their activation following liver injury, which are important for HSC proliferation and migration.25–27 MMP-9 is produced by various cell types in CCl4-induced liver injury, the activity of which is not associated with the degree of fibrosis but usually links to the histologically derived level of tissue inflammation.21 MMP-9 can activate latent TGF-β,28 which is vital in earlier stages of fibrogenesis, as collagen production of HSCs is stimulated by TGF-β.29 MMP-9’s important role in the initial phase of HSC activation is identified by French SW’s group, as IL-1α-induced HSC activation is completely prevented by deprivation of MMP-9.30 MMP13 has the ability to destroy normal liver tissue for newly synthesized ECM deposition and ECM-bound cytokines, i.e. TGFβ1 releasing, subsequently induces fibrogenesis.31, 32 At the same time, studies emphasized on the fibrolysis process using fibrosis regression mice models indicated that an elevation in MMPs expression could enhance liver recovery from fibrosis. For instance, during regression from liver fibrosis, MMP-13 and MMP-14 have the ability to degrade abnormal fibrillar collagen, thus might be responsible for key events in ECM degradation.33, 34 Other studies implied that MMP-2 and MMP-9 might contribute indirectly to fibrolysis for their association with HSCs apoptosis.35, 36 In our study, mice were sacrificed 3 days after last CCl4 injection and thus might be in the fibrogenesis developing stage not in the fibrosis regression stage. It is therefore possible that the significant decrease in the MMPs mRNA expression levels in SRC-3−/− mice compared to the similarly treated wild type mice could contribute to the resistance to chronic hepatic fibrosis in SRC-3−/− mice. Moreover, HSC activation, inflammatory infiltration, collagen accumulation as well as TGFβ1 secretion were all suppressed in SRC-3−/− mice compare to SRC-3+/+ mice, which were in consistent with the reduction levels of MMPs and their roles in fibrogenesis. SRC-3 is reported to directly regulate transcription of MMP-2 and MMP-13 through its coactivation of AP-1 and PEA3,37 which suggested that alterations in the MMPs expression levels caused by SRC-3 ablation could be one of the possible reasons for the fibrosis resistance in SRC-3−/− mice.
During the development of hepatic fibrosis, aberrant activity of TGF-β1 and its downstream signal transducers Smads have been proved to be vital to the progression of fibrogenesis at both cellular and molecular levels.38 Upon TGF-β1 binding to its receptor, the receptor associated Smads (Smads 1, 2, 3, 5, and 8) are phosphorylated, form a complex with the co-Smad (Smad4), and translocate into the nucleus to regulate transcription of profibrotic target genes.38 TGF-β1/Smad signaling pathway can influence various aspects in the fibrogenesis process, including inhibition of hepatocyte proliferation,39 induction of hepatocyte apoptosis 30, 41, and of utmost importance, mediation of HSC acitivation 42 and the subsequent stimulation of HSC collagen secretion29. In our CCl4 treated mice models, TGF-β1 serum level was significantly decreased in SRC-3−/− mice compared to SRC-3+/+ mice. Further detailed examinations revealed that protein expression levels of liver TGF-β1, phospho-Smad2 and phospho-Smad3 were sharply reduced in the absence of SRC-3. Changes in the Collagen family, Cyclin D1 and Bax expression levels, which have been reported to be responsive to TGF-β1/Smad regulation,43–45 confirmed our discovery. These results indicated that TGF-β1/Smad signaling was impaired in SRC-3−/− mice, which in turn causing enhanced hepatic damage repair, reduced hepatocyte apoptosis, suppressed HSC activation and decreased collagen production, thus protected SRC-3−/− mice against CCl4-induced acute liver necrosis and, more importantly, chronic hepatic fibrosis.
SRC-3 plays a critical role in gene transcription through its interaction with coactivator p300/CBP and P/CAF10, and it is clear that coactivator p300/CBP6 and P/CAF7 are essential for Smads-mediated transcriptional activity. As a family member to SRC-3, SRC-1 is demonstrated to be able to enhance TGFβ-induced, Smad-mediated transcription in vitro through its interaction with p300/CBP.8 Therefore, SRC-3 can exert its influence on TGF-β1/Smad signaling through its interaction with p300/CBP and/or P/CAF or by forming a functional coactivator complex with them, and regulate hepatic fibrogenesis independently or synergistically with its family members during different pathologic phases. The detailed molecular mechanism of SRC-3’s regulation on TGF-β1/Smad signaling will be the focus of our future work. It will also be useful to create a SRC-1/SRC-3 double knockout mouse strain to study the specificity and redundancy of the p160 family members during hepatic fibrogenesis.
Finally, we evaluated the SRC-3 expression levels in liver specimens from patients with hepatic fibrosis, cirrhosis or hepatocellular carcinomas by immunohistochemistry analysis. For fibrosis and cirrhosis samples, the intensity of SRC-3 immunostaining in nucleus was positively linked to the severity of hepatic fibrosis extent, with strong intensity in cirrhosis specimens and moderate in fibrosis ones. These results were in consistent with the data obtained from our SRC-3 deficiency mice models, which suggested that SRC-3 also played a crucial role in the pathogenesis of human fibrosis and cirrhosis. Besides, we also investigated the expression of SRC-3 in hepatocellular carcinomas samples. The finding showed that SRC-3 was over-amplified in hepatocellular carcinoma, which was confirmed by previous data, indicating SRC-3 was a tumor-promoting oncogene in the liver17
In summary, our study demonstrates that SRC-3 plays a vital role in the progression of fibrosis using an in vivo mice model. In SRC-3−/− mice, acute liver injury and chronic hepatic fibrosis induced by CCl4 administration are less severe as compared with their wild type littermates. The protective effect of SRC-3 ablation is mediated, at least partially, through the impaired TGF-β1/Smad signaling in SRC-3−/− mice. Moreover, clinical samples show that SRC-3 expression levels are tightly correlated with extents of hepatic fibrosis. These findings establish an essential involvement of SRC-3 in liver fibrogenesis, which may provide new clues to the future treatment of hepatic fibrosis.
Supplementary Material
Supplementary Figure 1 Enhanced hepatocyte proliferation and suppressed hepatocyte apoptosis in 6 week-CCl4-treated SRC-3−/− mice. (a) Immunohistochemical analysis of PCNA expression in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 6 weeks (×100 magnification). Slides were stained with hematoxylin to show PCNA negative cells. Images were magnified digitally (below) to show details of the staining. Percentages of PCNA positive cells were calculated and shown in Supplementary Figure 1b. (c) TUNEL staining was performed on liver sections from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 6 weeks. Results were examined using fluorescence microscope (×100 magnification). Images were magnified digitally (below) to show details of the staining. TUNEL positive cells per field were calculated and shown in Supplementary Figure 1d. All data are mean ± standard error. **P<0.01 and *** P<0.001, WT vs KO.
Supplementary Figure 2 Impaired TGFβ1/Smad Signaling in 6 week-CCl4-treated SRC-3−/− mice. (a) Western blot analysis of TGFβ1, phospho-Smad2 (p-SMAD2), total Smad2 (t-SMAD2), phospho-Smad3 (p-SMAD3) and total Smad3 (t-SMAD3) expression levels in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 6 weeks. GAPDH was used as an invariant control. (b) Western blot analysis of Cyclin D1 and BAX expression levels in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 6 weeks. GAPDH was used as an invariant control.
Supplementary Figure 3 Human hepatocellular carcinoma specimens showed strong SRC-3 staining. (a) A representative image of SRC-3 nucleus immunoreactivity showed extensive SRC-3 nucleus staining in human hepatocellular carcinoma. (b) Immunohistochemistry study of the SRC-3 gene in human hepatocellular carcinoma samples. Note the high prevalence of strong SRC-3 nucleus staining (++ and +++) in HCC samples.
Supplementary Table 1 Sirius red staining of human samples of viral hepatitis and hepatic cirrhosis. Sirius Red staining showed stronger fibrosis extent in human liver samples of hepatic cirrhosis than in samples of viral hepatitis.
Acknowledgments
We gratefully acknowledge Jianming Xu (Baylor College of Medicine, USA) for inspiring advices. We also thank Jiulan Cui for maintaining the mouse colony. This study is supported by the grants from 973 Project (No.2006CB503904), Natural Science Foundation of China (No.30725037 and No.30570880) and Shanghai Education Commission (No. Y0204).
Abbreviations
- ALT
alanine aminotransferase
- CCl4
carbon tetrachloride
- ECM
extracellular matrix
- HSC
hepatic stellate cells
- MMP
matrix metalloproteinases
- PCNA
proliferating cell nuclear antigen
- α-SMA
smooth muscle α-actin
- SRC-3
steroid receptor coactivator-3
- TGFβ1
transforming growth factor β1
- HCC
hepatocellular carcinoma
Footnotes
The authors have no duality of interest to declare.
References
- 1.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 2.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539–548. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallace K, Burt AD, Wright MC. Liver fibrosis. Biochem J. 2008;411:1–18. doi: 10.1042/BJ20071570. [DOI] [PubMed] [Google Scholar]
- 4.Gressner AM, Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-β as major players and therapeutic targets. J Cell Mol Med. 2006;10:76–99. doi: 10.1111/j.1582-4934.2006.tb00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghosh AK. Factors Involved in the Regulation of Type I Collagen Gene Expression: Implication in Fibrosis. Exp Biol Med. 2002;227:301–314. doi: 10.1177/153537020222700502. [DOI] [PubMed] [Google Scholar]
- 6.Ghosh AK, Varga J. The Transcriptional Coactivator and Acetyltransferase p300 in Fibroblast Biology and Fibrosis. J Cell Physiol. 2007;213:663–671. doi: 10.1002/jcp.21162. [DOI] [PubMed] [Google Scholar]
- 7.Itoh S, Ericsson J, Nishikawa J, et al. The transcriptional co-activator P/CAF potentiates TGF-β/Smad signaling. Nucleic Acids Research. 2000;28:4291–4298. doi: 10.1093/nar/28.21.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dennler S, Pendaries V, Tacheau C, et al. The steroid receptor co-activator-1 (SRC-1) potentiates TGF-b/Smad signaling: role of p300/CBP. Oncogene. 2005;24:1936–1945. doi: 10.1038/sj.onc.1208343. [DOI] [PubMed] [Google Scholar]
- 9.Xu J, Li Q. Review of the in Vivo Functions of the p160 Steroid Receptor Coactivator Family. Mol Endocrinol. 2003;17:1681–1692. doi: 10.1210/me.2003-0116. [DOI] [PubMed] [Google Scholar]
- 10.Naeem H, Cheng D, Zhao Q, et al. The activity and stability of the transcriptional coactivator p/CIP/SRC-3 are regulated by CARM1-dependent methylation. Mol Cell Biol. 2007;27:120–134. doi: 10.1128/MCB.00815-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi YH, Ding WX, Zhou J, et al. Expression of X-linked inhibitor-of-apoptosis protein in hepatocellular carcinoma promotes metastasis and tumor recurrence. Hepatology. 2008;48:497–507. doi: 10.1002/hep.22393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu W, Hellerbrand C, Köhler UA, et al. The Nrf2 transcription factor protects from toxin-induced liver injury and fibrosis. Lab Invest. 2008;88:1068–1078. doi: 10.1038/labinvest.2008.75. [DOI] [PubMed] [Google Scholar]
- 13.Schuster N, Krieglstein K. Mechanisms of TGF-β-mediated apoptosis. Cell Tissue Res. 2002;307:1–14. doi: 10.1007/s00441-001-0479-6. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen LN, Furuya MH, Wolfraim LA, et al. Transforming Growth Factor-Beta Differentially Regulates Oval Cell and Hepatocyte Proliferation. Hepatology. 2007;45:31–41. doi: 10.1002/hep.21466. [DOI] [PubMed] [Google Scholar]
- 15.Hemmann S, Graf J, Roderfeld M, et al. Expression of MMPs and TIMPs in liver fibrosis: a systematic review with special emphasis on anti-fibrotic strategies. J Hepatol. 2007;46:955–975. doi: 10.1016/j.jhep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Parsons CJ, Takashima M, Rippe RA. Molecular mechanisms of hepatic fibrogenesis. J Gastroenterol Hepatol. 2007;22 (Suppl 1):S79–84. doi: 10.1111/j.1440-1746.2006.04659.x. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Wu MC, Sham JS, et al. Prognostic significance of c-myc and AIB1 amplification in hepatocellular carcinoma:A broad survey using high-throughput tissue microarray. Cancer. 2002;95:2346–2352. doi: 10.1002/cncr.10963. [DOI] [PubMed] [Google Scholar]
- 18.Bravo R. Synthesis of the nuclear protein cyclin (PCNA) and its relationship with DNA replication. Exp Cell Res. 1986;163:287–293. doi: 10.1016/0014-4827(86)90059-5. [DOI] [PubMed] [Google Scholar]
- 19.Yu C, York B, Wang S, et al. An essential function of the SRC-3 coactivator in suppression of cytokine mRNA translation and inflammatory response. Mol Cell. 2007;25:765–778. doi: 10.1016/j.molcel.2007.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe A, Hashmi A, Gomes DA, et al. Apoptotic Hepatocyte DNA Inhibits Hepatic Stellate Cell Chemotaxis via Toll-Like Receptor 9. Hepatology. 2007;46:1509–1518. doi: 10.1002/hep.21867. [DOI] [PubMed] [Google Scholar]
- 21.Strongin AY, Collier I, Bannikov G, et al. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem. 1995;270:5331–5338. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- 22.Zhou X, Hovell CJ, Pawley S, et al. Expression of matrix metalloproteinase-2 and -14 persists during early resolution of experimental liver fibrosis and might contribute to fibrolysis. Liver Int. 2004;24:492–501. doi: 10.1111/j.1478-3231.2004.0946.x. [DOI] [PubMed] [Google Scholar]
- 23.Parsons CJ, Bradford BU, Pan CQ, et al. Antifibrotic effects of a tissue inhibitor of metalloproteinase-1 antibody on established liver fibrosis in rats. Hepatology. 2004;40:1106–1115. doi: 10.1002/hep.20425. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe T, Niioka M, Ishikawa A, et al. Dynamic change of cells expressing MMP-2 mRNA and MT1-MMP mRNA in the recovery from liver fibrosis in the rat. J Hepatol. 2001;35:465–473. doi: 10.1016/s0168-8278(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 25.Knittel T, Mehde M, Kobold D, et al. Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: regulation by TNF-alpha and TGF-beta1. J Hepatol. 1999;30:48–60. doi: 10.1016/s0168-8278(99)80007-5. [DOI] [PubMed] [Google Scholar]
- 26.Ikeda K, Wakahara T, Wang YQ, et al. In vitro migratory potential of rat quiescent hepatic stellate cells and its augmentation by cell activation. Hepatology. 1999;29:1760–1767. doi: 10.1002/hep.510290640. [DOI] [PubMed] [Google Scholar]
- 27.Zhou X, Hovell CJ, Pawley S, et al. Expression of matrix metalloproteinase-2 and -14 persists during early resolution of experimental liver fibrosis and might contribute to fibrolysis. Liver Int. 2004;24:492–501. doi: 10.1111/j.1478-3231.2004.0946.x. [DOI] [PubMed] [Google Scholar]
- 28.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 29.Cao Q, Mak KM, Lieber CS. Dilinoleoylphosphatidylcholine prevents transforming growth factor-beta1-mediated collagen accumulation in cultured rat hepatic stellate cells. J Lab Clin Med. 2002;139:202–210. doi: 10.1067/mlc.2002.121853. [DOI] [PubMed] [Google Scholar]
- 30.Han YP, Zhou L, Wang J, et al. Essential role of matrix metalloproteinases in interleukin-1-induced myofibroblastic activation of hepatic stellate cell in collagen. J Biol Chem. 2004;279:4820–4828. doi: 10.1074/jbc.M310999200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watanabe T, Niioka M, Hozawa S, et al. Gene expression of interstitial collagenase in both progressive and recovery phase of rat liver fibrosis induced by carbon tetrachloride. J Hepatol. 2000;33:224–235. doi: 10.1016/s0168-8278(00)80363-3. [DOI] [PubMed] [Google Scholar]
- 32.Yan S, Chen GM, Yu CH, et al. Expression pattern of matrix metalloproteinases-13 in a rat model of alcoholic liver fibrosis. Hepatobiliary Pancreat Dis Int. 2005;4:569–572. [PubMed] [Google Scholar]
- 33.Benyon RC, Arthur MJ. Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis. 2001;21:373–384. doi: 10.1055/s-2001-17552. [DOI] [PubMed] [Google Scholar]
- 34.Issa R, Zhou X, Trim N, et al. Mutation in collagen-1 that confers resistance to the action of collagenase results in failure of recovery from CCl4-induced liver fibrosis, persistence of activated hepatic stellate cells, and diminished hepatocyte regeneration. FASEB J. 2003;17:47–49. doi: 10.1096/fj.02-0494fje. [DOI] [PubMed] [Google Scholar]
- 35.Preaux AM, D’ortho MP, Bralet MP, et al. Apoptosis of human hepatic myofibroblasts promotes activation of matrix metalloproteinase-2. Hepatology. 2002;36:615–622. doi: 10.1053/jhep.2002.35279. [DOI] [PubMed] [Google Scholar]
- 36.Roderfeld M, Weiskirchen R, Wagner S, et al. Inhibition of hepatic fibrogenesis by matrix metalloproteinase-9 mutants in mice. FASEB J. 2006;20:444–454. doi: 10.1096/fj.05-4828com. [DOI] [PubMed] [Google Scholar]
- 37.Yan J, Erdem H, Li R, et al. Steroid receptor coactivator-3/AIB1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer Res. 2008;68:5460–5468. doi: 10.1158/0008-5472.CAN-08-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ueda S, Yamanoi A, Hishikawa Y, et al. Transforming growth factor-beta1 released from the spleen exerts a growth inhibitory effect on liver regeneration in rats. Lab Invest. 2003;83:1595–1603. doi: 10.1097/01.lab.0000095686.10639.c8. [DOI] [PubMed] [Google Scholar]
- 40.Samson CM, Schrum LW, Bird MA, et al. Hepatocyte apoptosis: is transforming growth factor-B1 the kiss of death? Hepatology. 1993;18:1536–1537. doi: 10.1002/hep.1840180638. [DOI] [PubMed] [Google Scholar]
- 41.Samson CM, Schrum LW, Bird MA, et al. Transforming growth factor-beta1 induces hepatocyte apoptosis by a c-Jun independent mechanism. Surgery. 2002;132:441–449. doi: 10.1067/msy.2002.125321. [DOI] [PubMed] [Google Scholar]
- 42.Gressner AM, Weiskirchen R, Breitkopf K, et al. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:793–807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- 43.Cocolakis E, Dai M, Drevet L, et al. Smad Signaling Antagonizes STAT5-mediated Gene Transcription and Mammary Epithelial Cell Differentiation. J Biol Chem. 2008;283:1293–1307. doi: 10.1074/jbc.M707492200. [DOI] [PubMed] [Google Scholar]
- 44.Jungert K, Buck A, Buchholz M, et al. Smad-Sp1 complexes mediate TGFb-induced early transcription of oncogenic Smad7 in pancreatic cancer cells. Carcinogenesis. 2006;27:2392–2401. doi: 10.1093/carcin/bgl078. [DOI] [PubMed] [Google Scholar]
- 45.Mithani SK, Balch GC, Shiou SR, et al. Smad3 Has a Critical Role in TGF-γ-Mediated Growth Inhibition and Apoptosis in Colonic Epithelial Cells. J Surg Res. 2004;117:296–305. doi: 10.1016/S0022-4804(03)00335-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Enhanced hepatocyte proliferation and suppressed hepatocyte apoptosis in 6 week-CCl4-treated SRC-3−/− mice. (a) Immunohistochemical analysis of PCNA expression in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 6 weeks (×100 magnification). Slides were stained with hematoxylin to show PCNA negative cells. Images were magnified digitally (below) to show details of the staining. Percentages of PCNA positive cells were calculated and shown in Supplementary Figure 1b. (c) TUNEL staining was performed on liver sections from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 6 weeks. Results were examined using fluorescence microscope (×100 magnification). Images were magnified digitally (below) to show details of the staining. TUNEL positive cells per field were calculated and shown in Supplementary Figure 1d. All data are mean ± standard error. **P<0.01 and *** P<0.001, WT vs KO.
Supplementary Figure 2 Impaired TGFβ1/Smad Signaling in 6 week-CCl4-treated SRC-3−/− mice. (a) Western blot analysis of TGFβ1, phospho-Smad2 (p-SMAD2), total Smad2 (t-SMAD2), phospho-Smad3 (p-SMAD3) and total Smad3 (t-SMAD3) expression levels in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 6 weeks. GAPDH was used as an invariant control. (b) Western blot analysis of Cyclin D1 and BAX expression levels in livers from SRC-3+/+ and SRC-3−/− mice treated chronically with CCl4 for 6 weeks. GAPDH was used as an invariant control.
Supplementary Figure 3 Human hepatocellular carcinoma specimens showed strong SRC-3 staining. (a) A representative image of SRC-3 nucleus immunoreactivity showed extensive SRC-3 nucleus staining in human hepatocellular carcinoma. (b) Immunohistochemistry study of the SRC-3 gene in human hepatocellular carcinoma samples. Note the high prevalence of strong SRC-3 nucleus staining (++ and +++) in HCC samples.
Supplementary Table 1 Sirius red staining of human samples of viral hepatitis and hepatic cirrhosis. Sirius Red staining showed stronger fibrosis extent in human liver samples of hepatic cirrhosis than in samples of viral hepatitis.