

Activation of adenosine A2A receptor aggravates lung damage in a neurogenic mouse model of acute lung injury (ALI) but protects against nonneurogenic ALI.
Abstract
The bone marrow–derived cell (BMDC)–associated inflammatory response plays a key role in the development of acute lung injury (ALI). Activation of adenosine A2A receptor (A2AR) is generally considered to be antiinflammatory, inhibiting BMDC activities to protect against ALI. However, in the present study, we found that in a mouse model of neurogenic ALI induced by severe traumatic brain injury (TBI), BMDC A2AR exerted a proinflammatory effect, aggravating lung damage. This is in contrast to the antiinflammatory effect observed in the mouse oleic acid–induced ALI model (a nonneurogenic ALI model.) Moreover, the A2AR agonist CGS21680 aggravated, whereas the antagonist ZM241385 attenuated, the severe TBI-induced lung inflammatory damage in mice. Further investigation of white blood cells isolated from patients or mouse TBI models and of cultured human or mouse neutrophils demonstrated that elevated plasma glutamate after severe TBI induced interaction between A2AR and the metabotropic glutamate receptor 5 (mGluR5) to increase phospholipase C–protein kinase C signaling, which mediated the proinflammatory effect of A2AR. These results are in striking contrast to the well-known antiinflammatory and protective role of A2AR in nonneurogenic ALI and indicate different therapeutic strategies should be used for nonneurogenic and neurogenic ALI treatment when targeting A2AR.
Traumatic brain injury (TBI) is a life-threatening syndrome that can be complicated by the dysfunction of peripheral organs such as lung, liver, kidney, or intestine. Among these, the association between TBI and subsequent acute lung injury (ALI) has been increasingly recognized (Dettbarn and Davidson, 1989; Bratton and Davis, 1997; Contant et al., 2001; Bronchard et al., 2004; Mascia et al., 2008). Mackersie et al. (1983) reported that 9 of 18 comatose victims with isolated TBI developed pulmonary edema, defined as increased extravascular lung fluid content measured by thermal green dye. Two studies have reported that 20–25% of patients with isolated TBI developed respiratory insufficiency (Fulton and Jones, 1975; Bratton and Davis, 1997). Moreover, Holland et al. (2003) investigated 137 patients with TBI and found that ∼31% developed ALI. In fact, TBI-induced ALI and its development may not only influence the lung epithelium, but may also impair brain function, aggravate the neurogenic injury, and cause higher mortality and worse prognosis. Although there is evidence to suggest that inflammation is the key pathological mechanism in TBI-induced ALI (Kalsotra et al., 2007; Jin et al., 2009), as it is in nonneurogenic ALI (such as ALI induced by lung trauma, shock, and sepsis; Wheeler and Bernard, 2007; Parekh et al., 2011; Qian et al., 2012), it is unclear how TBI triggers the lung inflammatory response.
BMDCs, including neutrophils, lymphocytes, monocytes, and eosinophils (also called WBCs), are the critical response cells for progression of inflammation in ALI/acute respiratory distress syndrome (Abraham, 2003; Nakajima et al., 2010; Grommes and Soehnlein, 2011). Adenosine A2A receptor (A2AR), one of four G protein–coupled adenosine receptors (A1R, A2AR, A2BR, and A3R), is found to be expressed on BMDCs and can regulate the function of BMDCs in several pathological conditions. Previous studies in multiple nonneurogenic ALI animal models such as LPS-induced ALI, lung ischemia-reperfusion injury, and lung injury in laparotomy-induced hemorrhagic shock have shown that activation of A2AR plays an antiinflammatory role via inhibition of BMDC activities (Thiel et al., 2005; Haskó et al., 2006; Reutershan et al., 2007; Sharma et al., 2010). Accordingly, this receptor is considered an attractive potential target for therapeutic approaches to human ALI (Schepp and Reutershan, 2008). Conversely, in TBI and some other central nervous system injury models, A2AR on BMDCs has been found to promote the inflammation of brain or spinal cord (Yu et al., 2004; Dai et al., 2010a). This leads us to speculate that in severe TBI-induced ALI (a neurogenic ALI), the role of BMDC A2AR may be different from that in nonneurogenic ALI and may be involved in the progression of TBI-induced ALI.
To confirm this hypothesis, we created BM chimeras to determine the role of BMDC A2AR in a mouse model of severe TBI-induced ALI, comparing it with the oleic acid–induced ALI model (a nonneurogenic model). In human and mouse WBCs and neutrophils, the major components of BMDCs and the key reactive cells in ALI, we further investigated the mechanisms of BMDC A2AR effects on inflammation associated with TBI-induced ALI.
RESULTS
Selective inactivation of BMDC A2AR aggravates lung damage in the oleic acid–induced ALI model but exerts a protective effect in the severe TBI-induced ALI model
In the severe TBI-induced ALI model, we found that selective inactivation of BMDC A2AR (KO→WT) significantly reduced lung water content (Fig. 1 A), elevated PaO2/FIO2 (P/F) ratios (Fig. 1 B), and attenuated histological signs of pulmonary injury (Fig. 1 C) at 24 h after injury when compared with WT littermates, consistent with the results observed in global A2AR KO mice. Conversely, in the oleic acid–induced ALI model, each of these measures of lung damage was exacerbated in the mice with selective inactivation of BMDC A2AR or global A2AR KO (Fig. 1). Reconstitution of BMDC A2AR in global A2AR KO mice (WT→KO) eliminated the A2AR KO-triggered protective effects in TBI-induced ALI and the deleterious effects in oleic acid–induced ALI. In addition, no significant difference was observed between WT mice and WT mice transplanted with WT BMDCs (WT→WT) or between KO and KO→KO transplanted mice, excluding the possibility that these results were induced nonspecifically by BM transplantation (BMT; Fig. 1). These data strongly suggest that in contrast to the protective effect of BMDC A2AR in oleic acid–induced ALI, BMDC A2AR plays a deleterious role in TBI-induced ALI.
Figure 1.

BMDC A2AR differently regulates the lung injury parameters in the severe TBI-induced ALI model and oleic acid–induced ALI model. At 24 h after injury, lung injury parameters were assessed. WT: WT mice; KO: global A2AR KO mice; WT→WT: WT recipient mice with BMDCs from WT mice; KO→KO: KO recipient mice with BMDCs from KO mice; KO→WT: WT recipient mice with BMDCs from KO mice (selective inactivation of A2AR in BMDCs of WT mice); WT→KO: KO recipient mice with BMDCs from WT mice (selective reconstitution of A2AR in BMDCs of A2AR KO mice). (A) The lung water content was assayed by the wet–dry method. (B) Blood gas analyses of arterial blood. (C) H&E staining for lung sections. Bars, 100 µm. (A and B) Data are expressed as mean ± SEM. *, P < 0.01, compared with the WT group; #, P < 0.01 compared with the KO group; NS, no significant difference between the two groups. For every parameter measured, n = 8–10 in each group, and each experiment was repeated three times.
BMDC-selective A2AR deficiency promotes inflammation in mice with oleic acid–induced ALI and suppresses inflammation in mice with severe TBI-induced ALI
As shown in Fig. 2 A, at 24 h after TBI, global A2AR KO mice and mice with selective inactivation of BMDC A2AR (KO→WT) displayed reduced numbers of pulmonary neutrophils for TBI-induced ALI but enhanced numbers for oleic acid–induced ALI. Similarly, global or BMDC-specific A2AR KO mice showed increased pulmonary levels of the proinflammatory cytokines TNF and IL-1β after TBI-induced ALI and reduced levels after oleic acid–induced TBI (Fig. 2 B). Reconstitution of BMDC A2AR in global A2AR KO mice (WT→KO) eliminated the A2AR KO–triggered antiinflammatory effect in TBI-induced ALI and the proinflammatory effect in oleic acid–induced ALI (Fig. 2). These results indicate that BMDC A2AR plays a significant proinflammatory role in the progression of TBI-induced ALI.
Figure 2.

BMDC A2AR inversely modulates the neutrophil infiltration and inflammatory cytokine expression in the severe TBI- and oleic acid–induced ALI models. (A) Alterations of neutrophil infiltration and A2AR expression on neutrophils in lung tissues. Neutrophil counting and statistical analysis were performed by Image-Pro plus version 4.5 and presented as an index of CD-177–positive cells. Bars, 100 µm. (B) Alterations of protein expression of TNF and IL-1β in lung tissues. Values are expressed as mean ± SEM of three independent experiments performed in triplicate. *, P < 0.01, compared with the WT group; #, P < 0.01 compared with the KO group; NS, no significant difference between the two groups; n = 8–10 for all groups. Each experiment was repeated three times.
Plasma glutamate level is increased more in severe TBI-induced ALI than in nonneurogenic ALI
In both the TBI- and oleic acid–induced ALI models, we measured plasma glutamate levels and found that they were much higher in mice with severe TBI-induced ALI (25–50 µmol/l) than in mice with oleic acid–induced ALI (8–15 µmol/l) at 6, 12, and 24 h after injury, even though the plasma glutamate level was elevated in both models (Fig. 3 A). To corroborate this, we also assayed the changes in plasma glutamate levels in patients suffering from TBI-induced ALI or lung trauma–induced ALI, which is also a nonneurogenic ALI. Consistent with the results in the animal models, the plasma glutamate concentrations of patients suffering from severe TBI-induced ALI were much higher than those of patients with lung trauma–induced ALI at 6, 12, and 24 h after injury (Fig. 3 B).
Figure 3.
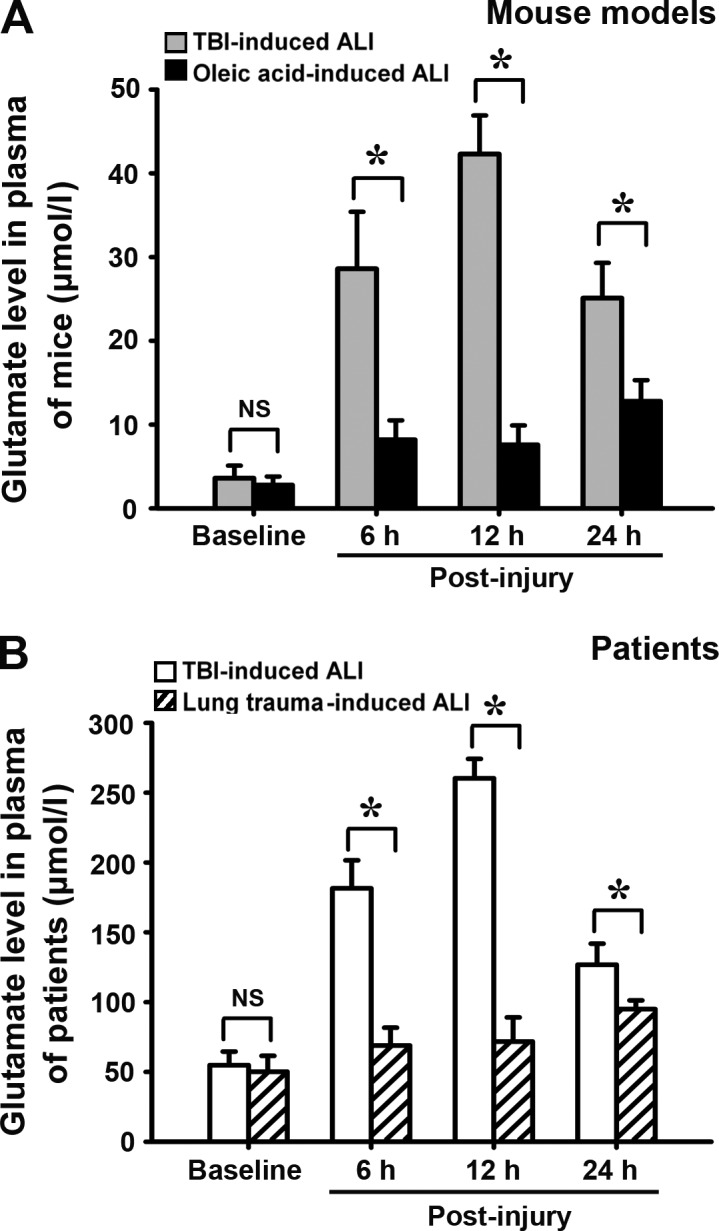
Plasma glutamate levels in severe TBI-induced ALI and nonneurogenic ALI over time. Plasma glutamate levels in mouse models and patients were detected at 6, 12, and 24 h after injury by HPLC. (A) Changes of plasma glutamate levels in mouse models (TBI- vs. oleic acid–induced ALI model). (B) Changes of plasma glutamate levels in patients (TBI- vs. lung trauma–induced ALI patients). Values are expressed as mean ± SEM, and each experiment was repeated three times. *, P < 0.01 between the two groups; NS, no significant difference between the two groups. 10 patients with TBI-induced ALI and 8 patients with lung trauma–induced ALI were investigated. The number of mice for the assay at each time point was 8–10 in each group.
A2AR–metabotropic glutamate receptor 5 (mGluR5) interaction in BMDCs is detected in severe TBI-induced ALI rather than in nonneurogenic ALI
Glutamate has been previously reported to regulate A2AR activity in brain injury, and heteromeric complexes of adenosine receptors and mGluRs were found in the brain under physiological or pathological conditions (Ciruela et al., 2001; Ferré et al., 2002; Nishi et al., 2003; Dai et al., 2010b). However, only mGluR5 and mGluR2/3 have been reported to be expressed on white blood inflammatory cells (Gill and Pulido, 2001; Liu et al., 2010b). Accordingly, using specific antibodies for A2AR, mGluR5, and mGluR2/3, we performed coimmunoprecipitation (co-IP) experiment to investigate the interaction between A2AR and these mGluRs. From WBC extracts of mice with TBI-induced ALI, the mouse antibody against A2AR coimmunoprecipitated a band corresponding to mGluR5 (Fig. 4 A). This band did not appear in immunoprecipitates from the cells of mice with oleic acid–induced ALI (Fig. 4 A). No IP of mGluR2/3 was obtained from WBCs in either of the two mouse ALI models (Fig. 4 A). Consistent with these results in animal models, analysis by SDS-PAGE and immunoblotting with anti-mGluR5 antibodies of immunoprecipitates obtained with human anti-A2AR showed a band corresponding to mGluR5 in WBCs of TBI-induced ALI patients but not those of lung trauma–induced ALI patients (Fig. 4 B). No mGluR2/3 band was detected in WBCs of either group (Fig. 4 A). In addition, using anti-mGluR5 antibody as the immunoprecipitating antibody and anti-A2AR antibody as the immunoblotting antibody, the interaction of A2AR and mGluR5 was confirmed in the WBCs from mice and patients (Fig. 4, C and D). These results indicate that A2AR–mGluR5 interaction is induced in BMDCs in severe TBI-induced ALI.
Figure 4.

A2AR–mGluR5 interaction is detected in WBCs of severe TBI-induced ALI mouse model or severe TBI-induced ALI patients. (A) Co-IP of A2AR and mGluR2/3 or mGluR5 in WBCs of mouse models using anti-A2AR antibody as the immunoprecipitating antibody. (B) Co-IP of A2AR and mGluR2/3 or mGluR5 in WBCs of patients using anti-A2AR antibody as the immunoprecipitating antibody. (C and D) Interaction of A2AR and mGluR5 in WBCs of mouse models (C) or of patients (D) was confirmed by co-IP using anti-mGluR5 antibody as the immunoprecipitating antibody. Each experiment was repeated three times. IB, immunoblot.
Elevation of plasma glutamate level and interaction of A2AR and mGluR5 in BMDCs are also observed in a mouse model of moderate TBI-induced ALI
To confirm the links between plasma glutamate level, A2AR–mGluR5 interaction in BMDCs, and TBI-induced lung damage, we used a mouse model of moderate TBI. Of 40 experimental mice with moderate TBI, 38 displayed decreased plasma glutamate levels at 6 h after TBI and recovered almost to baseline at 12 h after injury, which did not exhibit ALI after moderate TBI. Only two mice displayed increased plasma glutamate levels after injury (Fig. 5 A). At 24 h after TBI, only these two mice suffered from ALI with increased lung water content (Fig. 5 B), pathological changes in lung tissues (Fig. 5 D), and deficiency of pulmonary gas exchange (P/F ratio was below 300 mmHg; Fig. 5 C). Moreover, A2AR–mGluR5 interaction in WBCs was observed only in these two mice (Fig. 5, E and F).
Figure 5.

Plasma glutamate level, lung injury parameters, and WBC A2AR–mGluR5 interaction in moderate TBI mouse model. Moderate TBI was induced in 40 mice via cortical impact with the weight-dropping method. Only two of these mice were diagnosed with ALI at 24 h after TBI. (A) Changes of plasma glutamate levels at 6, 12, and 24 h after moderate TBI. (B) Lung water content. (C) Blood gas analyses of arterial blood. (A–C) Values are expressed as mean ± SEM. (D) H&E staining for lung sections. Bars, 100 µm. (E) Co-IP of A2AR and mGluR5 in WBCs using anti-A2AR antibody as the immunoprecipitating antibody. (F) Co-IP of A2AR and mGluR5 in WBCs using anti-mGluR5 antibody as the immunoprecipitating antibody. IB, immunoblot. *, P < 0.01 when compared with the baseline. Each experiment was repeated three times.
Elevated glutamate concentration induces A2AR–mGluR5 interaction to promote the proinflammatory effect of A2AR in neutrophils
To explore whether the elevated plasma glutamate level in TBI-induced ALI is involved in inducing the A2AR–mGluR5 interaction and whether this interaction mediates the proinflammatory effect of BMDC A2AR, we investigated the receptor interaction and the induction of proinflammatory cytokines in human and mouse neutrophils in vitro in the presence of varying concentrations of glutamate (the given glutamate concentrations were relative to the plasma glutamate levels in vivo described above). In mouse neutrophils in the presence of 30 and 300 µmol/l glutamate, a band corresponding to mGluR5 was coimmunoprecipitated by anti-A2AR antibodies (Fig. 6 A), and an A2AR band was coimmunoprecipitated by anti-mGluR5 antibodies (Fig. 6 B), indicating the interaction between these receptors at these glutamate concentrations. At lower concentrations of glutamate, no co-IP was observed (Fig. 6, A and B). mGluR5 siRNA significantly decreased the expression of mGluR5 in mouse neutrophils, establishing our ability to knock down mGluR5 (Fig. 6 C). As shown in Fig. 6 D, the A2AR agonist CGS21680 inhibited LPS-induced IL-1β mRNA expression in neutrophils from WT mice in control medium and in the presence of 5 µmol/l glutamate but enhanced IL-1β mRNA levels in the presence of 30 and 300 µmol/l glutamate (Fig. 6 D). This glutamate concentration–dependent change was not observed in neutrophils from A2AR KO mice or when mGluR5 was knocked down by mGluR5 siRNA in WT neutrophils (Fig. 6 D).
Figure 6.

Effect of plasma glutamate levels on A2AR–mGluR5 interaction and associated inflammatory cytokine expressions. (A and B) Co-IP of A2AR and mGluR5 at varying glutamate concentrations in mouse neutrophils using anti-A2AR antibody or anti-mGluR5 as the immunoprecipitating antibody. IB, immunoblot. (C) Western blot for mGluR5 expression in mouse neutrophils to detect the efficiency of mGluR5 RNAi. (D) Real-time PCR for IL-1β relative mRNA levels in mouse neutrophils. CGS21680: an A2AR agonist; A2AR KO: A2AR gene KO; mGluR5 RNAi: RNA interference of mGluR5 expression. (E and F) Co-IP of A2AR and mGluR5 at varying glutamate concentrations in human neutrophils using anti-A2AR antibody or anti-mGluR5 as the immunoprecipitating antibody. (G) Real-time PCR for IL-1β relative mRNA levels in human neutrophils. To confirm the effect of A2AR–mGluR5 interaction on IL-1β expression, the LPS-stimulated human neutrophils were treated with the selective A2AR agonist CGS21680, A2AR antagonist ZM241385, and mGluR5 antagonist MPEP. Data for real-time PCR are expressed as mean ± SEM. Each experiment was repeated three times. *, P < 0.01 between the two groups; NS, no significant difference between the two groups.
Similar results were obtained in human neutrophils: in the presence of 300 µmol/l glutamate (similar to the plasma glutamate level in patients with TBI-induced ALI), an mGluR5 band was coimmunoprecipitated by anti-A2AR antibody (Fig. 6 E) and an A2AR band was coimmunoprecipitated by anti-mGluR5 antibody (Fig. 6 F). And no co-IP was observed at lower concentrations of glutamate (Fig. 6, E and F). Moreover, CGS21680 switched from inhibiting (in the presence of 0, 5, or 30 µmol/l glutamate) to promoting (at 300 µmol/l glutamate) LPS-induced IL-1β mRNA expression in human neutrophils, dependent on glutamate concentration (Fig. 6 G). The antiinflammatory effect of CGS21680 in the presence of 0, 5, and 30 µmol/l glutamate was completely blocked by the selective A2AR antagonist ZM241385, whereas the proinflammatory effect of CGS21680 in the presence of 300 µmol/l glutamate was suppressed to some extent by ZM241385 or mGluR5 antagonist 2-methyl-6-(phenylethynyl)pyridine (MPEP) alone but completely suppressed by a combination of ZM241385 and MPEP (Fig. 6 G). These findings suggest that the increased plasma glutamate level caused by TBI-induced ALI promotes the interaction between A2AR and mGluR5 in BMDCs and that this interaction is critical for the proinflammatory effect exerted by activation of BMDC A2AR.
Phospholipase C (PLC)–protein kinase C (PKC) signaling mediates the proinflammatory effect of A2AR–mGluR5 interaction in the presence of elevated glutamate levels
We next investigated the signaling pathway associated with the A2AR–mGluR5 interaction–associated proinflammatory effect. As shown in Fig. 7 A, the stimulatory effect of CGS21680 on LPS-induced IL-1β mRNA expression in mouse neutrophils in the presence of 30 and 300 µmol/l glutamate was suppressed by the PKC inhibitor GF109203X but not by the PKA inhibitor H-89. Consistent with this, Western blots showed increased PLC expression in the presence of 30 or 300 µmol/l glutamate, whereas adenylyl cyclase (AC) expression was higher at lower glutamate concentrations (Fig. 7 B). However, in neutrophils from A2AR KO mice or neutrophils pretreated with mGluR5 siRNA, this glutamate level–dependent change in expression of signaling molecules was not observed (Fig. 7 B). We obtained similar results in human neutrophils: H-89 blocked the antiinflammatory effect of A2AR activation in the presence of 0, 5, and 30 µmol/l glutamate but had no effect on its proinflammatory effect in the presence of 300 µmol/l glutamate, which was instead inhibited by GF109203X (Fig. 7 C). The increase in AC expression detected at lower glutamate concentrations was blocked by the A2AR antagonist ZM241385, whereas the significantly elevated PLC levels detected in the presence of high concentrations of glutamate were largely inhibited by a combination of ZM241385 and the mGluR5 antagonist MPEP (Fig. 7 D). These data indicate that the A2AR–mGluR5 complex–dependent proinflammatory effect of A2AR activation that occurs in the presence of elevated glutamate levels is mediated by a PLC–PKC signaling pathway.
Figure 7.

Signaling of the A2AR–mGluR5 interaction–associated proinflammatory effect. (A) Real-time PCR for IL-1β relative mRNA levels in mouse neutrophils. CGS21680: an A2AR agonist; H-89: a PKA inhibitor; GF109203X: a PKC inhibitor. (B) Western blot for AC and PLC expression changes in mouse neutrophils. (C) Real-time PCR for IL-1β relative mRNA levels in human neutrophils. (D) Western blot for AC and PLC expression changes in human neutrophils. Data for real-time PCR are reported as mean ± SEM of three independent experiments with three replications in each experiment. *, P < 0.01 between the two groups; NS, no significant difference between the two groups.
The A2AR agonist CGS21680 aggravates and A2AR antagonist ZM241385 attenuates lung damage and neutrophil infiltration in a mouse model of severe TBI-induced ALI
To verify the plasma glutamate–modulated proinflammatory role of BMDC A2AR described above, we investigated the effects of A2AR agonist and antagonist on lung damage in a mouse model of severe TBI-induced ALI. As shown in Fig. 8, in WT mice suffering from severe TBI-induced ALI, the A2AR agonist CGS21680 aggravated lung damage as measured by lung water content (Fig. 8 A), P/F ratio (Fig. 8 B), pulmonary pathological changes (Fig. 8 C), and neutrophil infiltration into lung tissues (Fig. 8 D). The A2AR antagonist ZM241385 attenuated the lung damage as measured by these parameters (Figs. 8, A–D). These effects of CGS21680 and ZM241385 were not observed in A2AR KO mice (Fig. 8). These data confirm the aggravating effect of A2AR activation in severe TBI-induced ALI and suggest that A2AR antagonist is protective in the treatment of neurogenic ALI.
Figure 8.

A2AR agonist aggravates, whereas antagonist attenuates lung damage and neutrophil infiltration in the mouse severe TBI-induced ALI model. Effects of A2AR agonist CGS21680 and antagonist ZM241385 on the treatment of severe TBI-induced ALI. (A) Lung water content. (B) Blood gas analyses of arterial blood. (C) H&E staining for lung sections. (D) Immunofluorescence for neutrophil infiltration with anti–CD-177 antibody; nucleoli were stained by DAPI in WT or A2AR KO mice. Neutrophil counting and statistical analysis were performed by Image-Pro plus version 4.5 and presented as an index of CD-177–positive cells. (A, B, and D) Values are expressed as mean ± SEM. *, P < 0.01 when compared with the vehicle treatment; NS, no significant difference between the two groups. Each experiment was repeated three times. Bars, 100 µm.
DISCUSSION
Inflammatory response plays an important role in the progression of ALI. Activation of A2AR is widely considered to be an antiinflammatory event in nonneurogenic ALI, as A2AR inhibits BMDC activation and inflammatory cytokine release. Consistent with this perspective, our findings in an oleic acid–induced ALI model show that global A2AR KO and selective inactivation of BMDC A2AR accelerate lung inflammatory damage. In contrast, in the severe TBI-induced ALI mouse model, selective inactivation of BMDC A2AR significantly attenuated lung damage and expression of inflammatory cytokines TNF and IL-1β. This result provides strong evidence that BMDC A2AR activation has opposite effects on nonneurogenic ALI and the neurogenic ALI induced by severe TBI.
Our previous study in mouse models of moderate TBI and in isolated microglia demonstrated that high concentrations of glutamate in the environment can switch the antiinflammatory effect of A2AR activation to a proinflammatory effect (Dai et al., 2010b). This prompted us to measure glutamate levels in blood plasma during BMDC A2AR activation and to investigate whether glutamate levels also play a role in the progression of TBI-induced ALI. Our results show that both in patients with severe TBI-induced ALI and in mouse models of TBI-induced ALI, plasma glutamate levels during the 24 h after injury were much higher than in lung trauma–induced ALI patients or in mice with oleic acid–induced ALI. Moreover, we found a significant A2AR–mGluR5 interaction in WBCs from patients with severe TBI-induced ALI and WBCs from the mouse model of severe TBI-induced ALI but not in WBCs from patients with trauma-induced ALI or the oleic acid–induced ALI mouse model. This association between plasma glutamate level, lung injury, and BMDC A2AR–mGluR5 interaction was confirmed by the investigation in the mouse model of moderate TBI. In the very small minority of mice suffering from ALI after moderate TBI, we also detected elevated plasma glutamate levels, lung damage, and A2AR–mGluR5 interaction in WBCs. Numerous experimental and clinical studies have demonstrated that CSF or extracellular fluid glutamate concentration increases significantly after TBI, reflecting neuronal and glial glutamate release, diminished glial glutamate uptake, and a damaged blood–brain barrier (Baker et al., 1993; Palmer et al., 1993, 1994; Tomkins et al., 2008). However, further study is required to determine whether the deficiency of glutamate transporter, insufficient capacity of the blood glutamate scavenger oxaloacetate, muscle integrity and muscle breakdown (Lee et al., 1998; O’Kane et al., 1999; Hyde et al., 2003; Hawkins et al., 2006; Zlotnik et al., 2010), or some other mechanism accounts for the increase of plasma glutamate we observed in TBI-induced ALI in the present study.
Previously, functional A2AR–mGluR5 heteromeric complexes had only been reported in the central nervous system (Ferré et al., 2002; Nishi et al., 2003; Brown et al., 2012). The present study provides the first evidence for this interaction in BMDCs and the first evidence that the complex is induced in ALI. Infiltration of neutrophils associated with inflammatory cytokine release is a hallmark event in the progression of ALI and its severe form, acute respiratory distress syndrome (Baughman et al., 1996; Abraham et al., 2000; Azoulay et al., 2002; Reutershan and Ley, 2004). To confirm that TBI-induced high plasma glutamate levels trigger the A2AR–mGluR5 interaction and that the complex is an important mediator of the proinflammatory effect of BMDC A2AR, we performed additional experiments in isolated human and mouse neutrophils. These in vitro experiments revealed that elevated glutamate levels (300 µmol/l for human neutrophils; 30 and 300 µmol/l for mouse neutrophils) induced the A2AR–mGluR5 interaction. The glutamate levels required to trigger the A2AR–mGluR5 interaction in neutrophils are similar to those detected in patients with TBI-induced ALI and in the mouse model, respectively. In elevated glutamate environments, neutrophil A2AR activation exerted a proinflammatory effect rather than the antiinflammatory effect observed in the presence of lower glutamate concentrations. Moreover, this effect was largely suppressed by either A2AR antagonist ZM241385 or mGluR5 antagonist MPEP and completely blocked by their combination, confirming that A2AR–mGluR5 interaction mediated this proinflammatory effect.
Both A2AR and mGluR5 are G protein–coupled receptors (GPCRs) comprised of a bundle of seven transmembrane (7TM) α-helices connected by three extracellular loops and three intracellular loops (Katritch et al., 2012). Some previous studies have reported that agonists of GPCRs can affect the formation and stability of GPCR homo- or heterocomplexes (Hebert et al., 1996; Angers et al., 2000; Rocheville et al., 2000), provoke changes in downstream signaling, and regulate their functions (Jordan and Devi, 1999; Gomes et al., 2001; Rios et al., 2001; Cheng et al., 2003). These reports provide an explanation for our results. First, A2AR–mGluR5 interaction and this interaction-mediated proinflammatory effect require both A2AR agonist CGS21680 and the presence of a certain concentration of glutamate, the endogenous agonist of mGluR5. Second, this proinflammatory effect is mediated by PLC–PKC signaling rather than by the AC–PKA pathway responsible for the antiinflammatory effect of A2AR activation in low concentrations of glutamate. It is believed that mGluR5 is coupled with Gq, which classically activates the PLC–PKC pathway (Ozawa et al., 1998). A2AR is coupled with Gs, although AC-cAMP-PKA activation is the predominant downstream signaling pathway, provoking PKA-dependent or -independent PKC signaling (Pinto-Duarte et al., 2005; Fredholm et al., 2007). In the present study, we demonstrate that the PLC–PKC signaling pathway contributes to the glutamate-induced proinflammatory effect of A2AR–mGluR5 interaction in a PKA-independent manner, which accounts for the deleterious effect of BMDC A2AR in severe TBI-induced ALI. Confirming this finding, A2AR antagonist rather than agonist attenuated lung inflammatory damages in severe TBI-induced ALI. Previously, injection of enough glutamate was reported to cause excitotoxic ALI by activating NMDA receptors in lung tissue (Said et al., 1996; Shen et al., 2007), but our results suggest that plasma glutamate–modulated interaction of A2AR and mGluR5 on BMDCs aggravates TBI-induced ALI. It not only presents a novel view of the effect of A2AR in ALI, but also of the role of glutamate in ALI.
In summary, we demonstrate that high levels of glutamate induce the A2AR–mGluR5 interaction in BMDCs to trigger a proinflammatory effect of A2AR activation via PLC–PKC signaling. This finding offers new insight into the clinical use of A2AR modulators (agonist or antagonist) and glutamate inhibitor for TBI-induced ALI treatment. When plasma glutamate levels are elevated, A2AR antagonist rather than agonist is a potentially beneficial treatment for attenuating lung damage in severe TBI-induced ALI. Importantly, A2AR agonist may actually exacerbate the damage in these conditions.
MATERIALS AND METHODS
Animals.
Global A2AR homozygous KO mice and their WT littermates used in this study were developed in the laboratory of J.-F. Chen (Chen et al., 1999) and were generated as previously described (Chen et al., 1999; Dai et al., 2010a,b). In brief, congenic global A2AR KO mice on a C57BL/6 background were generated by backcrossing global A2AR KO on mixed (129-Steel×C57BL/6) genetic background to C57BL/6 mice for 13–15 generations. Heterozygous crossbreeding was used to generate global A2AR KO mice and their WT littermates. All procedures used in this study were approved by the Institutional Animal Care and Use Committee of the Third Military Medical University.
Patients.
ALI is defined as an acute noncardiogenic pulmonary edema with a P/F gradient below 300 mmHg. Hospitalized patients with TBI-induced ALI or lung trauma–induced ALI and their blood cell cultures were eligible for investigation. This study was approved by the Third Military Medical University Human Studies Committee. The clinical information for these patients is presented in Tables S1 and S2.
BMT.
To selectively inactivate or reconstitute A2AR on BMDCs, BMT between WT mice and A2AR KO mice was performed as previously described (Yu et al., 2004; Dai et al., 2010a).
Induction of severe TBI-induced ALI and oleic acid–induced ALI.
A severe cortical impact was performed by the weight-dropping method (Li et al., 2008, 2009). This reproducible and consistent model is generally associated with 30% mortality within the first 5 min after injury and neurological deficit scores of III within the 24 h after injury (Jin et al., 2008). As in patients, ALI in mice was defined as lung edema with a P/F gradient below 300 mmHg, ruling out heart failure as a mechanism. The nonneurogenic ALI model was induced by intravenous injection of 0.6 ml/kg oleic acid (Sigma-Aldrich). In addition, to confirm the effect of TBI severity on lung damage, a moderate TBI model in mice was also induced via the weight-dropping method. This model yielded a neurological deficit score of II within the 24 h after injury. To investigate the effect of A2AR agonist and antagonist in the mouse severe TBI-induced ALI model, A2AR agonist CGS21680 (0.1 mg/kg) or antagonist ZM241385 (1 mg/kg) was given to the mice at 3 h after TBI.
Assay of lung injury parameters.
The lung injury parameters of mice such as lung water content, hematoxylin and eosin (H&E) staining, and TNF and IL-1β protein levels were assessed as previously described (Liu et al., 2010a). Using goat anti–mouse A2AR antibody (Everest Biotech Ltd.), rabbit anti–mouse CD177 antibody (Beijing Biosynthesis Biotechnology Co., Ltd.), and FITC- or TRITC-conjugated secondary antibody (Beijing Cowin Biotech Co., Ltd.), A2AR location and neutrophil infiltration in lung tissue were determined with standard immunofluorescence immunohistochemistry (Donadieu et al., 2007). Nuclei were then stained by DAPI. The results were analyzed by Image-Pro plus version 4.5 (Media Cybernetics) as described previously (Li et al., 2008). Arterial blood samples were withdrawn into a heparinized syringe by percutaneous left ventricular sampling of lightly anesthetized mice spontaneously breathing room air (Fagan et al., 1999; Xu et al., 2006). Blood gas analysis was immediately performed using the I-STAT Analyzer (Abbott).
Measurement of glutamate level in blood plasma.
Glutamate levels in blood plasma of the patients (Tables S1 and S2) or mice were analyzed by HPLC using ortho-phthaldialdehyde precolumn derivatization and fluorescence detection as previously reported (Aliprandi et al., 2005). Samples were run through a C18 column (Supelcosil) detected by a 157 spectrofluorometric detector (Beckman Coulter) with the excitation wavelength set at 330 nm and the emission wavelength fixed at 450 nm.
WBC isolation and protein extraction.
Blood samples from patients or the mouse models were collected in EDTA tubes, and WBCs were prepared by isolating the buffy coat layer after centrifugation at 3,500 rpm for 10 min at 4°C. For the co-IP experiment, WBC proteins were extracted with TRIzol reagent (Invitrogen) from the interphase and lower phenol phase after first removing the upper aqueous phase according to the manufacturer’s instructions (Chung et al., 2009).
Pharmacological treatments of isolated neutrophil.
Using a discontinuous Percoll gradient (Henson and Oades, 1975; Graham et al., 2007), human neutrophils from healthy donors were isolated from heparinized blood, whereas mouse neutrophils were isolated from BM cells according to the method described previously (Tsai and Collins, 1993; Magalhães et al., 2007). Neutrophils were pretreated with 0, 5, 30, or 300 µM glutamate for 30 min, followed by a combination of 1,000 ng/ml LPS and 100 nM A2AR agonist CGS21680 for 6 h (Dai et al., 2010b). To elucidate the associated signaling, 1 µM A2AR selective antagonist ZM241385, 10 µM PKA inhibitor H-89, 5 µM PKC inhibitor GF109203X, and mouse mGluR5 siRNA (Santa Cruz Biotechnology, Inc.) and 1 µM mGluR5 antagonist MPEP (Pacheco et al., 2006) were used. For these cotreatments, CGS21680, ZM241385, and MPEP were added 10 min before LPS treatment. H-89 or GF109203X was added to the cultures 30 min before glutamate was added (Dai et al., 2010b). For the experiments involving mGluR5 siRNA, mouse neutrophils were transfected using the Nucleofection technology (Amaxa) for 24 h: 1.4 µg of each siRNA was transfected into 1 × 106 cells according to the manufacturer’s recommendations. Controls underwent the same transfection procedure. Transfection efficiency was confirmed by Western blot for mGluR5 expression.
Assay of IL-1β mRNA expressions in neutrophils.
6 h after agents were added, total RNA of neutrophils was isolated using TRIzol reverse transcribed. IL-1β mRNA expression in neutrophils was evaluated with real-time PCR (Takara Bio Inc.) using the primers described previously (Dai et al., 2010b; Keita et al., 2010). All reported values are presented as mean ± SEM of three independent experiments conducted in triplicate.
Detection of A2AR and mGluR5 interaction.
By using protein A–agarose beads (EMD Millipore) and with the anti-A2AR (Santa Cruz Biotechnology, Inc.), anti-mGluR2/3 (Abcam), or anti-mGluR5 antibodies (Abcam) for human and mouse, co-IP was performed as described previously (Schröder et al., 2009). The immunoreactive bands were developed with the enhanced chemiluminescence detection kit (SuperSignal; Thermo Fisher Scientific).
Western blot for signal molecule expressions.
To investigate the associated signaling of A2AR activation, Western blot analysis was also performed using antibodies against AC (Abcam) and PLC (Santa Cruz Biotechnology, Inc.). In these experiments, β-actin (Santa Cruz Biotechnology, Inc.) served as the endogenous control.
Statistical analysis.
Statistical comparisons of more than two groups were performed by factorial ANOVA followed by Bonferroni’s post hoc test. Graphic data represent mean ± SEM. A value of P < 0.01 was considered statistically significant.
Online supplemental material.
Table S1 lists characteristics of patients with severe TBI-induced ALI, including age, initial Glasgow coma scale, and the time of ALI onset. Table S2 lists characteristics of patients with lung trauma–induced ALI. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20122196/DC1.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr. Susan E. Lewis for critically reading and editing the manuscript. We also thank Dr. Ping Li, Dr. Xing-Yun Chen, Mr. Ren-Ping Xiong, Mr. Zi-Ai Zhao, and Xing Chen for technical assistance.
This work was supported by grants from the National Natural Science Foundation of China (nos. 30900587, 81172817, and 31171022), the Natural Science Foundation of Chongqing, China (no. CSTC2009BB5317), and the Science Foundation of Third Military Medical University (to S.-S. Dai).
The authors have no conflicting financial interests.
Author contributions: S.-S. Dai and Y.-G. Zhou designed the research; S.-S. Dai, H. Wang, N. Yang, J.-H. An, W. Li, and Y.-L. Ning performed the research; Y.-G. Zhou and P.-F. Zhu contributed analytical tools; S.-S. Dai, J.-F. Chen, and Y.-G. Zhou analyzed the data; and S.-S. Dai and Y.-G. Zhou wrote the paper.
Footnotes
Abbreviation used:
- A2AR
- adenosine A2A receptor
- AC
- adenylyl cyclase
- ALI
- acute lung injury
- BMT
- BM transplantation
- GPCR
- G protein–coupled receptor
- IP
- immunoprecipitation
- MPEP
- 2-methyl-6-(phenylethynyl)pyridine
- PLC
- phospholipase C
- TBI
- traumatic brain injury
References
- Abraham E. 2003. Neutrophils and acute lung injury. Crit. Care Med. 31:S195–S199 10.1097/01.CCM.0000057843.47705.E8 [DOI] [PubMed] [Google Scholar]
- Abraham E., Carmody A., Shenkar R., Arcaroli J. 2000. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 279:L1137–L1145 [DOI] [PubMed] [Google Scholar]
- Aliprandi A., Longoni M., Stanzani L., Tremolizzo L., Vaccaro M., Begni B., Galimberti G., Garofolo R., Ferrarese C. 2005. Increased plasma glutamate in stroke patients might be linked to altered platelet release and uptake. J. Cereb. Blood Flow Metab. 25:513–519 10.1038/sj.jcbfm.9600039 [DOI] [PubMed] [Google Scholar]
- Angers S., Salahpour A., Joly E., Hilairet S., Chelsky D., Dennis M., Bouvier M. 2000. Detection of beta 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. USA. 97:3684–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoulay E., Darmon M., Delclaux C., Fieux F., Bornstain C., Moreau D., Attalah H., Le Gall J.R., Schlemmer B. 2002. Deterioration of previous acute lung injury during neutropenia recovery. Crit. Care Med. 30:781–786 10.1097/00003246-200204000-00010 [DOI] [PubMed] [Google Scholar]
- Baker A.J., Moulton R.J., MacMillan V.H., Shedden P.M. 1993. Excitatory amino acids in cerebrospinal fluid following traumatic brain injury in humans. J. Neurosurg. 79:369–372 10.3171/jns.1993.79.3.0369 [DOI] [PubMed] [Google Scholar]
- Baughman R.P., Gunther K.L., Rashkin M.C., Keeton D.A., Pattishall E.N. 1996. Changes in the inflammatory response of the lung during acute respiratory distress syndrome: prognostic indicators. Am. J. Respir. Crit. Care Med. 154:76–81 [DOI] [PubMed] [Google Scholar]
- Bratton S.L., Davis R.L. 1997. Acute lung injury in isolated traumatic brain injury. Neurosurgery. 40:707–712 10.1097/00006123-199704000-00009 [DOI] [PubMed] [Google Scholar]
- Bronchard R., Albaladejo P., Brezac G., Geffroy A., Seince P.F., Morris W., Branger C., Marty J. 2004. Early onset pneumonia: risk factors and consequences in head trauma patients. Anesthesiology. 100:234–239 10.1097/00000542-200402000-00009 [DOI] [PubMed] [Google Scholar]
- Brown R.M., Duncan J.R., Stagnitti M.R., Ledent C., Lawrence A.J. 2012. mGlu5 and adenosine A2A receptor interactions regulate the conditioned effects of cocaine. Int. J. Neuropsychopharmacol. 15:995–1001 10.1017/S146114571100126X [DOI] [PubMed] [Google Scholar]
- Chen J.F., Huang Z., Ma J., Zhu J., Moratalla R., Standaert D., Moskowitz M.A., Fink J.S., Schwarzschild M.A. 1999. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J. Neurosci. 19:9192–9200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z.J., Harikumar K.G., Holicky E.L., Miller L.J. 2003. Heterodimerization of type A and B cholecystokinin receptors enhance signaling and promote cell growth. J. Biol. Chem. 278:52972–52979 10.1074/jbc.M310090200 [DOI] [PubMed] [Google Scholar]
- Chung L., Nelson A.E., Ho K.K., Baxter R.C. 2009. Proteomic profiling of growth hormone-responsive proteins in human peripheral blood leukocytes. J. Clin. Endocrinol. Metab. 94:3038–3043 10.1210/jc.2009-0778 [DOI] [PubMed] [Google Scholar]
- Ciruela F., Escriche M., Soloviev M.M., Canela E.I., Burgeño J., Mallol J., Chan W.-Y., Lluis C., Jeffrey McIlhinney R.A., Franco R. 2001. Adenosine-glutamate receptor–receptor interactions in the central nervous system. Drug Dev. Res. 52:316–322 10.1002/ddr.1129 [DOI] [Google Scholar]
- Contant C.F., Valadka A.B., Gopinath S.P., Hannay H.J., Robertson C.S. 2001. Adult respiratory distress syndrome: a complication of induced hypertension after severe head injury. J. Neurosurg. 95:560–568 10.3171/jns.2001.95.4.0560 [DOI] [PubMed] [Google Scholar]
- Dai S.S., Li W., An J.H., Wang H., Yang N., Chen X.Y., Zhao Y., Li P., Liu P., Chen J.F., Zhou Y.G. 2010a. Adenosine A2A receptors in both bone marrow cells and non-bone marrow cells contribute to traumatic brain injury. J. Neurochem. 113:1536–1544 [DOI] [PubMed] [Google Scholar]
- Dai S.S., Zhou Y.G., Li W., An J.H., Li P., Yang N., Chen X.Y., Xiong R.P., Liu P., Zhao Y., et al. 2010b. Local glutamate level dictates adenosine A2A receptor regulation of neuroinflammation and traumatic brain injury. J. Neurosci. 30:5802–5810 10.1523/JNEUROSCI.0268-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettbarn C.L., Davidson L.J. 1989. Pulmonary complications in the patient with acute head injury: neurogenic pulmonary edema. Heart Lung. 18:583–589 [PubMed] [Google Scholar]
- Donadieu E., Hamdi W., Deveze A., Lucciano M., Lavieille J.P., Magnan J., Riva C. 2007. Improved cryosections and specific immunohistochemical methods for detecting hypoxia in mouse and rat cochleae. Acta Histochem. 109:177–184 10.1016/j.acthis.2007.01.004 [DOI] [PubMed] [Google Scholar]
- Fagan K.A., Fouty B.W., Tyler R.C., Morris K.G., Jr, Hepler L.K., Sato K., LeCras T.D., Abman S.H., Weinberger H.D., Huang P.L., et al. 1999. The pulmonary circulation of homozygous or heterozygous eNOS-null mice is hyperresponsive to mild hypoxia. J. Clin. Invest. 103:291–299 10.1172/JCI3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S., Karcz-Kubicha M., Hope B.T., Popoli P., Burgueño J., Gutiérrez M.A., Casadó V., Fuxe K., Goldberg S.R., Lluis C., et al. 2002. Synergistic interaction between adenosine A2A and glutamate mGlu5 receptors: implications for striatal neuronal function. Proc. Natl. Acad. Sci. USA. 99:11940–11945 10.1073/pnas.172393799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm B.B., Chern Y., Franco R., Sitkovsky M. 2007. Aspects of the general biology of adenosine A2A signaling. Prog. Neurobiol. 83:263–276 10.1016/j.pneurobio.2007.07.005 [DOI] [PubMed] [Google Scholar]
- Fulton R.L., Jones C.E. 1975. The cause of post-traumatic pulmonary insufficiency in man. Surg. Gynecol. Obstet. 140:179–186 [PubMed] [Google Scholar]
- Gill S.S., Pulido O.M. 2001. Glutamate receptors in peripheral tissues: current knowledge, future research, and implications for toxicology. Toxicol. Pathol. 29:208–223 10.1080/019262301317052486 [DOI] [PubMed] [Google Scholar]
- Gomes I., Jordan B.A., Gupta A., Rios C., Trapaidze N., Devi L.A. 2001. G protein coupled receptor dimerization: implications in modulating receptor function. J. Mol. Med. 79:226–242 10.1007/s001090100219 [DOI] [PubMed] [Google Scholar]
- Graham D.B., Robertson C.M., Bautista J., Mascarenhas F., Diacovo M.J., Montgrain V., Lam S.K., Cremasco V., Dunne W.M., Faccio R., et al. 2007. Neutrophil-mediated oxidative burst and host defense are controlled by a Vav-PLCgamma2 signaling axis in mice. J. Clin. Invest. 117:3445–3452 10.1172/JCI32729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grommes J., Soehnlein O. 2011. Contribution of neutrophils to acute lung injury. Mol. Med. 17:293–307 10.2119/molmed.2010.00138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskó G., Xu D.Z., Lu Q., Németh Z.H., Jabush J., Berezina T.L., Zaets S.B., Csóka B., Deitch E.A. 2006. Adenosine A2A receptor activation reduces lung injury in trauma/hemorrhagic shock. Crit. Care Med. 34:1119–1125 10.1097/01.CCM.0000206467.19509.C6 [DOI] [PubMed] [Google Scholar]
- Hawkins R.A., O’Kane R.L., Simpson I.A., Viña J.R. 2006. Structure of the blood-brain barrier and its role in the transport of amino acids. J. Nutr. 136:218S–226S [DOI] [PubMed] [Google Scholar]
- Hebert T.E., Moffett S., Morello J.P., Loisel T.P., Bichet D.G., Barret C., Bouvier M. 1996. A peptide derived from a beta2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J. Biol. Chem. 271:16384–16392 10.1074/jbc.271.27.16384 [DOI] [PubMed] [Google Scholar]
- Henson P.M., Oades Z.G. 1975. Stimulation of human neutrophils by soluble and insoluble immunoglobulin aggregates. Secretion of granule constituents and increased oxidation of glucose. J. Clin. Invest. 56:1053–1061 10.1172/JCI108152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland M.C., Mackersie R.C., Morabito D., Campbell A.R., Kivett V.A., Patel R., Erickson V.R., Pittet J.F. 2003. The development of acute lung injury is associated with worse neurologic outcome in patients with severe traumatic brain injury. J. Trauma. 55:106–111 10.1097/01.TA.0000071620.27375.BE [DOI] [PubMed] [Google Scholar]
- Hyde R., Taylor P.M., Hundal H.S. 2003. Amino acid transporters: roles in amino acid sensing and signalling in animal cells. Biochem. J. 373:1–18 10.1042/BJ20030405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W., Zhu L., Guan Q., Chen G., Wang Q.F., Yin H.X., Hang C.H., Shi J.X., Wang H.D. 2008. Influence of Nrf2 genotype on pulmonary NF-kappaB activity and inflammatory response after traumatic brain injury. Ann. Clin. Lab. Sci. 38:221–227 [PubMed] [Google Scholar]
- Jin W., Wang H., Ji Y., Zhu L., Yan W., Qiao L., Yin H. 2009. Genetic ablation of Nrf2 enhances susceptibility to acute lung injury after traumatic brain injury in mice. Exp. Biol. Med. (Maywood). 234:181–189 10.3181/0807-RM-232 [DOI] [PubMed] [Google Scholar]
- Jordan B.A., Devi L.A. 1999. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 399:697–700 10.1038/21441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A., Zhao J., Anakk S., Dash P.K., Strobel H.W. 2007. Brain trauma leads to enhanced lung inflammation and injury: evidence for role of P4504Fs in resolution. J. Cereb. Blood Flow Metab. 27:963–974 [DOI] [PubMed] [Google Scholar]
- Katritch V., Cherezov V., Stevens R.C. 2012. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 33:17–27 10.1016/j.tips.2011.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keita M., Bessette P., Pelmus M., Ainmelk Y., Aris A. 2010. Expression of interleukin-1 (IL-1) ligands system in the most common endometriosis-associated ovarian cancer subtypes. J. Ovarian Res. 3:3 10.1186/1757-2215-3-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.J., Hawkins R.A., Viña J.R., Peterson D.R. 1998. Glutamine transport by the blood-brain barrier: a possible mechanism for nitrogen removal. Am. J. Physiol. 274:C1101–C1107 [DOI] [PubMed] [Google Scholar]
- Li W., Dai S., An J., Li P., Chen X., Xiong R., Liu P., Wang H., Zhao Y., Zhu M., et al. 2008. Chronic but not acute treatment with caffeine attenuates traumatic brain injury in the mouse cortical impact model. Neuroscience. 151:1198–1207 10.1016/j.neuroscience.2007.11.020 [DOI] [PubMed] [Google Scholar]
- Li W., Dai S., An J., Xiong R., Li P., Chen X., Zhao Y., Liu P., Wang H., Zhu P., et al. 2009. Genetic inactivation of adenosine A2A receptors attenuates acute traumatic brain injury in the mouse cortical impact model. Exp. Neurol. 215:69–76 10.1016/j.expneurol.2008.09.012 [DOI] [PubMed] [Google Scholar]
- Liu X.C., Zhou Y.G., Wang Y., Chen J.Y., Hao L.B., Li J.D., Dong J.Y., Lin F. 2010a. Antibiotic-loaded cement articulating spacer made by a self-made mold system in the treatment of the infected hip replacement. Zhonghua Wai Ke Za Zhi. 48:1050–1054 [In Chinese.] [PubMed] [Google Scholar]
- Liu X.Y., Liu Y., Li J.F., Yue S.J., Shen L., Li C., Han J.Z., Xu J.P., Feng D.D., Liu H.J., Luo Z.Q. 2010b. Activation of mGluRI in neutrophils promotes the adherence of neutrophils to endothelial cells. Sheng Li Xue Bao. 62:219–224 [In Chinese.] [PubMed] [Google Scholar]
- Mackersie R.C., Christensen J.M., Pitts L.H., Lewis F.R. 1983. Pulmonary extravascular fluid accumulation following intracranial injury. J. Trauma. 23:968–975 10.1097/00005373-198311000-00002 [DOI] [PubMed] [Google Scholar]
- Magalhães M.A., Zhu F., Sarantis H., Gray-Owen S.D., Ellen R.P., Glogauer M. 2007. Expression and translocation of fluorescent-tagged p21-activated kinase-binding domain and PH domain of protein kinase B during murine neutrophil chemotaxis. J. Leukoc. Biol. 82:559–566 10.1189/jlb.0207126 [DOI] [PubMed] [Google Scholar]
- Mascia L., Mastromauro I., Viberti S. 2008. High tidal volume as a predictor of acute lung injury in neurotrauma patients. Minerva Anestesiol. 74:325–327 [PubMed] [Google Scholar]
- Nakajima T., Suarez C.J., Lin K.W., Jen K.Y., Schnitzer J.E., Makani S.S., Parker N., Perkins D.L., Finn P.W. 2010. T cell pathways involving CTLA4 contribute to a model of acute lung injury. J. Immunol. 184:5835–5841 10.4049/jimmunol.0903238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi A., Liu F., Matsuyama S., Hamada M., Higashi H., Nairn A.C., Greengard P. 2003. Metabotropic mGlu5 receptors regulate adenosine A2A receptor signaling. Proc. Natl. Acad. Sci. USA. 100:1322–1327 10.1073/pnas.0237126100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Kane R.L., Martínez-López I., DeJoseph M.R., Viña J.R., Hawkins R.A. 1999. Na(+)-dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) of the blood-brain barrier. A mechanism for glutamate removal. J. Biol. Chem. 274:31891–31895 10.1074/jbc.274.45.31891 [DOI] [PubMed] [Google Scholar]
- Ozawa S., Kamiya H., Tsuzuki K. 1998. Glutamate receptors in the mammalian central nervous system. Prog. Neurobiol. 54:581–618 10.1016/S0301-0082(97)00085-3 [DOI] [PubMed] [Google Scholar]
- Pacheco R., Oliva H., Martinez-Navío J.M., Climent N., Ciruela F., Gatell J.M., Gallart T., Mallol J., Lluis C., Franco R. 2006. Glutamate released by dendritic cells as a novel modulator of T cell activation. J. Immunol. 177:6695–6704 [DOI] [PubMed] [Google Scholar]
- Palmer A.M., Marion D.W., Botscheller M.L., Swedlow P.E., Styren S.D., DeKosky S.T. 1993. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem. 61:2015–2024 10.1111/j.1471-4159.1993.tb07437.x [DOI] [PubMed] [Google Scholar]
- Palmer A.M., Marion D.W., Botscheller M.L., Bowen D.M., DeKosky S.T. 1994. Increased transmitter amino acid concentration in human ventricular CSF after brain trauma. Neuroreport. 6:153–156 10.1097/00001756-199412300-00039 [DOI] [PubMed] [Google Scholar]
- Parekh D., Dancer R.C., Thickett D.R. 2011. Acute lung injury. Clin. Med. 11:615–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto-Duarte A., Coelho J.E., Cunha R.A., Ribeiro J.A., Sebastião A.M. 2005. Adenosine A2A receptors control the extracellular levels of adenosine through modulation of nucleoside transporters activity in the rat hippocampus. J. Neurochem. 93:595–604 10.1111/j.1471-4159.2005.03071.x [DOI] [PubMed] [Google Scholar]
- Qian F., Deng J., Gantner B.N., Flavell R.A., Dong C., Christman J.W., Ye R.D. 2012. Map kinase phosphatase 5 protects against sepsis-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 302:L866–L874 10.1152/ajplung.00277.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reutershan J., Ley K. 2004. Bench-to-bedside review: acute respiratory distress syndrome - how neutrophils migrate into the lung. Crit. Care. 8:453–461 10.1186/cc2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reutershan J., Cagnina R.E., Chang D., Linden J., Ley K. 2007. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J. Immunol. 179:1254–1263 [DOI] [PubMed] [Google Scholar]
- Rios C.D., Jordan B.A., Gomes I., Devi L.A. 2001. G-protein-coupled receptor dimerization: modulation of receptor function. Pharmacol. Ther. 92:71–87 10.1016/S0163-7258(01)00160-7 [DOI] [PubMed] [Google Scholar]
- Rocheville M., Lange D.C., Kumar U., Sasi R., Patel R.C., Patel Y.C. 2000. Subtypes of the somatostatin receptor assemble as functional homo- and heterodimers. J. Biol. Chem. 275:7862–7869 10.1074/jbc.275.11.7862 [DOI] [PubMed] [Google Scholar]
- Said S.I., Berisha H.I., Pakbaz H. 1996. Excitotoxicity in the lung: N-methyl-D-aspartate-induced, nitric oxide-dependent, pulmonary edema is attenuated by vasoactive intestinal peptide and by inhibitors of poly(ADP-ribose) polymerase. Proc. Natl. Acad. Sci. USA. 93:4688–4692 10.1073/pnas.93.10.4688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepp C.P., Reutershan J. 2008. Bench-to-bedside review: adenosine receptors—promising targets in acute lung injury? Crit. Care. 12:226 10.1186/cc6990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder H., Wu D.F., Seifert A., Rankovic M., Schulz S., Höllt V., Koch T. 2009. Allosteric modulation of metabotropic glutamate receptor 5 affects phosphorylation, internalization, and desensitization of the micro-opioid receptor. Neuropharmacology. 56:768–778 10.1016/j.neuropharm.2008.12.010 [DOI] [PubMed] [Google Scholar]
- Sharma A.K., Laubach V.E., Ramos S.I., Zhao Y., Stukenborg G., Linden J., Kron I.L., Yang Z. 2010. Adenosine A2A receptor activation on CD4+ T lymphocytes and neutrophils attenuates lung ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 139:474–482 10.1016/j.jtcvs.2009.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L., Han J.Z., Li C., Yue S.J., Liu Y., Qin X.Q., Liu H.J., Luo Z.Q. 2007. Protective effect of ginsenoside Rg1 on glutamate-induced lung injury. Acta Pharmacol. Sin. 28:392–397 10.1111/j.1745-7254.2007.00511.x [DOI] [PubMed] [Google Scholar]
- Thiel M., Chouker A., Ohta A., Jackson E., Caldwell C., Smith P., Lukashev D., Bittmann I., Sitkovsky M.V. 2005. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 3:e174 10.1371/journal.pbio.0030174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkins O., Shelef I., Kaizerman I., Eliushin A., Afawi Z., Misk A., Gidon M., Cohen A., Zumsteg D., Friedman A. 2008. Blood-brain barrier disruption in post-traumatic epilepsy. J. Neurol. Neurosurg. Psychiatry. 79:774–777 10.1136/jnnp.2007.126425 [DOI] [PubMed] [Google Scholar]
- Tsai S., Collins S.J. 1993. A dominant negative retinoic acid receptor blocks neutrophil differentiation at the promyelocyte stage. Proc. Natl. Acad. Sci. USA. 90:7153–7157 10.1073/pnas.90.15.7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler A.P., Bernard G.R. 2007. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet. 369:1553–1564 10.1016/S0140-6736(07)60604-7 [DOI] [PubMed] [Google Scholar]
- Xu T., Qiao J., Zhao L., Wang G., He G., Li K., Tian Y., Gao M., Wang J., Wang H., Dong C. 2006. Acute respiratory distress syndrome induced by avian influenza A (H5N1) virus in mice. Am. J. Respir. Crit. Care Med. 174:1011–1017 10.1164/rccm.200511-1751OC [DOI] [PubMed] [Google Scholar]
- Yu L., Huang Z., Mariani J., Wang Y., Moskowitz M., Chen J.F. 2004. Selective inactivation or reconstitution of adenosine A2A receptors in bone marrow cells reveals their significant contribution to the development of ischemic brain injury. Nat. Med. 10:1081–1087 10.1038/nm1103 [DOI] [PubMed] [Google Scholar]
- Zlotnik A., Gurevich B., Artru A.A., Gruenbaum S.E., Dubilet M., Leibowitz A., Shaked G., Ohayon S., Shapira Y., Teichberg V.I. 2010. The effect of hyperthermia on blood glutamate levels. Anesth. Analg. 111:1497–1504 10.1213/ANE.0b013e3181fc0112 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.