

Abstract
Gastrin-releasing peptide (GRP), secreted by pulmonary neuroendocrine cells, mediates oxidant-induced lung injury in animal models. Considering that GRP blockade abrogates pulmonary inflammation and fibrosis in hyperoxic baboons, we hypothesized that ionizing radiation triggers GRP secretion, contributing to inflammatory and fibrotic phases of radiation-induced lung injury (RiLI). Using C57BL/6 mouse model of pulmonary fibrosis developing ≥20 weeks after high-dose thoracic radiation (15 Gy), we injected small molecule 77427 i.p. approximately 1 hour after radiation then twice weekly for up to 20 weeks. Sham controls were anesthetized and placed in the irradiator without radiation. Lung paraffin sections were immunostained and quantitative image analyses performed. Mice exposed to radiation plus PBS had increased interstitial CD68+ macrophages 4 weeks after radiation and pulmonary neuroendocrine cells hyperplasia 6 weeks after radiation. Ten weeks later radiation plus PBS controls had significantly increased pSmad2/3+ nuclei/cm2. GRP blockade with 77427 treatment diminished CD68+, GRP+, and pSmad2/3+ cells. Finally, interstitial fibrosis was evident 20 weeks after radiation by immunostaining for α-smooth muscle actin and collagen deposition. Treatment with 77427 abrogated interstitial α-smooth muscle actin and collagen. Sham mice given 77427 did not differ significantly from PBS controls. Our data are the first to show that GRP blockade decreases inflammatory and fibrotic responses to radiation in mice. GRP blockade is a novel radiation fibrosis mitigating agent that could be clinically useful in humans exposed to radiation therapeutically or unintentionally.
Radiation fibrosis is a serious complication that affects normal lung following unintentional exposure or due to therapeutic ionizing radiation of thoracic tumors. Despite advances in radiobiology, precise mechanisms by which radiation induces lung injury remain controversial.1 Classically, radiation-induced lung injury (RiLI) is characterized by a latent period that can last for weeks to months after radiation exposure, followed by 2 stages of overt lung injury that can lead to life-threatening and debilitating pulmonary toxic effects.2,3 Acute inflammatory lung injury arises 1 to 6 months after radiation exposure, with diffuse alveolar damage, similar to acute respiratory distress syndrome. Later, chronic interstitial and intra-alveolar fibrosis develops, predominantly in irradiated segments, with myofibroblast proliferation and collagen deposition. It is unclear why only approximately 15% of radiation-exposed patients develop RiLI.1,4 General cytoprotective agents, such as a catalytic antioxidant metalloporphyrin (AEOL10113), can reduce the severity of RiLI by decreasing free radical injury after radiation.5
Our novel paradigm links gastrin-releasing peptide (GRP) to radiation lung injury. We hypothesized that GRP is a mediator of RiLI: promoting both macrophage accumulation and fibrosis. We propose that ionizing radiation triggers pulmonary neuroendocrine cell (PNEC) hyperplasia, leading to GRP secretion, which then mediates chronic lung injury. GRP receptor (GRPR) gene expression is detected and functional in pulmonary epithelial cells, fibroblasts, endothelial cells, and macrophages.6–10 GRP is a proinflammatory neuropeptide that functions as an inflammatory cell activator, mitogen, and cell differentiation factor.8,10,11 GRP is expressed at the highest levels in PNEC in fetal lung,12 where it can promote lung development.13 After birth, GRP production normally decreases, but elevated levels are associated with many inflammatory lung conditions, including chronic lung disease of newborns (bronchopulmonary dysplasia).14–17 PNEC hyperplasia can be triggered by inflammation or exposure to oxygen or ozone10,16,18 and can take weeks to reach peak levels.19
The present investigation tests the hypothesis that GRP contributes to radiation-induced pulmonary fibrosis in C57BL/6 mice. One hour post exposure to thoracic radiation (15 Gy), we treated mice i.p. with either PBS or GRP blockade by using small molecule 77427. We have quantified results of immunohistochemistry (IHC) by using image analysis with ImageJ version 1.46e (NIH, Bethesda, MD) to determine whether GRP contributes to radiation-induced inflammatory responses and/or fibrosis, specifically including assessment of active transforming growth factor (TGF)-β signaling.
Materials and Methods
Mice
C57BL/6 mice at 8 weeks of age (Charles River Laboratories, Morrisville, NC) were selected for these experiments because they are susceptible to radiation fibrosis.20–22 Females were used because GRPR is X-linked and females have twice the level of GRPR gene expression as males.23
Radiation and 77427 Treatment Regimen
Radiation dose and uniformity of distribution were determined before initiating the study as described.24 In brief, X-ray dosimetry was performed using a calibrated ionization chamber,25,26 with dose rate variation across the field being <6%. Validation and quality assurance of the set-up were performed in mice, and the total accumulated dose differed by ≤5% different from the calculated dose.
Before irradiation, mice were anesthetized with 100 mg/kg of ketamine and 10 mg/kg of xylazine. Ten mice were simultaneously irradiated in the prone position with 15 Gy of 320-kVp X-rays delivered to the thorax (Precision X-ray Inc., North Branford, CT; half value layer = 1.00 mm Cu, dose rate = 69 cGy/min) via adjustable apertures with 8-mm lead shielding of the head and abdomen. These dose rates produce equivalent lung damage without radiation dose sparing.27 Sham-irradiated animals were treated similarly, except that the radiation source was not turned on.
At approximately 1 hour after radiation, mice were injected i.p. with 0.1 mL of either PBS or 77427 (10 nmol per mouse = 500 nmol/L final). This dose was previously determined to be optimal in suppressing inflammatory responses in mouse lung.10 Mice were analyzed for all markers at 2, 4, 6, 8, 10, and 20 weeks after radiation.
Antibodies and IHC
Antibodies were all available commercially from vendors as listed in Table 1. Antibody dilutions and slide pretreatments are also listed in Table 1. The avidin-biotin complex technique of IHC was used as detailed elsewhere.28 For most antibodies, the peroxidase method was used with diaminobenzidine as substrate, except for α-smooth muscle actin (SMA) staining, which used alkaline phosphatase-conjugated anti-SMA with the Vector Red substrate (Vector Laboratories, Burlingame, CA).
Table 1.
Antibodies used for IHC
| Antibody | Clone/catalog no. | Source | Dilution | Reference |
|---|---|---|---|---|
| GRP | 2A11 (mouse) | F. Cuttitta | 1:200 | 10 |
| PGP9.5 | Z5116 (rabbit) | Dako (Carpinteria, CA) | 1:200 | 25 |
| CD68 | MCA1957 (rat) | AbD Serotec (Raleigh, NC) | 1:100 | 29 |
| pSmad2/3 | sc-11769 (goat) | Santa Cruz Biotechnology (Santa Cruz, CA) | 1:100 | 30 |
| α–SMA | Clone 1A4/A5691 (mouse) | Sigma-Aldrich (St. Louis, MO) | 1:60 | 31 |
Computerized Quantitative Image Analysis
Slides from the four groups (eight mice per group at each time point) of experimental mice (radiation versus sham, injected with PBS or 77427 in PBS) were viewed by a board-certified anatomical pathologist using bright field microscopy at all time points (weeks 2, 4, 6, 8, 10, and 20 after radiation). For CD68-, pSmad2/3-, SMA-, and Trichrome-positive cells, 10 to 15 random photomicrographs of the alveoli were taken at ×40 magnification within 1 mm of the pleural surface, including ≥3 to 4 images per lobe for all three lobes of lung that were paraffin embedded (left upper, left lower, and right upper).
For CD68 and pSmad2/3 image analysis, numbers of positive cells were quantified by thresholding the images, binary conversion, and counting the number of positive particles >2 and <20 μm by using ImageJ version 1.46e. The total area of tissue per photomicrograph was determined by thresholding all of the tissue, followed by binary conversion and measurement of the tissue area.
For GRP and PGP9.5, all positive cells per slide were counted manually because they most often occur in clusters that are not recognized as separate cells by ImageJ. This was achieved by counting numbers of nuclei present in cells with immunopositive cytoplasm. The numbers of foci (clusters) and total number of PNECs were normalized for the total lung tissue area on the slide because many PNECs are present in alveolar ducts. The total lung area was determined on nonoverlapping photomicrographs of the whole section at ×2 magnification, which then underwent thresholding, binary conversion, and area measurement.
SMA immunostaining (without any counterstain) was quantified in photomicrographs within 1 mm of the pleural surface (but excluding the pleura) by thresholding in the red visibility range (224 to 255), followed by binary conversion and measurement of area. These values were normalized for the total area of the alveolar tissue in each photomicrograph (excluding conducting airways and their associated blood vessels). Quantification of Trichrome staining was performed using blue collagen staining without any hematoxylin counterstain by thresholding in the blue visibility range (134 to 211), followed by binary conversion and area measurement, again normalized for total tissue in each photomicrograph.
Statistical Analysis
Groups were compared using the unpaired Student’s t-test, with P < 0.05 defined as the level of significance.
Results
To assess possible roles for GRP in tissue injury responses after radiation, mice were given 500 nmol/L final concentration of 77427, twice weekly, beginning 1 hour after radiation. Lung tissues were harvested for inflation fixation and staining at 2, 4, 6, 8, 10, and 20 weeks later. Bright field microscopy and ImageJ screening demonstrated altered numbers of immunopositive cells as follows: macrophages (CD68) only at 4 weeks, neuroendocrine (NE) cells (GRP, PGP9.5) mainly at 6 weeks, activated TGF-β signaling (pSmad2/3) only at 10 weeks, and myofibroblasts (SMA) at 20 weeks (Figures 1, 2, 3 and 4). Trichrome staining was performed only at the 20-week time point.
Figure 1.

GRP blockade with 77427 abrogates peak CD68+ macrophages in mouse lungs at 4 weeks after radiation. A: Sham plus PBS group. CD68 immunostaining detects few alveolar and tissue macrophages in Sham controls (arrows). B: Radiation plus PBS group. After radiation there was a significant increase in CD68+ cells (arrowheads) in the alveolar interstitium. C: Radiation plus 77427 group. CD68+ cells diminished to baseline values in mice given 77427 twice a week after radiation. D: Image analysis. ∗ P < 0.00001 for radiation plus PBS versus either sham plus PBS, sham plus 77427, or radiation plus 77427. Scale bars: 25 μm (A–C). V, blood vessel.
Figure 2.

GRP blockade reduces peak numbers of PNECs at 6 weeks after radiation. A: Sham plus PBS controls have rare GRP+ cells (black arrow) most often at branch points in the airways (GRP immunostaining). B: Six weeks after radiation, there are numerous GRP+ cells in distal bronchiolar epithelium (many indicated by arrows). Most GRP+ cells are also PGP9.5+ (D–F), consistent with a neuroendocrine phenotype (GRP immunostaining). Many of these cells in irradiated mice given PBS are also CC10+ (unpublished data), consistent with a multipotent epithelial phenotype emerging after radiation. C: Mice exposed to radiation and then injected with 77427 twice a week have fewer GRP+ cells in bronchiolar epithelium at 6 weeks (GRP immunostaining). D: Sham plus PBS controls have rare PGP9.5+ cells (none in this field) despite strong positive control staining for PGP9.5 in nerve fibers (white arrows in right lower corner) (PGP9.5 immunostaining). E: Six weeks after radiation, there are numerous PGP9.5+ cells in distal bronchiolar epithelium (arrows) (PGP9.5 immunostaining). PGP9.5+ cells are also positive for GRP (A–C) and CC10 (not shown), consistent with a multipotent epithelial phenotype after radiation. F: Mice exposed to radiation then injected with 77427 twice a week have fewer PGP9.5+ cells in bronchiolar epithelium at 6 weeks (PGP9.5 immunostaining). G: Quantitative image analysis shows a large increase in GRP+ and PGP9.5+ cells in lungs of mice exposed 6 weeks earlier to radiation, which is significantly decreased by treatment with 77427. Compared with sham controls, ∗P < 0.001 and ‡P < 0.01 for either #GRP+ foci per cm2 or #PGP9.5+ cells/cm2 in radiation-treated lung tissue area. Compared with radiation-treated mice; †P < 0.01 for either §P < 0.04 (PGP9.5) for mice exposed to radiation, then further injected with 77427 twice a week. Scale bars: 25 μm (A–F). L, airway lumen; V, blood vessel.
Figure 3.

GRP blockade diminishes pSmad2/3+ cells in mouse lung at 10 weeks after radiation. A: Sham plus PBS group. B: Radiation plus PBS group. C: Radiation plus 77427 group. D: Image analysis. A–C: Nuclear pSmad2/3 immunostaining (black arrows) was significantly elevated at 10 weeks after radiation (compared with negative nuclei counterstained with methyl green, white arrows). Sham controls had relatively few pSmad2/3+ cells; then the relative numbers of these cells increased more than fourfold after radiation. ∗P < 0.001 for radiation plus PBS versus sham plus PBS (fourfold increase) or radiation plus 77427; †P < 0.01 for radiation plus 77427 was only 30% elevated over sham plus 77427; ‡P < 0.001 for sham controls given only 77427 had a doubling of pSmad2/3+ cells versus the sham plus PBS group. Scale bars: 25 μm (A–C). V, blood vessel.
Figure 4.
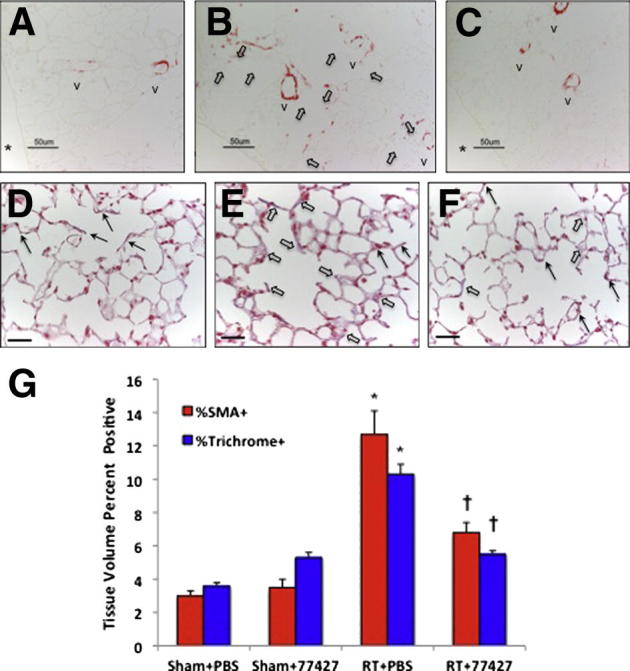
Increased SMA-immunostaining and collagen deposition 20 weeks after radiation is abrogated by GRP blockade. A: Normal SMA immunostaining (red) in sham plus PBS lung occurs in airway and vascular smooth muscle. In subpleural lung (photomicrographs for ImageJ), a few small blood vessels are present. B: Mice exposed to radiation plus PBS have widespread, patchy SMA+ cells throughout the alveolar interstitium (white arrows). C: In radiation plus 77427 mice, SMA immunostaining is mostly restricted to normal small blood vessels. D: Normal trichrome staining (blue to purplish blue) in sham plus PBS lung occurs around airways and blood vessels. In the subpleural region shown here and used for ImageJ analysis, blue outlines of small blood vessels are visible (black arrows). E: Mice exposed to radiation plus PBS have collagen deposition in the alveolar interstitium (some blue to purplish blue interstitium, white arrows), as well as blue outlines of small blood vessels (black arrows). F: In radiation plus 77427 mice, collagen is mostly restricted to small blood vessels (black arrows). G: Quantitative image analysis was performed as detailed in Materials and Methods. For both SMA and trichrome, ∗P < 0.0001 for radiation plus PBS versus sham plus PBS; †P < 0.001 for radiation plus PBS versus radiation plus 77427. Asterisks in A and C indicate pleural surface. Scale bars: 25 μm (A–F). V, blood vessel.
Increased numbers of alveolar macrophages have been reported to occur in mouse lung between 1 and 4 months after radiation as part of an inflammatory phase.29,30 In sham controls, we demonstrated scattered CD68+ cells both in alveolar spaces and in the distal lung interstitium (Figure 1A). At 4 weeks after radiation, CD68+ cells were increased, almost exclusively localized to the alveolar interstitium (Figure 1B). GRP blockade by 77427 reduced this macrophage accumulation in the pulmonary interstitium (Figure 1C). By quantitative image analysis using ImageJ, the CD68+ cells in radiation plus PBS mice were nearly threefold increased compared with sham plus PBS mice (Figure 1D). We did not observe any significant differences in numbers of macrophages at the other time points.
Rare GRP+ and/or PGP9.5+ cells were detected in sham controls (Figure 2, A and D), typically in bronchiolar epithelium near branch points, which is a niche for epithelial progenitor or stem cells in lung.31 At 4 weeks after radiation, three of the eight mice had increased numbers of GRP+ and PGP9.5+ PNECs (data not shown). By 6 weeks after radiation, all animals had elevated numbers of GRP+ (Figure 2B) and PGP9.5+ (Figure 2E) cells, increased up to 20-fold per square centimeter of lung (Figure 2G). This PNEC hyperplasia was linear, manifested primarily as ≥10-fold increase of small GRP+ foci with a mean of two cells per focus. There were significantly fewer PNECs when mice were treated with 77427 (Figure 2, C, F, and G). Both the GRP+ and PGP9.5+ cells were localized almost entirely to small bronchioles (Figure 2, B and E) and yielded similar results of quantitative image analysis (Figure 2G). There was no difference in PCNA labeling of these cells. Interestingly, many GRP+ cells also co-stained for CC10, a Clara cell–specific marker (not shown), indicating that these may fall into the category of regenerative multipotent cells after injury.31 There was no apparent difference in total numbers of CC10+ cells at any time point (data not shown).
Phospho-Smad2/3 (pSmad2/3) is a recognized marker of active TGF-β signaling, as occurs in bleomycin-induced pulmonary fibrosis,32 carbon tetrachloride–induced hepatic fibrosis, and glomerular fibrosis.33,34 Ten weeks after mice received 15 Gy of thoracic radiation, compared with sham plus PBS controls (Figure 3A), lungs had approximately fourfold increased numbers of pSmad2/3+ cells, including fibroblasts, macrophages, endothelium, and epithelial cells, all of which had nuclear immunostaining (Figure 3, B–D). GRP blockade of radiation-exposed mice with 77427 reduced pSmad2/3-immunopositive cells significantly compared with radiation plus PBS–treated mice, representing only a residual of 1.3-fold increased cells compared with the corresponding sham controls. It is unknown why mice given 77427 alone had higher pSmad2/3 immunostaining, but this was not associated with evidence of increased fibrosis (Figure 4).
Figure 4 presents results of two additional widely used markers of fibrosis: SMA (Figure 4, A–C and G), and Masson’s Trichrome (Figure 4, D–G). SMA is expressed by myofibroblasts35 during pulmonary fibrosis of diverse etiologies.36,37 In adult lung at baseline, SMA is normally present in smooth muscle cells surrounding pulmonary vasculature and airways (note “v” indicating small to medium blood vessels in Figure 4, A–C). At 20 weeks after radiation (but not before), mice demonstrated approximately three fold increased SMA, determined as volume percentage of immunostaining of the distal lung parenchyma (Figure 4, B and D). Treatment with 77427 abrogated this increased SMA immunostaining (Figure 4, C and D).
We also performed IHC staining for mature collagens using Masson’s trichrome method with blue stain for collagens (Figure 4, D–G). To permit ImageJ analysis by selecting blue wavelengths, we omitted the hematoxylin counterstain. Normal trichrome staining (blue to purplish blue) was seen in sham plus PBS lung, which was around airways and blood vessels. In the subpleural region, faint blue outlines of small blood vessels could be seen. Notably, mice exposed to radiation plus PBS had collagen deposition in the alveolar interstitium, as well as blue outlines of small blood vessels. Radiation plus 77427 mice had collagen staining mostly restricted to small blood vessels (Figure 4F).
Discussion
The present study demonstrates that GRP blockade significantly mitigates parameters of radiation-induced pulmonary fibrosis in an established mouse model.24 It has already been reported that PNEC triggered by reactive oxygen species secrete GRP in response to lung injury.38 Either of two GRP-blocking agents,39 small molecule 77427 or blocking antibody 2A11, can abrogate multiple parameters of oxidant-triggered lung injury in animal models of reactive airways disease or bronchopulmonary dysplasia.10,16 There is a logical precedent for determining whether GRP promotes radiation fibrosis. In the hyperoxic preterm baboon model of bronchopulmonary dysplasia, GRP blockade by 2A11 resulted in marked reduction in mesenchymal cell proliferation in the alveolar interstitium.16 GRP also induces fibroblast proliferation.40,41 This observation led to the hypothesis that GRP might contribute to interstitial fibrosis after thoracic radiation.
Radiation-induced fibrosis mediated by GRP represents the chronic phase of lung injury linked to a novel master inflammatory circuit that regulates responses of diverse inflammatory cell types.10 It has been known for decades that PNEC hyperplasia can develop with exposure to oxidants41 or chronic inflammation.42 In the setting of chronic inflammation, PNEC hyperplasia appears to be mainly a cell differentiation response, with minimal proliferation of PNECs.43 Furthermore, tumor necrosis factor (TNF)-α treatment of undifferentiated small cell lung carcinoma cells in vitro can induce rapid NE differentiation.44
Despite the large body of evidence for oxidant-induced PNEC hyperplasia and abrogation of PNEC hyperplasia by antioxidant therapy in animal models38 and the induction of NE cell differentiation by TNF-α,44 there has been no prior report of increased numbers of PNECs after radiation exposure. In the current study, PNEC hyperplasia occurred in a predominantly linear pattern 4 to 6 weeks after a single exposure to high-dose RT, peaking at 6 weeks. In hamsters exposed to continuous 65% hyperoxia plus diethylnitrosamine, induction of profound PNEC hyperplasia occurred after 8 to 12 weeks.19 It remains unclear exactly why it takes weeks for the PNEC hyperplasia to develop after radiation exposure and in turn why so many months lapse before radiation fibrosis develops. As with other types of pulmonary fibrosis, we speculate that this delay may be related to the timing of inflammatory cell accumulation and specific cytokine production in the lung.45–47 It is recognized that radiation fibrosis may not be clinically evident for 5 months to even a year after radiation exposure.48–50
In Figure 5, we present our modified working hypothesis about how GRP might mediate lung injury after radiation. Multiple chronic effects of radiation on lung are abrogated by GRP blockade. At 4 weeks after radiation, treatment with 77427 abrogates accumulation of CD68+ macrophages in the alveolar interstitium. Activated macrophages have a high level of GRPR gene expression.10 Functionally, both GRP and bombesin have been shown to promote macrophage activation, chemotaxis, phagocytosis, and oxidative burst.9,51,52 Macrophage-derived cytokines, such as TNF-α, IL-1, and IL-6, are recognized as playing roles in the pathogenesis of radiation fibrosis.30,46,53 The detection of increased numbers of GRP+/PGP9.5+ PNECs at 6 weeks after radiation suggests that macrophage-derived cytokines could contribute in part to the observed PNEC hyperplasia.
Figure 5.

GRP blockade diminishes or abrogates many cellular mechanisms after radiation. At 4 weeks after radiation, the data indicate that GRP increases CD68+ macrophages because GRP blockade with 77427 abrogates the increase and macrophages are GRPR+. At 6 weeks after radiation, GRP promotes increased numbers of GRP+/PGP9.5+ pulmonary neuroendocrine cells (PNECs) consistent with multipotent (plastic) epithelial cells [airway (AR) epithelium is GRPR+]. At 10 weeks after radiation, there is increased expression of pSmad2/3, indicating activation of TGF-β signaling. At 20 weeks after radiation, early interstitial fibrosis is evident as SMA+ myofibroblasts and collagen deposition in alveolar septa (pulmonary fibroblasts are GRPR+). All these functional and/or phenotypic changes are reversed by GRP blockade.
In the present study, many of the interstitial macrophages are positive for pSmad2/3, indicating activation of TGF-β signaling. TGF-β has been widely implicated in the etiology of radiation fibrosis.54 At 20 weeks after radiation, we demonstrate early interstitial fibrosis as SMA+ myofibroblasts and collagen deposition in the alveolar interstitium. All of the functional and phenotypic changes observed in the present study are diminished by GRP blockade. We previously reported that pulmonary fibroblasts are GRPR+7 and fibroblasts from multiple sources proliferate in response to GRP or bombesin.40,41 Peak levels of radiation fibrosis have been documented to occur at 5 to 9 months after radiation.46,55
In conclusion, we here demonstrate that radiation-induced PNEC hyperplasia occurs as a novel cellular response during the acute lung injury phase of radiation fibrosis. Chronologically, this cellular alteration in airway epithelial NE cell differentiation follows peak macrophage accumulation and leads to increased TGF-β signaling and the onset of interstitial fibrosis with myofibroblast infiltration and collagen deposition in the alveolar interstitium. By using GRP blockade in a definitive experiment, we have determined that GRP plays a significant role in vivo in diminishing or abrogating multiple significant parameters on the pathway to radiation fibrosis. It is likely that the ultimate outcome of pulmonary fibrosis is influenced by many other cellular alterations and innate immune responses, including altered regulation of angiogenesis, apoptosis, and antioxidant defenses after radiation exposure.45,48–50,56,57 The identification of PNECs and GRP as key players in radiation fibrosis should lead to new roadmaps for scientific exploration in pursuit of mitigating or protective agents to protect patients from therapeutic or unintentional radiation injury.
Footnotes
Supported by a RadCCore Pilot grant AI-067798-02 (M.E.S.) (program director: M. Dewhirst, Center for Medical Countermeasures Against Radiation Injury, University of Pittsburgh, Pittsburgh, PA, and Duke University, Durham, NC).
Disclosure: F.C. has a patent on 77427 as a treatment for cancer.
Current address of I.L.J. & Z.V., Department of Radiation Oncology, University of Maryland, Baltimore, Maryland.
Contributor Information
Shutang Zhou, Email: shutang.zhou@duke.edu.
Mary E. Sunday, Email: mary.sunday@duke.edu.
References
- 1.Vujaskovic Z., Marks L.B., Anscher M.S. The physical parameters and molecular events associated with radiation-induced lung toxicity. Semin Radiat Oncol. 2000;10:296–307. doi: 10.1053/srao.2000.9424. [DOI] [PubMed] [Google Scholar]
- 2.Dorr W., Baumann M., Herrmann T. Radiation-induced lung damage: a challenge for radiation biology, experimental and clinical radiotherapy. Int J Radiat Biol. 2000;76:443–446. doi: 10.1080/095530000138420. [DOI] [PubMed] [Google Scholar]
- 3.Morgan G.W., Breit S.N. Radiation and the lung: a reevaluation of the mechanisms mediating pulmonary injury. Int J Radiat Oncol Biol Phys. 1995;31:361–369. doi: 10.1016/0360-3016(94)00477-3. [DOI] [PubMed] [Google Scholar]
- 4.Marks L.B., Yu X., Vujaskovic Z., Small W., Jr., Folz R., Anscher M.S. Radiation-induced lung injury. Semin Radiat Oncol. 2003;13:333–345. doi: 10.1016/S1053-4296(03)00034-1. [DOI] [PubMed] [Google Scholar]
- 5.Vujaskovic Z., Batinic-Haberle I., Rabbani Z.N., Feng Q.F., Kang S.K., Spasojevic I., Samulski T.V., Fridovich I., Dewhirst M.W., Anscher M.S. A small molecular weight catalytic metalloporphyrin antioxidant with superoxide dismutase (SOD) mimetic properties protects lungs from radiation-induced injury. Free Radic Biol Med. 2002;33:857–863. doi: 10.1016/s0891-5849(02)00980-2. [DOI] [PubMed] [Google Scholar]
- 6.Siegfried J.M., DeMichele M.A.A., Hunt J.D., Davis A.G., Vohra K.P., Pilewski J.M. Expression of mRNA for gastrin-releasing peptide receptor by human bronchial epithelial cells: association with prolonged tobacco exposure and responsiveness to bombesin-like peptides. Am J Respir Crit Care Med. 1997;156:358–366. doi: 10.1164/ajrccm.156.2.9608047. [DOI] [PubMed] [Google Scholar]
- 7.Subramaniam M., Bausch C., Twomey A., Andreeva S., Yoder B.A., Chang L.Y., Crapo J.D., Pierce R.A., Cuttitta F., Sunday M.E. Bombesin-like peptides modulate alveolarization and angiogenesis in bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2007;176:902–912. doi: 10.1164/rccm.200611-1734OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jensen R.T., Battey J.F., Spindel E.R., Benya R.V. International Union of Pharmacology. LXVIII. Mammalian bombesin receptors: nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol Rev. 2008;60:1–42. doi: 10.1124/pr.107.07108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De la Fuente M., Del Rio M., Ferrandez M.D., Hernanz A. Modulation of phagocytic function in murine peritoneal macrophages by bombesin, gastrin-releasing peptide and neuromedin C. Immunology. 1991;73:205–211. [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou S., Potts E.N., Cuttitta F., Foster W.M., Sunday M.E. Gastrin-releasing peptide blockade as a broad-spectrum anti-inflammatory therapy for asthma. Proc Natl Acad Sci U S A. 2011;108:2100–2105. doi: 10.1073/pnas.1014792108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Majumdar I.D., Weber H.C. Biology of mammalian bombesin-like peptides and their receptors. Curr Opin Endocrinol Diabetes Obes. 2010;18:68–74. doi: 10.1097/MED.0b013e328340ff93. [DOI] [PubMed] [Google Scholar]
- 12.Wharton J., Polak J.M., Bloom S.R., Ghatei M.A., Solcia E., Brown M.R., Pearse A.G.E. Bombesin-like immunoreactivity in the lung. Nature. 1978;273:769–770. doi: 10.1038/273769a0. [DOI] [PubMed] [Google Scholar]
- 13.Sunday M.E., Cutz E. Role of neuroendocrine cells in fetal and postnatal lung. In: Mendelson C.R., editor. Humana Press; Totowa, NJ: 2000. pp. 299–336. [Google Scholar]
- 14.Johnson D.E., Lock J.E., Elde R.P., Thompson T.R. Pulmonary neuroendocrine cells in hyaline membrane disease and bronchopulmonary dysplasia. Pediatr Res. 1982;16:446–454. doi: 10.1203/00006450-198206000-00009. [DOI] [PubMed] [Google Scholar]
- 15.Meloni F., Ballabio P., Pistorio A., Todarello C., Montoli C., Berrayah L., Meloni C., Grassi C., Aguayo S.M. Urinary levels of bombesin-related peptides in a population sample from northern Italy: potential role in the pathogenesis of chronic obstructive pulmonary disease. Am J Med Sci. 1998;315:258–265. doi: 10.1097/00000441-199804000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Sunday M.E., Yoder B.A., Cuttitta F., Haley K.J., Emanuel R.L. Bombesin-like peptide mediates lung injury in a baboon model of bronchopulmonary dysplasia. J Clin Invest. 1998;102:584–594. doi: 10.1172/JCI2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cullen A., Van Marter L.J., Moore M., Parad R., Sunday M.E. Urine bombesin-like peptide elevation precedes clinical evidence of bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2002;165:1093–1097. doi: 10.1164/ajrccm.165.8.2108044. [DOI] [PubMed] [Google Scholar]
- 18.Sunday M.E., Shan L., Subramaniam M. Immunomodulatory functions of the diffuse neuroendocrine system: implications for bronchopulmonary dysplasia. Endocr Pathol. 2004;15:91–106. doi: 10.1385/ep:15:2:091. [DOI] [PubMed] [Google Scholar]
- 19.Sunday M.E., Willett C.G. Induction and spontaneous regression of intense pulmonary neuroendocrine cell differentiation in a model of preneoplastic lung injury. Cancer Res. 1992;52:2677s–2686s. [PubMed] [Google Scholar]
- 20.Johnston C.J., Williams J.P., Elder A., Hernady E., Finkelstein J.N. Inflammatory cell recruitment following thoracic irradiation. Exp Lung Res. 2004;30:369–382. doi: 10.1080/01902140490438915. [DOI] [PubMed] [Google Scholar]
- 21.Adawi A., Zhang Y., Baggs R., Rubin P., Williams J., Finkelstein J., Phipps R.P. Blockade of CD40-CD40 ligand interactions protects against radiation-induced pulmonary inflammation and fibrosis. Clin Immunol Immunopathol. 1998;89:222–230. doi: 10.1006/clin.1998.4606. [DOI] [PubMed] [Google Scholar]
- 22.Epperly M.W., Sikora C.A., DeFilippi S.J., Gretton J.E., Bar-Sagi D., Archer H., Carlos T., Guo H., Greenberger J.S. Pulmonary irradiation-induced expression of VCAM-I and ICAM-I is decreased by manganese superoxide dismutase-plasmid/liposome (MnSOD-PL) gene therapy. Biol Blood Marrow Transplant. 2002;8:175–187. doi: 10.1053/bbmt.2002.v8.pm12014807. [DOI] [PubMed] [Google Scholar]
- 23.Shriver S.P., Bourdeau H.A., Gubish C.T., Tirpak D.L., Davis A.L., Luketich J.D., Siegfried J.M. Sex-specific expression of gastrin-releasing peptide receptor: relationship to smoking history and risk of lung cancer. J Natl Cancer Inst. 2000;92:24–33. doi: 10.1093/jnci/92.1.24. [DOI] [PubMed] [Google Scholar]
- 24.Dileto C.L., Travis E.L. Fibroblast radiosensitivity in vitro and lung fibrosis in vivo: comparison between a fibrosis-prone and fibrosis-resistant mouse strain. Radiat Res. 1996;146:61–67. [PubMed] [Google Scholar]
- 25.Beddar A.S., Salehpour M., Briere T.M., Hamidian H., Gillin M.T. Preliminary evaluation of implantable MOSFET radiation dosimeters. Phys Med Biol. 2005;50:141–149. doi: 10.1088/0031-9155/50/1/011. [DOI] [PubMed] [Google Scholar]
- 26.Briere T.M., Lii J., Prado K., Gillin M.T., Sam Beddar A. Single-use MOSFET radiation dosimeters for the quality assurance of megavoltage photon beams. Phys Med Biol. 2006;51:1139–1144. doi: 10.1088/0031-9155/51/5/006. [DOI] [PubMed] [Google Scholar]
- 27.Down J.D., Easton D.F., Steel G.G. Repair in the mouse lung during low dose-rate irradiation. Radiother Oncol. 1986;6:29–42. doi: 10.1016/s0167-8140(86)80107-4. [DOI] [PubMed] [Google Scholar]
- 28.Kong Y., Glickman J., Subramaniam M., Shahsafaei A., Allamneni K.P., Aster J.C., Sklar J., Sunday M.E. Functional diversity of notch family genes in fetal lung development. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1075–L1083. doi: 10.1152/ajplung.00438.2002. [DOI] [PubMed] [Google Scholar]
- 29.Zhang H., Han G., Liu H., Chen J., Ji X., Zhou F., Zhou Y., Xie C. The development of classically and alternatively activated macrophages has different effects on the varied stages of radiation-induced pulmonary injury in mice. J Radiat Res (Tokyo) 2011;52:717–726. doi: 10.1269/jrr.11054. [DOI] [PubMed] [Google Scholar]
- 30.Chiang C.S., Liu W.C., Jung S.M., Chen F.H., Wu C.R., McBride W.H., Lee C.C., Hong J.H. Compartmental responses after thoracic irradiation of mice: strain differences. Int J Radiat Oncol Biol Phys. 2005;62:862–871. doi: 10.1016/j.ijrobp.2005.02.037. [DOI] [PubMed] [Google Scholar]
- 31.Reynolds S.D., Giangreco A., Power J.H., Stripp B.R. Neuroepithelial bodies of pulmonary airways serve as a reservoir of progenitor cells capable of epithelial regeneration. Am J Pathol. 2000;156:269–278. doi: 10.1016/S0002-9440(10)64727-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Higashiyama H., Yoshimoto D., Okamoto Y., Kikkawa H., Asano S., Kinoshita M. Receptor-activated Smad localisation in bleomycin-induced pulmonary fibrosis. J Clin Pathol. 2007;60:283–289. doi: 10.1136/jcp.2006.037606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flanders K.C. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaimori A., Potter J., Kaimori J.Y., Wang C., Mezey E., Koteish A. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem. 2007;282:22089–22101. doi: 10.1074/jbc.M700998200. [DOI] [PubMed] [Google Scholar]
- 35.Gu L., Zhu Y.J., Yang X., Guo Z.J., Xu W.B., Tian X.L. Effect of TGF-beta/Smad signaling pathway on lung myofibroblast differentiation. Acta Pharmacol Sin. 2007;28:382–391. doi: 10.1111/j.1745-7254.2007.00468.x. [DOI] [PubMed] [Google Scholar]
- 36.Westergren-Thorsson G., Larsen K., Nihlberg K., Andersson-Sjoland A., Hallgren O., Marko-Varga G., Bjermer L. Pathological airway remodelling in inflammation. Clin Respir J. 2010;4(Suppl 1):1–8. doi: 10.1111/j.1752-699X.2010.00190.x. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt M., Sun G., Stacey M.A., Mori L., Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. 2003;171:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 38.Chang L., Subramaniam M., Yoder B.A., Day B.J., Coalson J.J., Sunday M., Crapo J.D. A catalytic antioxidant attenuates alveolar structural remodeling in bronchopulmonary dysplasia. Am J Respir Cell Mol Biol. 2003;167:57–64. doi: 10.1164/rccm.200203-232OC. [DOI] [PubMed] [Google Scholar]
- 39.Martinez A., Zudaire E., Julian M., Moody T.W., Cuttitta F. Gastrin-releasing peptide (GRP) induces angiogenesis and the specific GRP blocker 77427 inhibits tumor growth in vitro and in vivo. Oncogene. 2005;24:4106–4113. doi: 10.1038/sj.onc.1208581. [DOI] [PubMed] [Google Scholar]
- 40.Rozengurt E., Sinnett-Smith J. Bombesin stimulation of DNA synthesis and cell division in cultures of Swiss 3T3 cells. Proc Natl Acad Sci U S A. 1983;80:2936–2940. doi: 10.1073/pnas.80.10.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rozengurt E., Sinnett-Smith J. Bombesin stimulation of fibroblast mitogenesis: specific receptors, signal transduction and early events. Philos Trans R Soc Lond B Biol Sci. 1990;327:209–221. doi: 10.1098/rstb.1990.0055. [DOI] [PubMed] [Google Scholar]
- 42.Dozor A.J. The role of oxidative stress in the pathogenesis and treatment of asthma. Ann N Y Acad Sci. 2010;1203:133–137. doi: 10.1111/j.1749-6632.2010.05562.x. [DOI] [PubMed] [Google Scholar]
- 43.Sunday M.E., Willett C.G., Patidar K., Graham S.A. Modulation of oncogene and tumor suppressor gene expression in a hamster model of chronic lung injury with varying degrees of pulmonary neuroendocrine cell hyperplasia. Lab Invest. 1994;70:875–888. [PubMed] [Google Scholar]
- 44.Haley K.J., Patidar K., Zhang F., Emanuel R.L., Sunday M.E. Tumor necrosis factor induces neuroendocrine differentiation in small cell lung carcinoma cell lines. Am J Physiol. 1998;275:L311–L321. doi: 10.1152/ajplung.1998.275.2.L311. [DOI] [PubMed] [Google Scholar]
- 45.Jackson I.L., Chen L., Batinic-Haberle I., Vujaskovic Z. Superoxide dismutase mimetic reduces hypoxia-induced O2∗-, TGF-beta, and VEGF production by macrophages. Free Radic Res. 2007;41:8–14. doi: 10.1080/10715760600913150. [DOI] [PubMed] [Google Scholar]
- 46.Thornton S.C., Walsh B.J., Bennett S., Robbins J.M., Foulcher E., Morgan G.W., Penny R., Breit S.N. Both in vitro and in vivo irradiation are associated with induction of macrophage-derived fibroblast growth factors. Clin Exp Immunol. 1996;103:67–73. doi: 10.1046/j.1365-2249.1996.898598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wynn T.A. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roswit B., White D.C. Severe radiation injuries of the lung. AJR Am J Roentgenol. 1977;129:127–136. doi: 10.2214/ajr.129.1.127. [DOI] [PubMed] [Google Scholar]
- 49.Vergara J.A., Raymond U., Thet L.A. Changes in lung morphology and cell number in radiation pneumonitis and fibrosis: a quantitative ultrastructural study. Int J Radiat Oncol Biol Phys. 1987;13:723–732. doi: 10.1016/0360-3016(87)90291-4. [DOI] [PubMed] [Google Scholar]
- 50.Rosiello R.A., Merrill W.W. Radiation-induced lung injury. Clin Chest Med. 1990;11:65–71. [PubMed] [Google Scholar]
- 51.Del Rio M., Hernanz A., de la Fuente M. Bombesin, gastrin-releasing peptide, and neuromedin C modulate murine lymphocyte proliferation through adherent accessory cells and activate protein kinase C. Peptides. 1994;15:15–22. doi: 10.1016/0196-9781(94)90164-3. [DOI] [PubMed] [Google Scholar]
- 52.Meloni F., Ballabio P., Bianchi L., Mangiarotti P., Grassi G., Bignamini A., Grassi G.G. Bombesin enhances monocyte and macrophage activities: possible role in the modulation of local pulmonary defenses in chronic bronchitis. Respiration. 1996;63:28–34. doi: 10.1159/000196512. [DOI] [PubMed] [Google Scholar]
- 53.Saito-Fujita T., Iwakawa M., Nakamura E., Nakawatari M., Fujita H., Moritake T., Imai T. Attenuated lung fibrosis in interleukin 6 knock-out mice after C-ion irradiation to lung. J Radiat Res (Tokyo) 2011;52:270–277. doi: 10.1269/jrr.10094. [DOI] [PubMed] [Google Scholar]
- 54.Fleckenstein K., Zgonjanin L., Chen L., Rabbani Z., Jackson I.L., Thrasher B., Kirkpatrick J., Foster W.M., Vujaskovic Z. Temporal onset of hypoxia and oxidative stress after pulmonary irradiation. Int J Radiat Oncol Biol Phys. 2007;68:196–204. doi: 10.1016/j.ijrobp.2006.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jackson I.L., Vujaskovic Z., Down J.D. Revisiting strain-related differences in radiation sensitivity of the mouse lung: recognizing and avoiding the confounding effects of pleural effusions. Radiat Res. 2010;173:10–20. doi: 10.1667/RR1911.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jackson J.R., Seed M.P., Kircher C.H., Willoughby D.A., Winkler J.D. The codependence of angiogenesis and chronic inflammation. FASEB J. 1997;11:457–465. [PubMed] [Google Scholar]
- 57.Yakovlev V.A., Rabender C.S., Sankala H., Gauter-Fleckenstein B., Fleckenstein K., Batinic-Haberle I., Jackson I., Vujaskovic Z., Anscher M.S., Mikkelsen R.B., Graves P.R. Proteomic analysis of radiation-induced changes in rat lung: modulation by the superoxide dismutase mimetic MnTE-2-PyP(5+) Int J Radiat Oncol Biol Phys. 2010;78:547–554. doi: 10.1016/j.ijrobp.2010.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]