

Abstract
These studies used bi-transgenic Clara cell secretory protein (CCSP)/IL-1β mice that conditionally overexpress IL-1β in Clara cells to determine whether IL-1β can promote angiogenesis and lymphangiogenesis in airways. Doxycycline treatment induced rapid, abundant, and reversible IL-1β production, influx of neutrophils and macrophages, and conspicuous and persistent lymphangiogenesis, but surprisingly no angiogenesis. Gene profiling showed many up-regulated genes, including chemokines (Cxcl1, Ccl7), cytokines (tumor necrosis factor α, IL-1β, and lymphotoxin-β), and leukocyte genes (S100A9, Aif1/Iba1). Newly formed lymphatics persisted after IL-1β overexpression was stopped. Further studies examined how IL1R1 receptor activation by IL-1β induced lymphangiogenesis. Inactivation of vascular endothelial growth factor (VEGF)-C and VEGF-D by adeno-associated viral vector-mediated soluble VEGFR-3 (VEGF-C/D Trap) completely blocked lymphangiogenesis, showing its dependence on VEGFR-3 ligands. Consistent with this mechanism, VEGF-C immunoreactivity was present in some Aif1/Iba1-immunoreactive macrophages. Because neutrophils contribute to IL-1β–induced lung remodeling in newborn mice, we examined their potential role in lymphangiogenesis. Triple-transgenic CCSP/IL-1β/CXCR2−/− mice had the usual IL-1β-mediated lymphangiogenesis but no neutrophil recruitment, suggesting that neutrophils are not essential. IL1R1 immunoreactivity was found on some epithelial basal cells and neuroendocrine cells, suggesting that these cells are targets of IL-1β, but was not detected on lymphatics, blood vessels, or leukocytes. We conclude that lymphangiogenesis triggered by IL-1β overexpression in mouse airways is driven by VEGF-C/D from macrophages, but not neutrophils, recruited by chemokines from epithelial cells that express IL1R1.
CME Accreditation Statement: This activity (“ASIP 2013 AJP CME Program in Pathogenesis”) has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint sponsorship of the American Society for Clinical Pathology (ASCP) and the American Society for Investigative Pathology (ASIP). ASCP is accredited by the ACCME to provide continuing medical education for physicians.
The ASCP designates this journal-based CME activity (“ASIP 2013 AJP CME Program in Pathogenesis”) for a maximum of 48 AMA PRA Category 1 Credit(s)™. Physicians should only claim credit commensurate with the extent of their participation in the activity.
CME Disclosures: The authors of this article and the planning committee members and staff have no relevant financial relationships with commercial interests to disclose.
IL-1β is a key inflammatory cytokine found in many pathologic conditions and is responsible for triggering multiple downstream inflammatory pathways.1 Inhibiting IL-1 signaling by neutralizing antibodies or by blocking IL1R1 receptors is effective in treating inflammation in numerous pathologic conditions.2 However, IL-1β can be a two-edged sword. Depending on the context, IL-1β is responsible for deleterious effects by amplifying inflammation and also for protective effects, for example, by activating the immune system during infection.3
IL-1β has a main role in the remodeling of many tissues, including the airways and lungs. Overexpression of IL-1β in adult mouse airways and lungs results in pulmonary inflammation and the recruitment of inflammatory cells, including neutrophils, enlargement of distal airspaces, and the induction of mucous metaplasia and airway fibrosis.4 In neonatal mice, overexpression of IL-1β results in the disruption of lung development characteristic of bronchopulmonary dysplasia,5,6 and this effect is mediated in part by integrins.7,8 Furthermore, in addition to its known effects on remodeling of many tissue types, IL-1β has been reported to induce angiogenesis in several experimental models and in human diseases, including the eye, arthritic joints, and tumors, mediated in part by recruitment of leukocytes that release other inflammatory mediators.9–14
Blood vessels and lymphatics of airways show a wide repertoire of responses to different inflammatory stimuli. Various patterns of blood vessel enlargement and angiogenic sprouting are found in mice with chronic airway inflammation.15–17 For the most part, the cellular and molecular mediators that drive vascular changes are still poorly understood, but numerous cytokines and chemokines, including IL-1β, are up-regulated in Mycoplasma pulmonis infection.17–20 M. pulmonis-infected mice also show profound lymphangiogenesis, mediated by vascular endothelial growth factor receptor (VEGFR)-3 signaling.21 Because IL-1β can activate NF-κB pathways to up-regulate vascular endothelial growth factor (VEGF)-C and -D, ligands for VEGFR-3,22,23 IL-1β could also be a candidate for driving lymphangiogenesis. IL-1β is also known to up-regulate VEGF-C in vitro, a VEGFR-3 ligand that can drive lymphangiogenesis.24 However, it has been difficult to dissect the effects of individual cytokines in bacterial infection, and the effects of IL-1β alone in airways have not been examined.
With this background, we took advantage of bi-transgenic (CCSP/IL-1β) mice in which IL-1β is overexpressed in airways by the rat Clara cell secretory protein (CCSP) promoter in a doxycycline (Dox)-inducible fashion.4 This model permitted us to study the effects of overexpression of IL-1β alone on lymphangiogenesis and angiogenesis.
The goal of this study was to determine whether selective overexpression of IL-1β in adult mouse airways would induce growth or remodeling of blood vessels or lymphatic vessels and to determine the involved cells and molecules. We also sought to learn if vessel remodeling persisted after IL-1β was turned off and if VEGFR-3 signaling drove the lymphangiogenesis. To approach these issues, we stained blood vessels and lymphatics immunohistochemically in whole mounts of tracheas from CCSP/IL-1β mice treated with Dox. We also used immunohistochemistry to identify airway cells that stained for IL1R1. Because IL-1β induced leukocyte influx, including abundant neutrophils, we tested whether neutrophils were essential for the effects of IL-1β on lymphatic vessels by examining lymphangiogenesis in CXCR2−/− mice crossed to CCSP/IL-1β mice.
We found that overexpression of IL-1β in mouse airways produced neutrophil and macrophage influx, expression of inflammatory cytokines and chemokines, and long-lasting lymphangiogenesis, but not angiogenesis. IL1R1 receptors were abundant on epithelial basal cells and neuroendocrine cells, but not on lymphatics. Inactivation of VEGFR-3 ligands by soluble VEGFR-3 (VEGF-C/D Trap) from an adeno-associated viral (AAV) vector completely blocked the lymphangiogenesis, indicative of the necessity of VEGFR-3 ligands, VEGF-C and/or VEGF-D. VEGF-C immunoreactivity was present in some recruited macrophages, but the lymphangiogenesis did not require the influx of neutrophils.
Materials and Methods
Mice
We used 8- to 10-week-old bi-transgenic mice with CCSP reverse tetracycline transactivator (rtTA) and tetracycline operator (tetO)-hIL-1 (abbreviated to CCSP/IL-1β) as previously described.4 The mice express human (h) IL-1 in the presence of Dox. Because expression of the rtTA gene or protein can have immunologic effects in mice,25,26 single-transgenic CCSP-rtTA littermates (The Jackson Laboratory, Bar Harbor, ME; strain no. 006222, line 127) were used as controls. For some experiments, we generated triple-transgenic CCSP/IL-1β/CXCR2−/− mice that can be induced to overexpress IL-1β but lack CXCR2 receptors by back-crossing mice that lacked CXCR2 receptors (The Jackson Laboratory; strain no. 002724,28) into the FVB/N background for nine generations and then crossing with CCSP/IL-1β mice.29 To assess the distribution of Clara cells in tracheas, we crossed CCSP mice to (tetO)-nuclear-targeted green fluorescent protein (GFP) reporter mice (The Jackson Laboratory; strain no. 00510430). All mice were genotyped by PCR analysis of genomic tail DNA with the use of primers specific for transgene constructs.4 Mice were housed in pathogen-free conditions and had free access to food and water. For experiments, mice were anesthetized with a mixture of ketamine, xylazine, and acepromazine injected intraperitoneally and were sacrificed by transection of abdominal vessels or by perfusion of fixative. The Institutional Animal Care and Use Committees of the University of Gothenburg, University of Helsinki, and the University of California at San Francisco approved all experimental procedures.
Doxycycline Treatment
To induce IL-1β overexpression, transgenic mice received Dox (Sigma-Aldrich, St Louis, MO) at 0.5 mg/mL in drinking water. In some experiments, we turned off gene expression by removing the Dox water and substituting with normal water.
Treatment with AAV Vector-Mediated Soluble VEGFR-3
To block VEGFR-3 signaling, CCSP/IL-1β mice were treated with a recombinant AAV-derived soluble VEGFR-3 receptor (VEGF-C/D Trap) that absorbs VEGF-C and VEGF-D, the ligands for VEGFR-3. Control mice received AAVs that encoded the inactive domains four to seven of VEGFR-3-Ig. The AAVs (serotype 9) were produced and administered as described previously.31,32 In brief, 150 μL of AAV particles (at a concentration of approximately 4.5 × 109 virus particles per μL) were injected via a tail vein 1 week in advance of Dox treatment, which then continued for a further 2 weeks.
ELISA
Tracheas were homogenized and underwent enzyme-linked immunosorbent assay (ELISA) for their concentration of hIL-1β with the use of a kit (DY201, R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions as described previously.5 The assay has a detection range of 4 to 250 pg/mL and no cross-reactivity to mouse IL-1β. hIL-1β concentrations were normalized to the total tracheal protein concentration, as measured with the bicinchoninic acid method (Sigma-Aldrich).
Tissue Preparation and Immunohistochemistry
The vasculature was perfused transcardially for 2 minutes with 1% paraformaldehyde in PBS, pH 7.4. Tissues were then immersed in fixative for 1 hour at 4°C, washed, and stained immunohistochemically by incubating whole mounts or sections with one or more of the following primary antibodies (Table 1) diluted at 1 μg/mL as described previously. 22 We stained macrophages and dendritic cells with the use of an antibody to allograft inflammatory factor 1, also known as Iba1, ionized calcium binding adapter molecule 1 (Aif1/Iba1),33 and neutrophils with S100A8 (also known as MRP8 or calgranulin A34), because these antibodies worked well on fixed whole mount preparations. Airway epithelial cells were identified by staining with antibodies to pan-cytokeratin or keratin 5, and neuroendocrine cells were stained for Protein Gene Product 9.5 (PGP9.5). Secondary antibodies were labeled with fluorescein isothiocyanate, Alexa dyes, cyanine 3, or cyanine 5 (Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Specimens were imaged with a Zeiss LSM-510 confocal microscope (Carl Zeiss, Thornwood, NY) with the use of AIM software version 4.0 (Carl Zeiss). Bronchial lymph nodes were weighed to assess the extent of immune activation in the respiratory tract.
Table 1.
Antibodies Used in Present Study
| Target | Supplier | Catalog No. | Species | Clone |
|---|---|---|---|---|
| LYVE-1 | AngioBio (Del Mar, CA) | 11-034 | Rabbit | Polyclonal |
| PECAM-1/CD31 | Thermo/Fisher (Pittsburgh, PA) | MA3105 | Armenian hamster | 2H8 |
| ZO-1 | Invitrogen/Zymed (Grand Island, NY) | 61-7300 | Rabbit | Polyclonal |
| S100A8 | R&D Systems | AF3059 | Goat | Polyclonal |
| Aif1/Iba1 | Wako Chemicals (Richmond, VA) | 019-19741 | Rabbit | Polyclonal |
| VEGF-C | Santa Cruz Biotechnology (Dallas, TX) | sc-7132 | Goat | Polyclonal |
| IL1R1 | R&D Systems | AF771 | Goat | Polyclonal |
| PGP9.5 | Ultraclone (Yarmouth, UK) | RA95101 | Rabbit | Polyclonal |
| Keratin 5 | Dr. Julia A. Segre | N/A | Rabbit | Polyclonal |
| Pan-cytokeratin | Dako (Carpinteria, CA) | Z0622 | Rabbit | Polyclonal |
| CD45 | BD Biosciences (San Jose, CA) | 550539 | Rat | 30-F11 |
Morphometric Measurements
Vessel diameter and length, width, and mucosal area occupied by blood vessels and lymphatics were measured in real-time images of tracheas stained for platelet endothelial cell adhesion molecule (PECAM)-1 and lymphatic endothelial hyaluronan receptor (LYVE)-1 by using a digitizing tablet linked to a video camera on a Zeiss Axiophot microscope with 10×/0.5 NA, 20×/0.75 NA, or 40×/1.0 NA objectives as described previously.22 Area densities for blood vessels and lymphatics are presented for regions of the mucosa overlying the tracheal cartilage rings, because these regions experienced the greatest lymphangiogenesis and most noticeable changes.
Gene Expression Profiling
We assessed the expression of selected genes in tracheas of three bi-transgenic CCSP/IL-1β mice and three single-transgenic control CCSP mice immediately after 2 weeks of Dox exposure (Dox-On) or 2 weeks after the end of Dox exposure (Dox-On/Dox-Off). Total RNA was isolated as described.5 For gene array experiments, 300 ng of total RNA extracted from each trachea was amplified, transcribed to complementary RNA, and biotinylated with an Illumina Total Prep RNA Amplification kit (Ambion, Austin, TX). RNA concentrations and quality was checked with a Nanodrop ND-1000 spectrophotometer (Bio-Rad, Hercules, CA) and an Experion electrophoresis station (Bio-Rad). For each sample, 1500 ng of complementary RNA was hybridized to Sentrix Mouse WG-6 v2 Expression Bead Chips (Illumina, San Diego, CA) at 58°C overnight according to the manufacturer’s instructions. Gene chips were scanned with Illumina Bead Array Reader, and numerical results were extracted with GenomeStudio software version 1.6.0 (Illumina).
Statistical Analysis
Data are presented as means ± SEMs with four to five mice per group unless otherwise indicated. Differences between means were assessed by analysis of variance followed by Dunn-Bonferroni test for multiple comparisons, and P values <0.05 were considered significant. For gene expression studies, data were analyzed by the Limma package of BioConductor software version 2.6 and expressed as fold changes between groups. Adjusted P values were calculated and corrected to adjust for false discovery rates in large samples.
Results
Distribution and Amount of Transgenic IL-1β in Tracheal Epithelium
To understand the distribution of the sources of IL-1β production in tracheas of bi-transgenic CCSP/IL-1β mice, we first examined Clara cells in bi-transgenic CCSP/EGFP mice. These mice express nuclear-targeted GFP wherever CCSP-rtTA promoters are activated by Dox (Figure 1A). Numerous strongly fluorescent nuclei were present in the epithelium, as recognized by cell borders stained for ZO-1 (Figure 1B). Triple antibody-labeling experiments showed that the greatest density of Clara cells with fluorescent GFP nuclei was in the epithelium overlying the intercartilage regions. The mucosa between cartilages contained most of the lymphatic vessels stained for LYVE-1 and blood vessels stained for PECAM-1. Regions of mucosa over the cartilages were almost completely free of lymphatic vessels (Figure 1A). However, a few Clara cells with fluorescent nuclei were also found over the cartilages (Figure 1, A and B).
Figure 1.

Distribution of Clara cells in mouse tracheal epithelium. Confocal images of whole mount of trachea from pathogen-free CCSP/EGFP mouse given Dox for 2 days. A: Green fluorescent nuclei of Clara cells are more abundant in regions of epithelium between cartilages than over cartilages. Blood vessels (PECAM-1, cyan) and lymphatics (LYVE-1, red) form segmented networks between the cartilage rings. B: Some epithelial cells outlined by borders stained with tight junction protein ZO-1 (red) have green fluorescent nuclei representing CCSP/EGFP (green). Scale bars: 200 μm (A); 50 μm (B).
To examine the time course and amount of the transgene expression in mouse tracheas, we assayed hIL-1β by ELISA (Figure 2). Two weeks after induction of transgene activity by exposure to Dox, CCSP/IL-1β mouse tracheas had 168 pg of hIL-1β/μg of total protein. Four weeks after continuous Dox exposure, levels of hIL-1β were significantly lower in tracheas of bi-transgenic mice (only 9 pg/μg of total protein) than after a 2-week exposure. In Dox-On/Dox-Off reversal experiments, 2 weeks after removal of Dox after a 2-week exposure, hIL-1β levels dropped to baseline levels. hIL-1β was undetectable in tracheas of single-transgenic CCSP mice.
Figure 2.
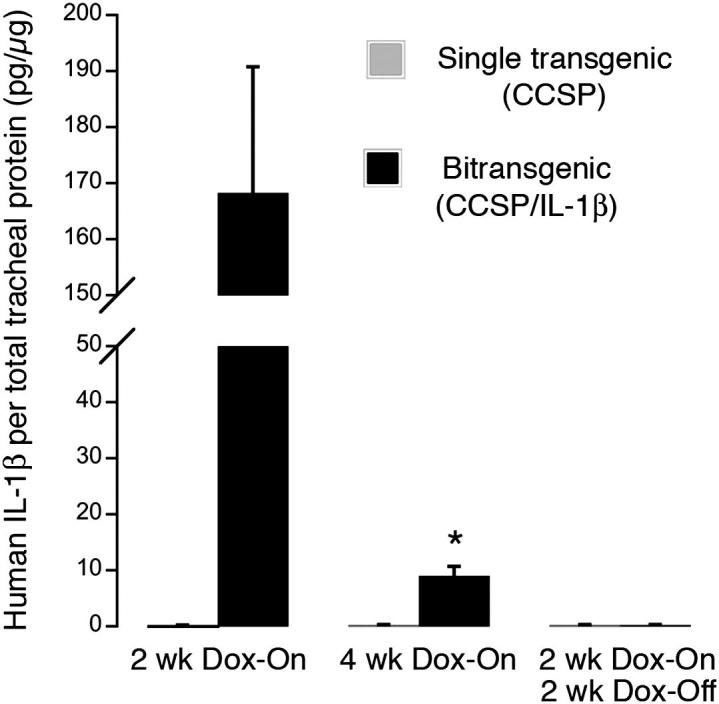
Concentration of IL-1β in trachea after transgenic overexpression in tracheas of CCSP/IL-β mice. ELISA measurements of hIL-1β in bi-transgenic CCSP/IL-1β mice and in control single-transgenic CCSP mice after exposure to Dox. hIL-1β level is significantly lower at 4 weeks of Dox than at 2 weeks and undetectable in control mice and in bi-transgenic mice given Dox for 2 weeks, then normal water for 2 weeks. Values normalized to total protein concentration. Values in single-transgenic mice were at zero. Note break in y axis; n = 4 to 6 mice/group.
Induction of Lymphatic Growth by IL-1β Overexpression
Tracheal lymphatics formed a regular stereotyped segmentally repeated network of vessels in CCSP/IL-1β mice (Figure 3A) under baseline conditions (normal water), and in control single-transgenic CCSP mice exposed to Dox (not shown). These vessels closely resembled normal lymphatics in wild-type mice. Almost all of the lymphatics were confined to the regions of mucosa located between the cartilage rings. One week after transgene induction by exposure to Dox, no obvious differences were found between control and CCSP/IL-1β groups (not shown). However, after 2 weeks of Dox, numerous lymphatic sprouts were present over the cartilages (Figure 3B). The profiles of these new lymphatic sprouts were generally smooth in outline and were smaller in diameter than their parent lymphatics (19.4 ± 0.5 μm versus 51.0 ± 3.1 μm; P < 0.05). The newly formed lymphatics persisted for at least 2 weeks after the cessation of Dox (see Reversability of Lymphatic Growth, Lymph Node Hypertrophy, Leukocyte Influx, and Gene Expression and Figure 3C).
Figure 3.

Effect of IL-1β expression on lymphatics and blood vessels. A–F: Whole mounts of tracheas from bi-transgenic CCSP/IL-1β mice stained for lymphatics (LYVE-1, red) and blood vessels (PECAM-1, green). Boxed areas located in A, B, and C are enlarged in D, E, and F. A and D: Baseline (normal water, no Dox). B and E: Two weeks after Dox (Dox-On). C and F: After 2 weeks of Dox, then 2 weeks of normal water (Dox-On/Off). G–I: Quantitation of lymphatic area density (G) and blood vessel area density (H), both measured over cartilage regions and weights of draining bronchial lymph nodes (I). Lymphatics grow after Dox but do not regress after cessation of Dox. No change detected in blood vessels. *P < 0.05, single-transgenic mice or mice not given Dox. †P < 0.05, mice given Dox for 2 weeks. Four or more mice per group. J and K: Tracheal whole mounts from CCSP/IL-1β mice treated in advance with either a control adeno-associated null virus (AAV-null; J) or AAV-soluble VEGFR-3 (K) to absorb VEGFR-3 ligands then treated for 2 week with Dox to induce overexpression of IL-1β. Lymphatics were stained for LYVE-1 (red) and blood vessels for PECAM-1 (green). L: Quantitation of lymphatic area density over cartilage rings. *P < 0.05, mice not given Dox. Five or more mice per group. Scale bars: 200 μm (A); 50 μm (D and J).
Lack of Airway Blood Vessel Growth after IL-1β Overexpression
Under the same conditions as above, we could not detect any obvious differences in the network of blood vessels (Figure 3, D–F). The visual impressions of a large increase in lymphatics but no change in blood vessels were confirmed statistically by measurements of vessel area density made over the tracheal cartilages (Figure 3, G and H).
Lymph Node Hypertrophy and Leukocyte Influx by IL-1β Overexpression
In addition to the changes in lymphatic vessels noted above, IL-1β transgene expression induced an immune response and caused the bronchial lymph nodes that drain the trachea and lungs to hypertrophy. Compared with baseline values, the bronchial lymph node weights of CCSP/IL-1β mice increased more than sixfold after 2 weeks of exposure to Dox (Figure 3I). However, after 4 weeks of continuous exposure, the increase was significantly less (only approximately fourfold). Lymph nodes were still enlarged (approximately threefold) in CCSP/IL-1β mice exposed to Dox for 2 weeks followed by 2 weeks of normal water (Figure 3I).
Mechanism of Lymphangiogenesis after IL-1β Overexpression
In many inflammatory situations, the growth of new lymphatic vessels is mediated by the ligands VEGF-C or VEGF-D signaling via VEGFR-3 receptors located on existing lymphatics.35 To determine whether this was also the case in the present model of IL-1β overexpression in adult airways, we absorbed VEGFR-3 ligands with an excess of AAV-mediated soluble VEGFR-3 receptor, given systemically 1 week in advance of a 2-week overexpression of IL-1β. In mice treated with control AAV-null, IL-1β overexpression induced lymphatic growth in the mucosa over the tracheal cartilage rings (Figure 3J), but it did not induce any lymphangiogenesis in mice treated with soluble VEGFR-3 (Figure 3, K and L).
Because IL-1β overexpression induces an influx of immune cells into the lungs, in particular neutrophils and macrophages,8,29 we assessed whether a similar phenomenon occurs in the airways. Two populations of endogenous macrophages and dendritic cells immunohistochemically stained for Aif1/Iba1 were present under baseline conditions (Figure 4A). Dendritic cells with long slender process were located just below the tracheal epithelium, and larger, fatter macrophages with shorter processes were found in deeper layers of the tracheal mucosa, as determined by focusing up and down. In contrast, neutrophils stained for S100A8 were scarce. In our preparations, staining for Aif1/Iba1 and S100A8 was not colocalized in the same inflammatory cells; that is, these markers labeled separate populations of macrophages/dendritic cells and neutrophils. Two weeks after exposure of CCSP/IL-1β mice to Dox, the number of neutrophils and rounded macrophages was greatly increased, but the number of dendritic cells with long elaborate processes near the epithelium appeared to be decreased (Figure 4B). Two weeks after the withdrawal of Dox, the number and appearance of macrophages/dendritic cells and neutrophils returned almost to baseline (Figure 4C).
Figure 4.

Effect of IL-1β expression on leukocytes in trachea of CCSP/IL-1β mice. A–C: Whole mounts of tracheas stained for macrophages/dendritic cells (Aif1/Iba1, red) and neutrophils (S100A8, green). A: At baseline (no Dox), few leukocytes stain for AIf1/Iba1, but some (arrows) have clear dendritic morphology. Scale bar = 50 μm. B: Large increase in leukocytes stained for S100A8 (green) and Aif1/Iba1 (red), mostly with rounded phenotype (arrowheads) at 2 weeks of Dox. C: Reduction in leukocyte number after 2 weeks of Dox, followed by 2 weeks of normal water. Cells staining for Aif1/Iba1 and with dendritic morphology re-appear (arrows). D–I: Staining for macrophages/dendritic cells (red) and VEGF-C (green) in single- or double-transgenic mice given Dox for 2 weeks. Diffuse granular staining with the polyclonal antibody to VEGF-C was found in the epithelium of all tracheas. D–F: Single-transgenic CCSP mice. D: Many cells staining for Aif1/Iba1 have a dendritic morphology E: Only a few (arrow) have weak staining for VEGF-C (arrow). F: Merged image. G–I: Double-transgenic CCSP/IL-1β mice. G: Most cells staining for Aif1/Iba1 have a rounded macrophage-like phenotype. H: Some of these macrophages have stronger VEGF-C staining (arrowheads). I: Merged image.
To determine whether the macrophages were a potential source of VEGF-C, we examined the distribution of Aif1/Iba1+ macrophages/dendritic cells and VEGF-C immunoreactivity in tracheas of double-transgenic CCSP/IL-1β mice after Dox treatment for 2 weeks (Figure 4, D–I). In single-transgenic CCSP mice used as controls, VEGF-C immunoreactivity was weak and granular in some epithelial cells that resembled dendritic cells (Figure 4, D–F). In double-transgenic CCSP/IL-1β mice, VEGF-C immunoreactivity was strong in some Aif1/Iba1+ cells in the lamina propria but was weak and granular in epithelial cells (Figure 4, G–I).
Effect of IL-1β Overexpression on Gene Expression
IL-1β overexpression has been shown to up-regulate the expression of inflammatory genes in the developing mouse lungs.8,29 We wanted to determine whether similar genes were up-regulated in the tracheas of adult mice. As expected, IL-1β up-regulated many genes for chemokines and their receptors (Table 2). For example, the neutrophil chemoattractant chemokine Cxcl1 was up-regulated 20.8-fold, as were other markers of inflammation (eg, serum amyloid A3, 34.6-fold), genes associated with neutrophils (S100a8 and S100a9; 6.7 and 14.5-fold, respectively), macrophages (Aif1/Iba1; 4.4-fold), other genes related to leukocyte-related genes, and genes up-regulated in asthma (Muc5ac). Genes for mouse IL-1β, tumor necrosis factor α (TNFα), and lymphotoxin-β were up-regulated (11.5-, 3.5-, and 7.3-fold, respectively).
Table 2.
Effect of IL-1β Overexpression in Mouse Trachea on Expression of Selected Genes for Chemokines and Their Receptors, Leukocyte-Related Genes, Extracellular Matrix Proteases, Inflammation-Related Genes, and Lymphatic Markers
| Symbol | Name | Fold change∗ | Adjusted P value† |
|---|---|---|---|
| Chemokines and receptors | |||
| Cxcl1 | Chemokine (C-X-C motif) ligand 1 | 20.80 | 0.00009 |
| Ccl7 | Chemokine (C-C motif) ligand 7 | 7.06 | 0.00008 |
| Ccl17 | Chemokine (C-C motif) ligand 17 | 4.91 | 0.00036 |
| Ccl9 | Chemokine (C-C motif) ligand 9 | 4.76 | 0.00001 |
| Cxcr2/Il8r1 | Chemokine (C-X-C motif) receptor 2 | 3.57 | 0.00190 |
| Cxcl14 | Chemokine (C-X-C motif) ligand 14 | 3.07 | 0.00004 |
| Ccr6 | Chemokine (C-C motif) receptor 6 | 3.01 | 0.00001 |
| Cxcl16 | Chemokine (C-X-C motif) ligand 16 | 2.53 | 0.00001 |
| Leukocyte related | |||
| Lcn2 | Lipocalin 2 (neutrophil-gelatinase associated lipocalin) | 37.81 | 5.67×10−8 |
| S100a9 | S100 calcium binding protein A9 (calgranulin B) | 14.46 | 0.00002 |
| S100a8 | S100 calcium binding protein A8 (calgranulin A) | 6.65 | 0.00051 |
| Ncf4 | Neutrophil cytosolic factor 4 | 6.08 | 5.42×10−6 |
| Selp | P-selectin | 5.14 | 0.00037 |
| Selplg | P-selectin ligand | 4.62 | 4.75×10−6 |
| Aif1 | Allograft inflammatory factor 1 (Aif1/Iba1) | 4.36 | 0.00001 |
| Cd68 | CD68 antigen | 2.98 | 0.00009 |
| Extracellular matrix proteases (remodeling related) | |||
| Mmp3 | Matrix metallopeptidase 3 (stromelysin, progelatinase) | 8.90 | 0.00012 |
| Ctse | Cathepsin E | 3.86 | 0.00079 |
| Mmp12 | Matrix metallopeptidase 12 | 2.79 | 0.00460 |
| Inflammation and asthma related | |||
| Clca3 | Chloride channel calcium activated 3 | 61.91 | 4.85×10−5 |
| Chi3l4 | Chitinase 3-like 4 | 42.46 | 6.39×10−7 |
| Saa3 | Serum amyloid A3 | 34.57 | 1.73×10−6 |
| Chi3l1 | Chitinase 3-like 1 | 16.19 | 2.13×10−5 |
| Muc5ac | Mucin 5, subtypes A and C, tracheobronchial/gastric | 13.18 | 1.25×10−6 |
| Muc13 | Mucin 13 | 11.03 | 7.82×10−7 |
| Clca4 | Chloride channel calcium activated 4 | 10.74 | 3.65×10−6 |
| Interleukin 1 related | |||
| Il1b | Interleukin 1 beta (mouse gene) | 11.52 | 0.00002 |
| Il1rn | Interleukin 1 receptor antagonist | 4.86 | 0.00001 |
| Il1r2 | Interleukin 1 receptor, type II | 3.09 | 0.00005 |
| TNF family related | |||
| Ltb | Lymphotoxin-beta | 7.32 | 0.00002 |
| Tnf | Tumor necrosis factor alpha | 3.50 | 0.00005 |
| Lta | Lymphotoxin-alpha | 2.10 | 0.00377 |
Gene expression in tracheas of bi-transgenic CCSP/IL-1β mice given Dox for 2 weeks as determined by Illumina genome-wide gene array chips; n = 3 mice per group.
Fold changes compared with control bi-transgenic mice given normal water.
P values adjusted to compensate for false discovery rate in large microarray samples.
Lack of Dependence of Lymphatic Growth on Neutrophil Influx
Because overexpression of IL-1β recruits neutrophils, we sought to examine whether their recruitment was essential for the lymphangiogenesis. In mice, CXCR2 receptors (also known as IL8R1) are the dominant receptors responsible for chemotaxis of neutrophils to sites of inflammation.28 We cross-bred CXCR2−/− mice to bi-transgenic CCSP/IL-1β mice to generate triple-transgenic mice29 and examined whether lymphangiogenesis differed between wild-type CCSP/IL-1β and the triple-transgenic CCSP/IL-1β/CXCR2−/− mice after IL-1β overexpression (Figure 5). Tracheas stained for the neutrophil marker S100A8 showed that CXCR2−/− mice had 93% fewer neutrophils in the mucosa over cartilage rings (Figure 5, A–C). We found that after Dox treatment, the increased area density of lymphatics was not significantly different in CCSP/IL-1β and triple-transgenic CCSP/IL-1β/CXCR2−/− mice (Figure 5, D and E). Furthermore, the lack of CXCR2 receptors had no effect of lymphatics in baseline conditions or on blood vessels in any conditions examined (Figure 5, F and G).
Figure 5.

Lack of effect of deletion of CXCR2 receptors on changes in lymphatics and blood vessels induced by IL-1β overexpression. Tracheal whole mounts from CCSP/IL-1β bi-transgenic mice (A) and CCSP/IL-1β/CXCR2−/− triple-transgenic mice (B) given Dox for 4 weeks, then stained to detect neutrophils by their S100A8 immunoreactivity (green). C: Neutrophil numbers were 93% lower in CXCR2−/− mice than in CXCR2+/+ mice. ∗P < 0.05. CCSP/IL-1β bi-transgenic mice (D) and CCSP/IL-1β/CXCR2−/− triple-transgenic mice (E) were given Dox for 4 weeks. Lymphatics (LYVE-1, red) and blood vessels (PECAM-1, green) and were stained and quantified (F and G). Lymphangiogenesis (arrows) was equally abundant in both groups of mice given Dox (∗P < 0.05 compared with mice lacking the IL-1β transgene) and was not significantly different between mice with or without CXCR2 receptors. No obvious changes were found in blood vessels. Four to six mice per group. Scale bars: 50 μm (A); 200 μm (D).
Reversibility of Lymphatic Growth, Lymph Node Hypertrophy, Leukocyte Influx, and Gene Expression
To determine whether the effects of IL-1β transgene overexpression described above could be reversed, we examined CCSP/IL-1β mice that had been exposed to Dox for 2 weeks and then given normal water for 2 weeks (Dox-On/Dox-Off). A striking finding, already noted above (Figure 3, C, F, and G), was that the extent of lymphatic proliferation was largely unchanged after the cessation of Dox. When examined more closely by high-magnification confocal microscopy, a few lymphatics showed abnormal features that suggested degeneration and regression. Such lymphatics had some endothelial cells with much weaker LYVE-1 staining than the rest (Figure 6A). Occasional segments appeared as isolated “islands” with a nonuniform distribution of LYVE-1 and PECAM-1, including particularly dense regions (Figure 6B). Additional experiments with CCSP/IL-1β mice exposed to Dox for 4 weeks and then given normal water showed that newly formed lymphatics lasted up to 13 weeks (Supplemental Figure S1).
Figure 6.
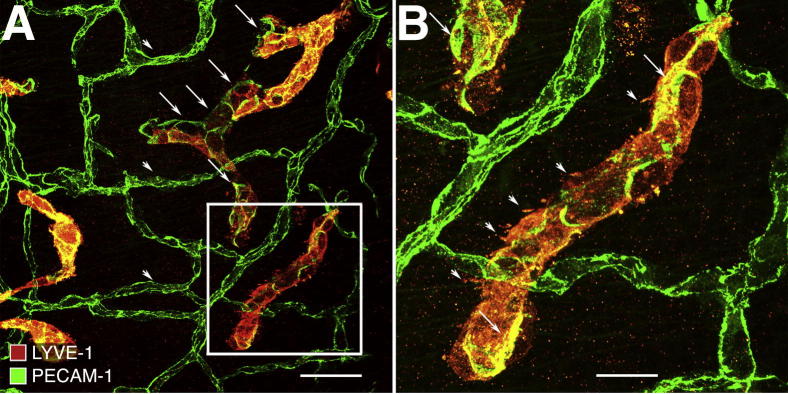
Persistence of newly formed lymphatics in trachea of CCSP/IL-1β bi-transgenic mouse after cessation of stimulus. Tracheal whole mount after 2 weeks of Dox, followed by 2 weeks of normal water. High magnification view of trachea treated similarly to that shown in Figure 3F. A: Some endothelial cells (arrowheads) in a few lymphatics have much weaker LYVE-1 staining (red) than other lymphatic endothelial cells. Their borders are demarcated by PECAM-1 (green). Blood vessels (arrowheads) have stronger PECAM-1 staining but no LYVE-1. B. White box in A is enlarged. Some endothelial cells in this isolated lymphatic island have a nonuniform distribution of LYVE-1 (red), including dense granular regions and irregular membrane protrusions (arrowheads) and intense PECAM-1 staining (arrows). Scale bars: 200 μm (A); 50 μm (B).
Blood vessels appeared normal in Dox-On/Dox-Off conditions (Figure 3, C, F, and H, and Figure 6). In contrast to the persistence of newly formed lymphatics, bronchial lymph node weights and the influx of macrophages and neutrophils were reduced toward baseline values in Dox-On/Dox-Off experiments (Figures 3H and 4C).
IL1R1 Receptor Localization in Mouse Trachea and Other Tissues
In an effort to gain insight into the mechanism by which IL-1β overexpression induces lymphangiogenesis, we sought to identify the target cells with the most abundant IL1R1 receptors, which are believed to be the only functional receptors for IL-1β (and IL-1α). We stained whole mounts of wild-type mouse tracheas for IL1R1 immunoreactivity and counterstained lymphatic vessels for LYVE-1 and blood vessels for PECAM-1. Two types of cells in the epithelium had IL1R1 immunoreactivity (Figure 7A). Scattered elongated cells stained strongly, and clusters of polygonal cells had weaker staining (Figure 7B). These cells were most abundant in the epithelium between cartilage rings, but some were present over the cartilages. Lymphatics (Figure 7C) and blood vessels (Figure 7D) or, for that matter, all other kinds of cells in the trachea had no detectable IL1R1 immunoreactivity (Figure 7E).
Figure 7.

IL1R1 receptor immunoreactivity in tracheas of wild-type mice. A: Low magnification confocal image of IL1R1 receptors (red), lymphatics (LYVE-1, green), and blood vessels (PECAM-1, cyan) in whole mount of trachea. White box is enlarged in B–E. B: Two types of epithelial cells stain for IL1R1 (red). Clustered polygonal cells stain weakly (arrows), and scattered elongated spindle-shaped cells stain strongly (arrowheads). C–E: Structures corresponding to lymphatics (green) or blood vessels (blue) have no detectable IL1R1 receptor staining. F–H: The elongated spindle-shaped cells (arrowheads) with strong red IL-RI staining (F) correspond to neuroendocrine cells (G) that also stain for protein gene product 9.5 (PGP9.5, green), also found in some fine varicose unmyelinated nerve fibers. H: Merged image. I–K: The clusters of polygonal cells (I) that stain weakly (arrows) for IL1R1 (red) correspond to a subset of a larger number (J) of epithelial basal cells that express keratin 5 (green). K: Merged image. Scale bars: 200 μm (A); 50 μm (B and F).
To identify the epithelial cells with IL1R1 immunoreactivity, we asked whether it was found in neuroendocrine cells, visualized by PGP9.5, or in basal cells, visualized by keratin 5. IL1R1 immunoreactivity was strong in many neuroendocrine cells (Figure 7, F–H) and was moderate in some groups of basal cells stained for keratin 5 in the tracheal epithelium (Figure 7, I–K). Little IL1R1 immunoreactivity was found in other cell types in the trachea.
The identity of IL1R1 immunoreactive cells was confirmed in histologic sections of trachea, and the specificity of the antibody to mouse IL1R1 was tested in brain and heart. IL1R1 immunoreactivity colocalized with pan-cytokeratin in some epithelial cells of the trachea but was not found in blood vessel or lymphatic endothelial cells or other cells in the trachea, including CD45-immunoreactive leukocytes (Supplemental Figure S2, A–D). In contrast to the trachea, IL1R1 immunoreactivity colocalized with PECAM-1 in some blood vessels in the brain and heart (Supplemental Figure S2 E–J). No IL1R1 staining was found in experiments in which the primary antibody was omitted or replaced by normal goat IgG (data not shown).
Discussion
In this study we sought to determine the effects of inducible, selective overexpression of IL-1β on blood vessels and lymphatics in the airways and the reversibility of these effects. By using CCSP/IL-1β double-transgenic mice, we found that overexpression of hIL-1β in the mouse trachea induced lymphangiogenesis without accompanying angiogenesis. The lymphangiogenesis was found to require VEGF-C and/or VEGF-D ligands that drive VEGFR-3 signaling. Overexpression of IL-1β also induced the influx of neutrophils and macrophages into the airways. Some macrophages contained VEGF-C, which drives lymphatic growth, but neutrophil influx was not essential for the lymphangiogenesis. The newly formed lymphatics persisted in the airways for several weeks after IL-1β overexpression was ended by withdrawal of Dox, but other changes returned to baseline.
Growth of Lymphatics and Presumptive Mechanism
IL-1β–induced lymphangiogenesis is likely to involve VEGF-C- or VEGF-D-mediated signaling of VEGFR-3,35 but site of action of IL-1β and the source of the VEGF-C/D had not been identified. We found that lymphangiogenesis in CCSP/IL-1β mice was completely prevented by sequestration of ligands that bind VEGFR-3. In search of the source of the ligands, we found VEGF-C immunoreactivity in some Aif1/Iba1+ macrophages in the trachea.
IL1R1 immunoreactivity was strong in some airway epithelial cells but not in lymphatics or blood vessels. The absence of IL1R1 immunoreactivity does not ensure the absence of the receptor, and only a few copies of receptor per cell can effectively mediate IL-1 signaling.36 However, the detection of IL1R1 immunoreactivity on epithelial basal cells and neuroendocrine cells of the trachea and on blood vessels in the brain and heart, which are known to respond to IL-1β, weigh against lymphatics as the main target of IL-1β in the trachea.
A possible alternative mechanism of lymphangiogenesis involves IL-1β activation of IL1R1-positive epithelial cells to release chemokines that then attract leukocytes that secrete lymphangiogenic factors. Macrophages have been reported to contain VEGF-C and neutrophils to contain VEGF-D.21,35 We asked whether neutrophils are responsible for lymphangiogenesis in CCSP/IL-1β mice, because these cells are thought to mediate lung remodeling after IL-1β overexpression in a model of bronchopulmonary dysplasia in infant mice.29 We found that lymphangiogenesis was not significantly different after IL-1β overexpression in CXCR2−/− mice, which lack the dominant pathway for neutrophil recruitment.28,37 This finding indicates that neutrophils are not essential for lymphangiogenesis in these mice and focuses attention on macrophages that produce VEGF-C.
Further work will be required to understand the link between IL-1β secretion and action in the epithelium and macrophage production of VEGF-C. According to one possible mechanism, epithelial basal cells and/or neuroendocrine cells activated by IL-1β secrete chemokines that attract the macrophages. Recent studies of basal cells isolated from mouse tracheas have reported a distinct gene expression profile38 that could give insight into the signaling pathways involved.
Another notable finding in the present study was the persistence of many new lymphatics after withdrawal of the Dox that drove IL-1β overexpression. Most other features accompanying IL-1β overexpression, including IL-1β mRNA expression in the trachea and number of recruited leukocytes and lymph node hypertrophy, returned to baseline after Dox was withdrawn. Some new lymphatics that appeared to undergo regression had little or no LYVE-1 immunoreactivity and were fragmented.
The persistence of the newly formed lymphatics mirrors that of lymphatics that grow in tracheas of mice after M. pulmonis bacterial infection, despite treatment with antibiotics or dexamethasone.17,21 Newly formed lymphatics also persist for many weeks in skin of transgenic mice after the overexpression of VEGF-C that drives the lymphangiogenesis.39 Similarly, persistence of abnormally numerous lymphatics has been reported in the skin of patients with breast cancer more than a year after radiation therapy.40
Persistence of new lymphatics after the stimulus is withdrawn contrasts with the rapid regression of angiogenic blood vessels after inhibition of VEGF-A. Pericytes and well-developed basement membrane are thought to contribute to the stabilization of blood vessels, but lymphatic vessels have neither.
Absence of Angiogenesis
Inflammation and angiogenesis often go together, but not always. IL-1β increases in inflammation and is well known to trigger the activation of inflammatory genes and proteins.1,41 In addition, IL-1β has been reported to down-regulate angiopoietin-1, which can favor blood vessel destabilization and angiogenesis.11 Therefore, contrary to our expectations, we were surprised to find that overexpression of hIL-1β in CCSP/IL-1β mice did not induce angiogenesis or blood vessel remodeling in the mouse trachea, despite clear signs of inflammation, as reflected by leukocyte influx and gene expression changes.
In the eye and in tumors, whereby IL-1β and angiogenesis can increase in parallel, most evidence suggests that the angiogenesis is linked to the recruitment of leukocytes and their production of VEGF and other growth factors.23,42–44 In these tissues, inhibition of IL-1β production or receptor signaling can reduce or prevent angiogenesis, presumably by reducing the influx of inflammatory cells.10,12,13,23 Why the recruited leukocytes did not promote angiogenesis in the trachea of CCSP/IL-1β mice is puzzling and deserves further study. The observation of robust expression of inflammation-related genes such as serum amyloid A3 weighs against the possibility that IL-1β in the airways did not reach the threshold required to promote angiogenesis, but this cannot be excluded.
Attributes and Limitations of the Tracheal Model
The present studies evolved from previous studies of transgenic overexpression of IL-1β in the mouse lung and from studies of the tracheal vasculature of mice with respiratory tract infections. IL-1β overexpression causes pulmonary inflammation, emphysema, and airway remodeling in the adult mouse lung4 and pulmonary dysplasia in neonatal lungs.5,8,29,45 Studies of the lung also showed that hIL-1β concentrations rapidly returned to baseline levels after cessation of Dox, indicative of a sharp off-response. Our earlier studies of tracheas in mice with M. pulmonis infection showed robust lymphangiogenesis and blood vessel remodeling and up-regulation of many inflammatory genes, some overlapping those found in the present study, including IL-1β.17,20 The present model permitted us to study the effects of IL-1β on the airway vasculature in a regulated and reversible fashion without the complications of bacterial infection.
The mouse trachea has several experimental advantages over the lungs for studies of the vasculature. Blood vessels and lymphatics are arranged in a regular two-dimensional plexus observable in its entirety. The vascular architecture is simpler than in the lung, facilitating the relation of vessel changes to the levels of IL-1β. The trachea can display several distinct patterns of vascular changes, including angiogenesis, vascular remodeling, and lymphangiogenesis, depending on the nature of the stimulus.21,46 For instance, transgenic overexpression of VEGF-A causes angiogenesis, but not lymphangiogenesis, exactly the opposite phenomenon than after IL-1β overexpression.47 The sensitivity and versatility of mouse tracheal whole-mount preparations for visualizing the vasculature makes it likely that angiogenesis would have detected had it occurred after IL-1β overexpression.
In the trachea, the degree of IL-1β overexpression was similar to that found previously in the lungs.4 Gene profiling also showed that mouse IL-1β was up-regulated. Our immunohistochemical and ELISA results confirmed that tracheal Clara cells are sufficiently abundant to drive robust transgene overexpression via their Dox-regulated CCSP promoters.48,49 We also found that IL-1β levels were significantly lower at 4 weeks than at 2 weeks of continuous Dox exposure, perhaps suggesting an immune response to hIL-1β protein. Overall, our gene expression studies supported our immunohistochemical studies and were broadly consistent with the up-regulated genes reported in the lungs after overexpression of IL-1β, including several chemokines and leukocyte markers.29 Consistent with previous notions that IL-1β stimulated its own production in a feedback loop,36 we found that transgenic overexpression of hIL-1β in mouse trachea up-regulated mouse IL-1β, and also TNFα and lymphotoxin-β. Interestingly, both IL-1β and TNFα are known to up-regulate VEGF-C gene expression in vitro.24 Consistently, our finding that the lymphangiogenesis induced by IL-1β was completely blocked by soluble VEGFR-3 also provides strong evidence for VEGFR-3 signaling in driving the lymphatic growth.
Our findings of effects of IL-1β overexpression in the trachea of CCSP/IL-1β transgenic mice differ in several key respects from those of studies of corneal implants containing VEGF, VEGF-C, fibroblast growth factor-2, or other growth factors.23,50,51 These factors all induce angiogenesis and lymphangiogenesis, but to different extents. The reasons for the different results in the trachea are not clear but could involve multiple issues, including differences in the route of administration and local concentrations of mediators, time course and threshold of inflammatory effects, and cellular composition of cornea and trachea. The differences between the effects of IL-1β on the vasculature of the cornea and trachea suggest that examination of other tissues is warranted. Another issue for consideration is that IL-1β acts differently in organs such as the heart and brain where the vasculature has clear IL1R1 immunoreactivity. Therefore, it is premature to generalize the present findings to other organs.
In summary, we found that overexpression of hIL-1β in mouse airways resulted in lymphangiogenesis, driven by VEGFR-3 signaling, but not angiogenesis. The newly formed lymphatics persisted long after IL-1β overexpression was switched off. In addition, IL-1β overexpression induced the influx of neutrophils and macrophages; hypertrophy of draining lymph nodes; and up-regulation of genes for chemokines, leukocyte markers, and some molecules associated with allergic inflammation. Neutrophil influx was not essential for IL-1β-induced lymphangiogenesis. The strong staining for IL1R1 receptor immunoreactivity on airway epithelial cells, apparent absence on lymphatics, and presence of VEGF-C in some macrophages is consistent with an indirect mechanism of IL-1β–induced lymphangiogenesis, whereby IL1R1 signaling in epithelial cells leads to chemokine recruitment of macrophages that release VEGF-C.
Acknowledgments
We thank Dr. Jeff Whitsett (Cincinnati Children’s Hospital Medical Center, Cincinnati, OH) for donating the original CCSP-rtTA mice, Dr. Urpo Lappalainen (University of Gothenburg, Gothenburg, Sweden) for making the tetO-IL-1β mice, the AAV Gene Transfer and Cell Therapy Core Facility of Biocentrum, Finland, for providing the viral vectors, Dr. Julia A. Segre (National Human Genome Research Institute, NIH, Bethesda, MD) and Jason Rock (University of California San Francisco, San Francisco, CA) for the kind gift of antibody to keratin 5, Jennifer Feng (University of California San Francisco) for help in morphometric measurements, and Drs. Jason Rock and Li-Chin Yao (University of California San Francisco) for helpful discussions.
P.B., K.B., K.A., and D.M. designed research; P.B., A.H., and M.B. performed research, collected, analyzed, and interpreted data and statistical analysis; and P.B., K.B., and D.M. wrote the manuscript.
Footnotes
See next page for support information.
Supported in part by National Heart, Lung, and Blood Institute grants HL-24136 and HL-59157 (D.M.), National Cancer Institute grant CA-82923 (D.M.), and the Leducq Foundation (D.M.), the Academy of Finland Collaborative Research Consortium grant 262976 (K.A.), European Research Council (TX-Factors, grant 268804 to K.A.), Swedish Medical Research Council (K.B.), Swedish Heart and Lung Foundation (K.B.), the Frimurare Barnhus Foundation (K.B.), the Swedish Government Grants for Medical Research (K.B.), and the Queen Silvia Children’s Hospital Research Foundation (K.B.).
See related Commentary on page 1087.
Supplemental Data
Newly formed lymphatics persist after cessation of stimulus. A and B: Low magnification views of whole mounts of tracheas from CCSP and CCSP/IL-1β mice after 4 weeks of Dox, followed by 13 weeks of normal water. Top two thirds of A and B show regions containing cartilages and intercartilage spaces and bottom thirds show membranous portion of the trachea containing trachealis muscle. Lymphatics stained for LYVE-1 (red) and blood vessels for PECAM-1 (green). A subset of macrophages (arrowheads) also stains weakly for LYVE-1. C and D: Insets from A and B. Many newly formed lymphatics (absent in C) persist in D. Some have a fragmented appearance (arrows). Scale bars: 400 μm (A and B); 200 μm (C and D).
IL1R1 receptor immunoreactivity in trachea, brain, and heart. A–D: IL1R1-immunoreactivity (arrows, red) colocalizes with some epithelial cells (pan-cytokeratin, green) but not with immune cells (CD45, blue) or other cells in confocal images of sections of wild-type trachea. E–J: However, IL1R1-immunoreactivity (red) colocalizes with PECAM-1 (green) in some blood vessels (arrows) in the brain (E–G) and heart (H–J). Scale bars: 50 μm (D, G, and J).
References
- 1.Dinarello C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2010;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dinarello C.A., Simon A., van der Meer J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Apte R.N., Voronov E. Is interleukin-1 a good or bad ‘guy’ in tumor immunobiology and immunotherapy? Immunol Rev. 2008;222:222–241. doi: 10.1111/j.1600-065X.2008.00615.x. [DOI] [PubMed] [Google Scholar]
- 4.Lappalainen U., Whitsett J.A., Wert S.E., Tichelaar J.W., Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32:311–318. doi: 10.1165/rcmb.2004-0309OC. [DOI] [PubMed] [Google Scholar]
- 5.Bry K., Whitsett J.A., Lappalainen U. IL-1beta disrupts postnatal lung morphogenesis in the mouse. Am J Respir Cell Mol Biol. 2007;36:32–42. doi: 10.1165/rcmb.2006-0116OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lukkarinen H., Hogmalm A., Lappalainen U., Bry K. Matrix metalloproteinase-9 deficiency worsens lung injury in a model of bronchopulmonary dysplasia. Am J Respir Cell Mol Biol. 2009;41:59–68. doi: 10.1165/rcmb.2008-0179OC. [DOI] [PubMed] [Google Scholar]
- 7.Bry K., Hogmalm A., Backstrom E. Mechanisms of inflammatory lung injury in the neonate: lessons from a transgenic mouse model of bronchopulmonary dysplasia. Semin Perinatol. 2011;34:211–221. doi: 10.1053/j.semperi.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Hogmalm A., Sheppard D., Lappalainen U., Bry K. beta6 Integrin subunit deficiency alleviates lung injury in a mouse model of bronchopulmonary dysplasia. Am J Respir Cell Mol Biol. 2010;43:88–98. doi: 10.1165/rcmb.2008-0480OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Norrby K. Interleukin-1-alpha and de novo mammalian angiogenesis. Microvasc Res. 1997;54:58–64. doi: 10.1006/mvre.1997.2024. [DOI] [PubMed] [Google Scholar]
- 10.Coxon A., Bolon B., Estrada J., Kaufman S., Scully S., Rattan A., Duryea D., Hu Y.L., Rex K., Pacheco E., Van G., Zack D., Feige U. Inhibition of interleukin-1 but not tumor necrosis factor suppresses neovascularization in rat models of corneal angiogenesis and adjuvant arthritis. Arthritis Rheum. 2002;46:2604–2612. doi: 10.1002/art.10546. [DOI] [PubMed] [Google Scholar]
- 11.Fan F., Stoeltzing O., Liu W., McCarty M.F., Jung Y.D., Reinmuth N., Ellis L.M. Interleukin-1beta regulates angiopoietin-1 expression in human endothelial cells. Cancer Res. 2004;64:3186–3190. doi: 10.1158/0008-5472.can-03-0407. [DOI] [PubMed] [Google Scholar]
- 12.Voronov E., Carmi Y., Apte R.N. Role of IL-1-mediated inflammation in tumor angiogenesis. Adv Exp Med Biol. 2007;601:265–270. doi: 10.1007/978-0-387-72005-0_28. [DOI] [PubMed] [Google Scholar]
- 13.Carmi Y., Voronov E., Dotan S., Lahat N., Rahat M.A., Fogel M., Huszar M., White M.R., Dinarello C.A., Apte R.N. The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J Immunol. 2009;183:4705–4714. doi: 10.4049/jimmunol.0901511. [DOI] [PubMed] [Google Scholar]
- 14.Lavalette S., Raoul W., Houssier M., Camelo S., Levy O., Calippe B., Jonet L., Behar-Cohen F., Chemtob S., Guillonneau X., Combadiere C., Sennlaub F. Interleukin-1beta inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration. Am J Pathol. 2011;178:2416–2423. doi: 10.1016/j.ajpath.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thurston G., Murphy T.J., Baluk P., Lindsey J.R., McDonald D.M. Angiogenesis in mice with chronic airway inflammation: strain-dependent differences. Am J Pathol. 1998;153:1099–1112. doi: 10.1016/S0002-9440(10)65654-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ezaki T., Baluk P., Thurston G., La Barbara A., Woo C., McDonald D.M. Time course of endothelial cell proliferation and microvascular remodeling in chronic inflammation. Am J Pathol. 2001;158:2043–2055. doi: 10.1016/S0002-9440(10)64676-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao L.C., Baluk P., Feng J., McDonald D.M. Steroid-resistant lymphatic remodeling in chronically inflamed mouse airways. Am J Pathol. 2010;176:1525–1541. doi: 10.2353/ajpath.2010.090909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faulkner C.B., Simecka J.W., Davidson M.K., Davis J.K., Schoeb T.R., Lindsey J.R., Everson M.P. Gene expression and production of tumor necrosis factor alpha, interleukin 1, interleukin 6, and gamma interferon in C3H/HeN and C57BL/6N mice in acute Mycoplasma pulmonis disease. Infect Immun. 1995;63:4084–4090. doi: 10.1128/iai.63.10.4084-4090.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis C.C., Yang J.Y., Huang X., Banerjee S.K., Blackburn M.R., Baluk P., McDonald D.M., Blackwell T.S., Nagabhushanam V., Peters W., Voehringer D., Erle D.J. Disease-specific gene expression profiling in multiple models of lung disease. Am J Respir Crit Care Med. 2008;177:376–387. doi: 10.1164/rccm.200702-333OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baluk P., Yao L.C., Feng J., Romano T., Jung S.S., Schreiter J.L., Yan L., Shealy D.J., McDonald D.M. TNF-alpha drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice. J Clin Invest. 2009;119:2954–2964. doi: 10.1172/JCI37626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baluk P., Tammela T., Ator E., Lyubynska N., Achen M.G., Hicklin D.J., Jeltsch M., Petrova T.V., Pytowski B., Stacker S.A., Yla-Herttuala S., Jackson D.G., Alitalo K., McDonald D.M. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest. 2005;115:247–257. doi: 10.1172/JCI22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maruyama K., Asai J., Ii M., Thorne T., Losordo D.W., D’Amore P.A. Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing. Am J Pathol. 2007;170:1178–1191. doi: 10.2353/ajpath.2007.060018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watari K., Nakao S., Fotovati A., Basaki Y., Hosoi F., Bereczky B., Higuchi R., Miyamoto T., Kuwano M., Ono M. Role of macrophages in inflammatory lymphangiogenesis: enhanced production of vascular endothelial growth factor C and D through NF-kappaB activation. Biochem Biophys Res Commun. 2008;377:826–831. doi: 10.1016/j.bbrc.2008.10.077. [DOI] [PubMed] [Google Scholar]
- 24.Ristimaki A., Narko K., Enholm B., Joukov V., Alitalo K. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J Biol Chem. 1998;273:8413–8418. doi: 10.1074/jbc.273.14.8413. [DOI] [PubMed] [Google Scholar]
- 25.Sisson T.H., Hansen J.M., Shah M., Hanson K.E., Du M., Ling T., Simon R.H., Christensen P.J. Expression of the reverse tetracycline-transactivator gene causes emphysema-like changes in mice. Am J Respir Cell Mol Biol. 2006;34:552–560. doi: 10.1165/rcmb.2005-0378OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perl A.K., Zhang L., Whitsett J.A. Conditional expression of genes in the respiratory epithelium in transgenic mice: cautionary notes and toward building a better mouse trap. Am J Respir Cell Mol Biol. 2009;40:1–3. doi: 10.1165/rcmb.2008-0011ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tichelaar J.W., Lu W., Whitsett J.A. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J Biol Chem. 2000;275:11858–11864. doi: 10.1074/jbc.275.16.11858. [DOI] [PubMed] [Google Scholar]
- 28.Cacalano G., Lee J., Kikly K., Ryan A.M., Pitts-Meek S., Hultgren B., Wood W.I., Moore M.W. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994;265:682–684. doi: 10.1126/science.8036519. [DOI] [PubMed] [Google Scholar]
- 29.Hogmalm A., Backstrom E., Bry M., Lappalainen U., Lukkarinen H.P., Bry K. Role of CXC chemokine receptor-2 in a mouse model of bronchopulmonary dysplasia. Am J Respir Cell Mol Biol. 2012;46:746–758. doi: 10.1165/rcmb.2011-0394OC. [DOI] [PubMed] [Google Scholar]
- 30.Tumbar T., Guasch G., Greco V., Blanpain C., Lowry W.E., Rendl M., Fuchs E. Defining the epithelial stem cell niche in skin. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He Y., Rajantie I., Pajusola K., Jeltsch M., Holopainen T., Yla-Herttuala S., Harding T., Jooss K., Takahashi T., Alitalo K. Vascular endothelial cell growth factor receptor 3-mediated activation of lymphatic endothelium is crucial for tumor cell entry and spread via lymphatic vessels. Cancer Res. 2005;65:4739–4746. doi: 10.1158/0008-5472.CAN-04-4576. [DOI] [PubMed] [Google Scholar]
- 32.Bry M., Kivela R., Holopainen T., Anisimov A., Tammela T., Soronen J., Silvola J., Saraste A., Jeltsch M., Korpisalo P., Carmeliet P., Lemstrom K.B., Shibuya M., Yla-Herttuala S., Alhonen L., Mervaala E., Andersson L.C., Knuuti J., Alitalo K. Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation. 2010;122:1725–1733. doi: 10.1161/CIRCULATIONAHA.110.957332. [DOI] [PubMed] [Google Scholar]
- 33.Kohler C. Allograft inflammatory factor-1/ionized calcium-binding adapter molecule 1 is specifically expressed by most subpopulations of macrophages and spermatids in testis. Cell Tissue Res. 2007;330:291–302. doi: 10.1007/s00441-007-0474-7. [DOI] [PubMed] [Google Scholar]
- 34.Passey R.J., Xu K., Hume D.A., Geczy C.L. S100A8: emerging functions and regulation. J Leukoc Biol. 1999;66:549–556. doi: 10.1002/jlb.66.4.549. [DOI] [PubMed] [Google Scholar]
- 35.Tammela T., Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 2010;140:460–476. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 36.Dinarello C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 37.McDonald B., Pittman K., Menezes G.B., Hirota S.A., Slaba I., Waterhouse C.C., Beck P.L., Muruve D.A., Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 38.Rock J.R., Onaitis M.W., Rawlins E.L., Lu Y., Clark C.P., Xue Y., Randell S.H., Hogan B.L. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci U S A. 2009;106:12771–12775. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lohela M., Helotera H., Haiko P., Dumont D.J., Alitalo K. Transgenic induction of vascular endothelial growth factor-C is strongly angiogenic in mouse embryos but leads to persistent lymphatic hyperplasia in adult tissues. Am J Pathol. 2008;173:1891–1901. doi: 10.2353/ajpath.2008.080378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackowski S., Janusch M., Fiedler E., Marsch W.C., Ulbrich E.J., Gaisbauer G., Dunst J., Kerjaschki D., Helmbold P. Radiogenic lymphangiogenesis in the skin. Am J Pathol. 2007;171:338–348. doi: 10.2353/ajpath.2007.060589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chung K.F. Cytokines in chronic obstructive pulmonary disease. Eur Respir J Suppl. 2001;34:50s–59s. [PubMed] [Google Scholar]
- 42.Germano G., Allavena P., Mantovani A. Cytokines as a key component of cancer-related inflammation. Cytokine. 2008;43:374–379. doi: 10.1016/j.cyto.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 43.Dinarello C.A. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010;29:317–329. doi: 10.1007/s10555-010-9229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Voronov E., Shouval D.S., Krelin Y., Cagnano E., Benharroch D., Iwakura Y., Dinarello C.A., Apte R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A. 2003;100:2645–2650. doi: 10.1073/pnas.0437939100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Backstrom E., Hogmalm A., Lappalainen U., Bry K. Developmental stage is a major determinant of lung injury in a murine model of bronchopulmonary dysplasia. Pediatr Res. 2011;69:312–318. doi: 10.1203/PDR.0b013e31820bcb2a. [DOI] [PubMed] [Google Scholar]
- 46.McDonald D.M., Yao L.C., Baluk P. Dynamics of airway blood vessels and lymphatics: lessons from development and inflammation. Proc Am Thorac Soc. 2011;8:504–507. doi: 10.1513/pats.201102-022MW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baluk P., Lee C.G., Link H., Ator E., Haskell A., Elias J.A., McDonald D.M. Regulated angiogenesis and vascular regression in mice overexpressing vascular endothelial growth factor in airways. Am J Pathol. 2004;165:1071–1085. doi: 10.1016/S0002-9440(10)63369-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whitsett J.A., Glasser S.W., Tichelaar J.W., Perl A.K., Clark J.C., Wert S.E. Transgenic models for study of lung morphogenesis and repair: Parker B. Francis lecture. Chest. 2001;120:27S–30S. doi: 10.1378/chest.120.1_suppl.s27. [DOI] [PubMed] [Google Scholar]
- 49.Perl A.K., Tichelaar J.W., Whitsett J.A. Conditional gene expression in the respiratory epithelium of the mouse. Transgenic Res. 2002;11:21–29. doi: 10.1023/a:1013986627504. [DOI] [PubMed] [Google Scholar]
- 50.Nakao S., Maruyama K., Zandi S., Melhorn M.I., Taher M., Noda K., Nusayr E., Doetschman T., Hafezi-Moghadam A. Lymphangiogenesis and angiogenesis: concurrence and/or dependence? Studies in inbred mouse strains. FASEB J. 2010;24:504–513. doi: 10.1096/fj.09-134056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakao S., Noda K., Zandi S., Sun D., Taher M., Schering A., Xie F., Mashima Y., Hafezi-Moghadam A. VAP-1-mediated M2 macrophage infiltration underlies IL-1beta- but not VEGF-A–induced lymph- and angiogenesis. Am J Pathol. 2011;178:1913–1921. doi: 10.1016/j.ajpath.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Newly formed lymphatics persist after cessation of stimulus. A and B: Low magnification views of whole mounts of tracheas from CCSP and CCSP/IL-1β mice after 4 weeks of Dox, followed by 13 weeks of normal water. Top two thirds of A and B show regions containing cartilages and intercartilage spaces and bottom thirds show membranous portion of the trachea containing trachealis muscle. Lymphatics stained for LYVE-1 (red) and blood vessels for PECAM-1 (green). A subset of macrophages (arrowheads) also stains weakly for LYVE-1. C and D: Insets from A and B. Many newly formed lymphatics (absent in C) persist in D. Some have a fragmented appearance (arrows). Scale bars: 400 μm (A and B); 200 μm (C and D).
IL1R1 receptor immunoreactivity in trachea, brain, and heart. A–D: IL1R1-immunoreactivity (arrows, red) colocalizes with some epithelial cells (pan-cytokeratin, green) but not with immune cells (CD45, blue) or other cells in confocal images of sections of wild-type trachea. E–J: However, IL1R1-immunoreactivity (red) colocalizes with PECAM-1 (green) in some blood vessels (arrows) in the brain (E–G) and heart (H–J). Scale bars: 50 μm (D, G, and J).