

Abstract
Alzheimer's disease (AD) is a neurodegenerative disease that proceeds with the age-dependent neuronal loss, an irreversible event which causes severe cognitive and psychiatric devastations. In the present study, we investigated whether the compound, AAD-2004 [2-hydroxy-5-[2-(4-trifluoromethylphenyl)-ethylaminobenzoic acid] which has anti-oxidant and anti-inflammatory properties, is beneficial for the brain of Tg-betaCTF99/B6 mice, a murine AD model that was recently developed to display age-dependent neuronal loss and neuritic atrophy in the brain. Administration of AAD-2004 in Tg-betaCTF99/B6 mice from 10 months to 18 months of age completely repressed the accumulation of lipid peroxidation in the brain. AAD-2004 markedly suppressed neuronal loss and neuritic atrophy, and partially reversed depleted expression of calbindin in the brain of Tg-beta-CTF99/B6. These results suggest that AAD-2004 affords neurodegeneration in the brain of AD mouse model.
Keywords: Alzheimer's disease, neuronal loss, neuritic atrophy, neuroprotection, small molecule
INTRODUCTION
Alzheimer's disease (AD) attacks 37 million people worldwide [1]. AD is a neurodegenerative disease that is characterized by the accumulation of senile plaque and neurofibrilary tangle formation in the brain including cerebral cortex and hippocampus. Although such changes are considered as important pathophenotypes of AD, more severe and ultimate problem of this devastating disease is believed to ascribe to the neuronal loss, an irreversible event which might cause severe cognitive decline, psychiatric symptoms, and other neurological deficits [2]. The drugs that are used on the clinics at present, such as acetylcholinesterase inhibitors, are basically anti-symptomatic for a limited aspect of AD pathology [3, 4]. The NMDA receptor antagonist, memantine, provides a new therapeutic choice for AD patients [4]. However, AD clinics and AD pharmaceutical fields are eagerly waiting for a new generation of drugs with a therapeutic activity to modify the AD pathogenesis per se, although some attempts are on human clinical trials [5, 6].
Transgenic mouse lines that overexpress mutant forms of APP, presenilin 1 (PS1), PS2, or tau mimic various features of AD including β-amyloidosis, tauopathy, neuronal loss, gliosis and/or cognitive deficits [7, 8]. However, none of the known AD models fully captures the complete spectrum of AD symptoms. For example, Tg2576, PDAPP, TgCRND8, and TgAPP23 mice have been used in many laboratories for the reason of robust age-dependent plaque deposition in the brain [7, 9]. These AD mouse lines do display plaque pathology, but no obvious neuronal loss or if any it is too subtle [7, 10], suggesting that physiological accumulation of Aβ and amyloid plaque do not rapidly cause neuronal loss and the underlying mechanism might be complex. Moreover, many preclinical studies for AD candidate drugs remain to test whether Aβ accumulation and plaque deposition were suppressed by a candidate drug of choices [11].
In regard to this issue, the AD mouse line, Tg-βCTF99/B6, deserves to draw an attention because this model displays profound age dependent neuronal loss in the brain. Tg-βCTF99/B6 mouse line was originally developed as a transgenic mouse expressing the βCTF99, the C-terminal fragment of human APP cleaved at the site by beta-secretase, in the brain of the inbred C57BL/6 strain [12]. Tg-βCTF99/B6 mice show a progressive neuronal loss and neuritic atrophy in the cerebral cortex along with various biochemical and transcriptional changes. The neuronal cell density in the cerebral cortex started to be lower than the non-transgenic control from the age of 12-14 months, and the reduction reached approximately 15% in 18 months. The agedependent histological and biochemical changes are accompanied with a prior onset of cognitive impairments and increased anxiety [12]. Accordingly, this AD model provides an opportunity to test therapeutic potentials of drugs of interests if they confer neuroprotection in the brain with AD-like pathogenesis.
AAD-2004 [2-hydroxy-5-[2-(4-trifluoromethylphenyl)-ethylaminobenzoic acid] is a derivative of 5-aminosalicylate that has anti-oxidant activity at nanomolar concentration in cultured primary cortical cells. Recently, it was demonstrated that AAD-2004 had an anti-oxidant property in primary cortical culture, and its anti-oxidant capacity was similar to that by its analog neu2000 [13]. AAD-2004 reduced autophagosome formation, axonopathy, and motor neuron degeneration, improving motor function and increasing life span in in a mouse model of amyotrophic lateral sclerosis [14]. In the present study, we investigated whether AAD-2004 produces a beneficial effect against neuronal loss in the brain of Tg-βCTF99/B6 mice.
MATERIALS AND METHODS
Animals and treatment with AAD-2004
Tg-βCTF99/B6 mice expressing the β-secretase-cut C-terminal fragment of human APP (βCTF99) in the brain of inbred C57BL/6 strain were maintained as previously described [12]. Mice were housed in clear plastic cages in pair in a temperature- and humidity-controlled environment with a 12 hour-light/dark cycle, and were maintained on an ad libitum diet of lab chow and water. Beginning from 10 months of age, Tg-βCTF99/B6 mice and their non-transgenic control mice were fed either lab chow containing AAD-2004 or no drug (control group). AAD-2004 was prepared as described previously [14] and was provided by GNT Pharma Inc. (Yongin, Korea). We prepared the lab chow containing AAD-2004 and control food. All animals were handled in accordance with the animal care guidelines of the Ewha Womans University School of Medicine.
Histological works
Tg-βCTF99/B6 mice and their control non-transgenic mice at the age of 18-18.5 months were sacrificed and perfused with 0.9% saline. The right and left hemispheres of the brain were used for, respectively, histologic and biochemical analyses. For histologic analysis, brain sections were stained with cresyl violet, and the numbers of stained cells were counted using the TOMORO ScopeEye 3.6 system (Olympus, Japan) as described previously [12, 15]. Because cresyl violet stained a part of glia, anti-NeuN staining was also applied as a supplemental approach. For immunohistochemistry, the right hemisphere was post-fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) at 4℃ overnight. Brain sections were prepared by cutting the brain at 40 µm intervals using a vibratome (Leica VT 1000S; Leica Instruments, Nussloch, Germany), as previously described [12]. Free-floating sections were blocked by 5% normal goat serum, 2% BSA, and 2% FBS. Brain sections were stained with anti-calbindin (Sigma, C9848; St. Louis, MO, USA) or anti-MAP2 (Lab Vision, MS-567-P0; Fremont, CA, USA) and a biotinylated HRP system was used for color development.
Malondialdehyde (MDA) assay
MDA assay was performed using the Bioxytech MDA-586 kit (Oxis Research, Portland, OR, USA) as described previously [15]. Briefly, brains were homogenized in 4 vol. of ice-cold 20 mM PBS (phosphate buffered saline; pH 7.4) containing 5 mM butylated hydroxytoluene. Homogenates were centrifuged at 3,000 g for 10 min at 4℃, and the supernatant was collected. For each reaction, 200 µl of the supernatant was mixed with 10 µl of probucol, 640 µl of diluted R1 reagent (1:3 of methanol:Nmethyl-2-phenylindole), and 150 µl of 12 N HCl. Each reaction was incubated at 45℃ for 60 min and centrifuged at 10,000 g for 10 min. The supernatant was taken and used to measure MDA formation at 586 nm in duplicate. MDA data were normalized to the protein concentration and expressed as a percentage of the sham control value.
Western blots
For the Western blot analysis, the hippocampus was homogenized in ice-cold lysis buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate) containing 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor (Complete™; Roche, Mannheim, Germany). Immunoblots were detected using ECL detection reagents (Santa Cruz, CA, USA). Each lane was loaded with 30 µg protein.
Anti-mouse APP antibody (51-2700) from Zymed Laboratories (South San Francisco, CA, USA); anti-COX-2 (22420) from Transduction Laboratory (Lexington, KY, USA); anti-TTR (sc-13098) from Santa Cruz Biotechnology, Inc.; anti-calbindin (C9848) from Sigma (St. Louis, MO, USA); anti-phospho-CREB (06-519) were obtained from Upstate Biotechnology (Lake placid, NY, USA); and anti-β-actin (ab6276) from Abcam (Cambridge, UK).
Statistical analyses
Regarding statistical analyses, two-sample comparisons were carried out using the Student's t-test and multiple comparisons were made using one-way ANOVA followed by the Newman-Keuls multiple range test. All data were presented as the mean±SEM and statistically significant differences were accepted at the 5% level, unless otherwise indicated.
RESULTS
Tg-βCTF99/B6 mice displayed enhanced anxiety at 7 months, and various biochemical and transcriptional changes related to AD pathology at 10 months of age, prior to the advent of obvious neuronal loss [12]. In consideration of this fact, Tg-βCTF99/B6 mice were fed chow containing AAD-2004 starting from -10 months to -18 months of age (Fig. 1A). The administration of AAD-2004 at 25 mg/kg/day of dose for 8 months did not give a significant influence on the body weight compared with the untreated non-transgenic control (data not shown).
Fig. 1.

Administration of AAD-2004 suppressed the increase of oxidative stress build-up in the brain of aged Tg-βCTF99/B6 mice. (A) Schematic presentation of treatment with AAD-2004. AAD-2004 was administered at 25 mg/kg/day for 8 months from 10 months to ~18 months of age. (B) MDA levels in the superior prefrontal cortex of non-transgenic control and Tg-βCTF99/B6 mice with or without AAD-2004 treatment. WTCon, non-transgenic wild type control; WT-AAD, wild type mice treated with AAD-2004; Tg-Con, Tg-βCTF99/B6 mice; Tg-AAD, Tg-βCTF99/B6 mice treated with AAD-2004. Data are presented as the means±SEM (each group with 5~8 animals). * and **, differences from the control or between indicated group at p<0.05 and p<0.01, respectively.
Tg-βCTF99/B6 mice at 18-18.5 months of age, when severe neuronal loss occurred [12], showed an enhanced level of lipid peroxidation in the brain compared to the control brain, indicting that the brain of Tg-βCTF99/B6 mice at -18.5 months of age showed an increase of oxidative stress. In contrast, administration of AAD-2004 in Tg-βCTF99/B6 mice completely blocked the increase of lipid peroxidation in the brain (Fig. 1B).
Next, we examine whether AAD-2004 confers a protective effect against the neuronal loss. Histologic analysis of cresyl violetstained brain sections showed that brain sections of Tg-βCTF99/B6 mice were lightly stained compared with the non-transgenic aged-matched control brain, as reported in the previous study [12]. We examined the numbers of cresyl violet-stained cells in the cortex using a computer-aid cell counting system (Fig. 2A, B). The cell density of cresyl violet-stained cells in the prefrontal cortex and the parietal cortex of the brains of Tg-βCTF99/B6 mice was reduced to, respectively, 87% and 89% of the non-transgenic control mice (Fig. 2C-G, Table 1). Anti-NeuN staining showed a similar result (data not shown). These results are in agreement with the previous report [12]. Whereas the cell density in the prefrontal cortex and in the parietal cortex of Tg-βCTF99/B6 mice treated with AAD-2004 was, respectively, 94% and 96% of the non-transgenic control mice (Fig. 2C-G, Table 1), suggesting that AAD-2004 indeed afforded significant neuroprotection.
Fig. 2.

Administration of AAD-2004 protected the neuronal loss in the brain of Tg-βCTF99/B6 mice. (A, B) Cresyl violet-stained prefrontal and parietal cortices of control mice. The hatched areas on the two panels in (A) were the region used for cell counting shown in E-G and in Table 1, and the red rectangle on the hatched area was for the images in C-E. (B) An example of computer-aid cell counting stained with cresyl violet in the selected cortical region indicated (left panel). Right panel is the high magnification of the rectangle in the left. (C-E) Photomicrographs showing the cresyl violet-stained prefrontal cortices of non-transgenic control mice (WT; A), Tg-βCTF99/B6 control mice (TG; B), and Tg-βCTF99/B6 mice fed with AAD-2004 (TG+AAD-2004; C). Scale bar, 200 µm. (F, G) The cell density in the prefrontal cortex (pfCTX) and parietal cortex (pCTX) was presented in % values and also in the numbers per mm2. Data are presented as the means±SEM (each group with 5-8 animals). * and **, differences from the control or between indicated group at p<0.05 and p<0.01, respectively.
Table 1.
Summary of cell counts in the prefrontal and parietal cortices of Tg-βCTF99/B6 mice and of their non-transgenic control with or without AAD-2004 treatment
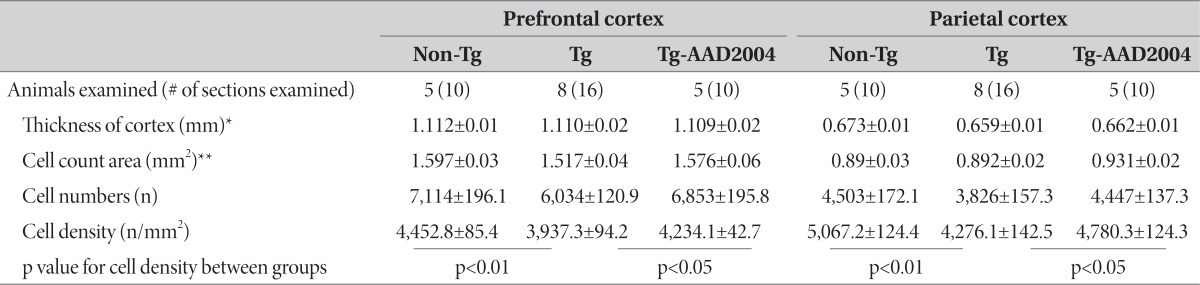
The parameters and values for the count of cresyl-violet stained cell densities in the prefrontal and parietal cortices of Tg-βCTF99/B6 mice at the age 18-18.5 M and of their non-transgenic control. Cell numbers and cell density in the respective region were determined for each brain section, and their averaged values (means±S.E.M.) were presented. *The thickness was measured at the primary sensory cortex in the prefrontal cortex and at the posterior parietal association area in the parietal cortex. **Cell count areas for each brain region were indicated in Fig. 2A.
Consistent with the previous study [12], the cerebral cortex in the brain of Tg-βCTF99/B6 mice showed the reduced expression of anti-MAP2 immunoreactivity and fewer long-running neuritic fibers, compared with the non-transgenic control (Fig. 3A-C). Whereas the reduced anti-MAP2 immunoreactivity and fewer long-running neuritic fibers in the brain of Tg-βCTF99/B6 mice were reversed to the levels of the non-transgenic control in mice treated with AAD-2004 (Fig. 3D).
Fig. 3.

AAD-2004 reverted the neuritic atrophy in the brain of Tg-βCTF99/B6 mice. (A) Photomicrograph showing anti-MAP-2 antibody-stained parietal cortex of non-transgenic control mouse. The rectangle is the region shown with high magnification in B-D. All animals analyzed were 18-18.5 months-old. (B-D) Photomicrographs showing anti-MAP-2 antibody-stained parietal cortex of non-transgenic control mice (WT; B), Tg-βCTF99/B6 control mice (TG; C), and Tg-βCTF99/B6 mice fed with AAD-2004 (TG+AAD-2004; D). Note the reduced numbers and morphology of long-running apical dendrites in Tg-βCTF99/B6 mice (C), while the protection was evident in the Tg-βCTF99/B6 mice fed with AAD-2004 (D). 5-8 animals were analyzed for each group. Scale bar, 200 µm.
We continued to examine the expression levels of other cellular factors which were altered in the expression in the brain of Tg-βCTF99/B6 mice. Immunohistochemical analysis revealed that calbindin expressions in the pyramidal cells of CA1 and CA 3 regions, in the granule cells of the dentate gyrus, in the stratum oriens and stratum lucidum of the hippocampus, and in the molecular layer of the dentate gyrus were severely down-regulated in Tg-βCTF99/B6 mice. These reduced expressions were partially restored following the administration of AAD-2004 (Fig. 4A-F). Western blot analysis showed that Tg-βCTF99/B6 mice showed enhanced expression of mouse APP (mAPP) and down-regulated expression of phosopho-CREB in the hippocampus compared to the non-transgenic control. Following the treatment with AAD-2004 in Tg-βCTF99/B6 mice, mAPP, calbindin, phosopho-CREB, and transthyretin (TTR) were weakly up-regulated (Fig. 4G).
Fig. 4.

AAD-2004 partially reversed the down-regulated expression of calbindin in the brain of Tg-βCTF99/B6 mice. (A-F) Photomicrographs showing anti-calbindin-stained hippocampus of non-transgenic control mice (WT; A, B), control Tg-βCTF99/B6 mice (TG; C, D), and Tg-βCTF99/B6 mice fed with AAD-2004 (TG+AAD-2004; E, F). Photomicrographs with high magnification of rectangles on the left panels (A, C, E) were shown (B, D, F). Note the anti-calbindin immunoreactivity not only in the granular layer (g) of the dentate gyrus, but also in the pyramidal cells (arrows), in the molecular layer (ml) of the dentate gyrus, and in the stratum oriens (so) and stratum radiatum (sr) of the hippocampus. p, pyramidal layer; hi, hilus. Scale bar, 200 µm. (G) Representative Western blot images showing the expression of mouse amyloid precursor protein (mAPP), COX-2, calbindin, transthyretin (TTR), phospho-CREB, and β-actin in the hippocampus. Total 5-8 animals were analyzed for each group.
DISCUSSION
In the present study, we demonstrated that administration of AAD-2004 in Tg-βCTF99/B6 mice suppressed the neuronal loss and neuritic atrophy in the brain, which otherwise occurs in an age-dependent manner (Fig. 2, 3, Table 1). Because the neuronal loss ultimately causes various devastating cognitive and psychiatric catastrophes, this data argues that AAD-2004 confers a therapeutic potential for AD-like brain. This interpretation is further supported by the finding that the administration of AAD-2004 up-regulated the expressions of calbindin and phospho-CREB in the brain of Tg-βCTF99/B6 mice (Fig. 4). Calbindin and phospho-CREB are two cellular markers that are distinctly downregulated in the brain of Tg-βCTF99/B6 mice [12], as well as in AD-like brains [16-18]. AAD-2004 up-regulated the expression of mAPP (Fig. 4G), although its physiological significance remains unknown.
Many known transgenic AD models display plaque loading at various times of the onset, while others show tau pathology [9, 19, 20]. Double or triple transgenic mice (ex, Tg-APPswe/PS1dE9 and 3xTg-AD) start to show plaque deposition in the brain at -6 months of age [21] or even at 2 months of age in 5xFAD mice [22]. Despite such dramatic plaque deposition in transgenic mice, none of the known AD models fully recapitulate the complete spectrum of AD symptoms including plaque pathology, tau pathology, the neuronal loss, and neuronal loss-related pathology within a single mouse line. Therefore, the evaluation of candidate drugs for AD has been relied upon plaque models in most cases [23] or tau pathology models in a few cases [24]. Because neuronal loss is the most critical event in the brain of AD, evaluation of AD candidate drugs needs to be carried out using proper neurodegeneration models, but these attempts can be barely found thus far. We speculate that the reason should not be attributed to the ignorance of physiological significance of neuronal loss in the AD brain, but it might be attributed to the availability and/or lack of proper animal models that allows to address this issue. In regard to these, Tg-βCTF99/B6 mice stand at a special niche among the numerous available plaque or tau pathology models, because Tg-βCTF99/B6 mice displayed a distinct age-dependent cell loss in the brain and related cognitive deficits [12]. Because of the availability of this neurodegenration model, we were able to evaluate the neuroprotective property of AAD-2004 in the present study. As demonstrated here, AAD-2004 indeed has a capability to suppress the age-dependent neuronal loss in vivo.
We provide evidence that the age-dependent neuronal loss in the brain of Tg-βCTF99/B6 mice proceeded with the accumulation of metabolic oxidative stress, which was blocked by treatment with AAD-2004 (Fig. 1). These results suggest that anti-AD-like effect of AAD-2004 is related to anti-ROS property or a property to suppress the ROS levels. This interpretation is supported by the fact that AAD-2004 is constructed from the parent chemicals, aspirin and sulfasalazine. Sulfasalazine produces an anti-ROS property in primary cortical neurons insulted with ferrous or H2O2 [25]. Nonetheless, we do not exclude the possibility that anti-AD-like effect of AAD-2004 is affected by the mechanism that needs to be studied in the future. Because AAD-2004 confers beneficial effects on AD-like brain, it will be of interests to understand the mechanism underlying the action of AAD-2004 on the AD-like brain.
ACKNOWLEDGEMENTS
This work was supported by a grant (GT5) from Ewha Womans University.
References
- 1.Lynch C, Eeckman F, Lynch Z. The neurotechnology industry 2012 report. St. San Francisco, CA: Neuroinsights; 2012. [Google Scholar]
- 2.Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corbett A, Pickett J, Burns A, Corcoran J, Dunnett SB, Edison P, Hagan JJ, Holmes C, Jones E, Katona C, Kearns I, Kehoe P, Mudher A, Passmore A, Shepherd N, Walsh F, Ballard C. Drug repositioning for Alzheimer's disease. Nat Rev Drug Discov. 2012;11:833–846. doi: 10.1038/nrd3869. [DOI] [PubMed] [Google Scholar]
- 4.Howard R, McShane R, Lindesay J, Ritchie C, Baldwin A, Barber R, Burns A, Dening T, Findlay D, Holmes C, Hughes A, Jacoby R, Jones R, Jones R, McKeith I, Macharouthu A, O'Brien J, Passmore P, Sheehan B, Juszczak E, Katona C, Hills R, Knapp M, Ballard C, Brown R, Banerjee S, Onions C, Griffin M, Adams J, Gray R, Johnson T, Bentham P, Phillips P. Donepezil and memantine for moderate-to-severe Alzheimer's disease. N Engl J Med. 2012;366:893–903. doi: 10.1056/NEJMoa1106668. [DOI] [PubMed] [Google Scholar]
- 5.Salloway S, Mintzer J, Weiner MF, Cummings JL. Disease-modifying therapies in Alzheimer's disease. Alzheimers Dement. 2008;4:65–79. doi: 10.1016/j.jalz.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Rafii MS, Aisen PS. Recent developments in Alzheimer's disease therapeutics. BMC Med. 2009;7:7. doi: 10.1186/1741-7015-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer's disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hall AM, Roberson ED. Mouse models of Alzheimer's disease. Brain Res Bull. 2012;88:3–12. doi: 10.1016/j.brainresbull.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eriksen JL, Janus CG. Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behav Genet. 2007;37:79–100. doi: 10.1007/s10519-006-9118-z. [DOI] [PubMed] [Google Scholar]
- 11.Higgins GA, Jacobsen H. Transgenic mouse models of Alzheimer's disease: phenotype and application. Behav Pharmacol. 2003;14:419–438. doi: 10.1097/01.fbp.0000088420.18414.ff. [DOI] [PubMed] [Google Scholar]
- 12.Lee KW, Im JY, Song JS, Lee SH, Lee HJ, Ha HY, Koh JY, Gwag BJ, Yang SD, Paik SG, Han PL. Progressive neuronal loss and behavioral impairments of transgenic C57BL/6 inbred mice expressing the carboxy terminus of amyloid precursor protein. Neurobiol Dis. 2006;22:10–24. doi: 10.1016/j.nbd.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 13.Cho J, Son SJ, Ko SY, Lee MJ, Byun SY, Lee YA, Park SM, Kim JH, Gwag BJ. Nonclinical safety and pharmacokinetic profile of AAD-2004 as a putative disease-modifying drug for neurodegenerative diseases; The 9th international conference AD/PD 2009; 2009 Mar 11-15; Prague, Czech Republic. 2009. [Google Scholar]
- 14.Shin JH, Lee YA, Lee JK, Lee YB, Cho W, Im DS, Lee JH, Yun BS, Springer JE, Gwag BJ. Concurrent blockade of free radical and microsomal prostaglandin E synthase-1-mediated PGE2 production improves safety and efficacy in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2012;122:952–961. doi: 10.1111/j.1471-4159.2012.07771.x. [DOI] [PubMed] [Google Scholar]
- 15.Seo JS, Kim TK, Leem YH, Lee KW, Park SK, Baek IS, Kim KS, Im GJ, Lee SM, Park YH, Han PL. SK-PC-B70M confers anti-oxidant activity and reduces Abeta levels in the brain of Tg2576 mice. Brain Res. 2009;1261:100–108. doi: 10.1016/j.brainres.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto-Sasaki M, Ozawa H, Saito T, Rösler M, Riederer P. Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res. 1999;824:300–303. doi: 10.1016/s0006-8993(99)01220-2. [DOI] [PubMed] [Google Scholar]
- 17.Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee KW, Lee SH, Kim H, Song JS, Yang SD, Paik SG, Han PL. Progressive cognitive impairment and anxiety induction in the absence of plaque deposition in C57BL/6 inbred mice expressing transgenic amyloid precursor protein. J Neurosci Res. 2004;76:572–580. doi: 10.1002/jnr.20127. [DOI] [PubMed] [Google Scholar]
- 19.Richardson JA, Burns DK. Mouse models of Alzheimer's disease: a quest for plaques and tangles. ILAR J. 2002;43:89–99. doi: 10.1093/ilar.43.2.89. [DOI] [PubMed] [Google Scholar]
- 20.Mineur YS, McLoughlin D, Crusio WE, Sluyter F. Genetic mouse models of Alzheimer's disease. Neural Plast. 2005;12:299–310. doi: 10.1155/NP.2005.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sabbagh JJ, Kinney JW, Cummings JL. Animal systems in the development of treatments for Alzheimer's disease: challenges, methods, and implications. Neurobiol Aging. 2013;34:169–183. doi: 10.1016/j.neurobiolaging.2012.02.027. [DOI] [PubMed] [Google Scholar]
- 22.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lahiri DK, Maloney B. Beyond the signaling effect role of amyloid-β42 on the processing of AβPP, and its clinical implications. Exp Neurol. 2010;225:51–54. doi: 10.1016/j.expneurol.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Himmelstein DS, Ward SM, Lancia JK, Patterson KR, Binder LI. Tau as a therapeutic target in neurodegenerative disease. Pharmacol Ther. 2012;136:8–22. doi: 10.1016/j.pharmthera.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryu BR, Lee YA, Won SJ, Noh JH, Chang SY, Chung JM, Choi JS, Joo CK, Yoon SH, Gwag BJ. The novel neuroprotective action of sulfasalazine through blockade of NMDA receptors. J Pharmacol Exp Ther. 2003;305:48–56. doi: 10.1124/jpet.102.042606. [DOI] [PubMed] [Google Scholar]