

Abstract
Hyperhomocysteinemia is considered to be a significant risk factor in atherosclerosis and plays an important role in it. The purpose of this study was to determine the molecular mechanism of blood monocyte chemoattractant protein-1 (MCP-1) promoter DNA hypomethylation in the formation of atherosclerosis induced by hyperhomocysteinemia, and to explore the effect of nuclear factor-κB (NF-κB)/DNA methyltransferase 1 (DNMT1) in this mechanism. The atherosclerotic effect of MCP-1 in apolipoprotein E–deficient (ApoE−/−) and wild-type C57BL/6J mice was evaluated using atherosclerotic lesion area; serum NF-κB, MCP-1, and DNMT1 levels; and MCP-1 promoter DNA methylation expression. In vitro, the mechanism responsible for the effect of NF-κB/DNMT1 on foam cells was investigated by measuring NF-κB and DNMT1 levels to determine whether NF-κB/DNMT1 had an effect on gene expression. Compared with the control group, atherosclerotic lesions in ApoE−/− mice fed a high methionine diet significantly increased, as did the expression of MCP-1. In vitro study showed that pyrrolidine dithiocarbamate treatment down-regulated levels of NF-κB and raised DNMT1 concentrations, confirming the effect of NF-κB/DNMT1 in the MCP-1 promoter DNA methylation process. In conclusion, our results suggest that through NF-κB/DNMT1, MCP-1 promoter DNA hypomethylation may play a key role in formation of atherosclerosis under hyperhomocysteinemia.
Key words: atherosclerosis, DNA methylation, hyperhomocysteinemia, monocyte chemoattractant protein-1
Introduction
Homocysteine (Hcy) is an intermediate in methionine metabolism.1 A large amount of epidemiological studies2 have confirmed that an increased plasma Hcy concentration is an independent risk factor for atherosclerosis leading to cardiovascular diseases. Though mechanisms such as inflammation and oxidative stress3 of hyperhomocysteinemia-induced atherosclerosis have been reported in related research, the mechanism itself is not definitive so far.
Atherosclerosis has been established as a complex inflammatory disease characterized by the accumulation of lipids, fibrous materials, and cell debris in the arteries,4 and it is mediated by complicated molecular interactions, in which chemokines play a key role.5 A significant and the most well-characterized chemokine is monocyte chemoattractant protein-1 (MCP-1/CCL2), which is increased in atherosclerotic lesions but not in normal arteries, suggesting its potential value in stimulating the migration of monocytes into the intima of the arterial wall and taking part in most processes during atherosclerosis.6,7 The role of MCP-1 appears to be more evident, and research has simultaneously demonstrated that anti-MCP-1 gene therapy not only limits progression of established atheroma but also limits transformation from destabilized plaque to stable plaque.8 This strongly shows a major role of MCP-1 in the pathogenesis of atherosclerosis and suggests that MCP-1 may be a promising therapeutic target against atherosclerosis. However, it is not clear whether MCP-1 comes into play in hyperhomocysteinemia-induced atherosclerosis. Therefore, our study explored the molecular mechanism of MCP-1 in atherosclerosis.
DNA methylation is an important epigenetic mechanism and may explain the relationships between an individual's genetic background, environment, and disease.9 DNA methylation patterns are inhibited by S-adenosylhomocysteine (SAH), and pattern maintenance depends on DNA methyltransferase 1 (DNMT1) and intracellular S-adenosylmethionine (SAM) levels.10,11 Aberrant DNA methylation regulates gene expression12 and is associated with advanced stages of disease, such as atherosclerosis. The presence of aberrant DNA methylation in an individual may indicate predisposition to atherosclerosis and related diseases.13 Pilot studies14–16 have suggested the relationship between atherosclerosis-related genes and DNA methylation in vascular smooth muscle cells, endothelial cells, and foam cells, however, there remain limitations in these in vitro experiments. Consequently, we chose to observe MCP-1 promoter DNA methylation of blood, which is closely related to an individual's atherosclerotic pathogenesis. DNA methylation takes part in transcription silencing, and hypomethylation activates the expression of genes via inhibition of the association of DNA promoter recognition site and special transcription factors such as nuclear factor-κB (NF-κB).17 Activated NF-κB binds to specific DNA sequences of target genes and regulates transcription of genes involved in growth regulation and inflammation.18 Because of various levels of regulation, the NF-κB signaling pathway can be potentially targeted at various levels including nuclear translocation, DNA binding, and methyltransferases.19 Contemporary research has pointed out the ability of NF-κB (RelA/p65) to directly recruit DNMT1 to chromatin, resulting in promoter-specific methylation,20 and suggested that bortezomib can result in down-regulation of DNMT1 via the SP1/NF-κB pathway and induce genomic DNA hypomethylation in human leukemia cells.21 We postulated that disruption of the NF-κB and DNMT1 interplay might in turn lead to DNMT1 down-regulation and DNA hypomethylation.
Marumo et al.22 showed that Hcy can induce elevated MCP-1 secretion via the NF-κB in human umbilical vein endothelial cells and human vascular smooth muscle cells. However, the connections between Hcy, MCP-1 promoter DNA methylation, and atherosclerosis remain largely unknown. The present study explored whether blood MCP-1 promoter DNA hypomethylation may promote formation of atherosclerosis under hyperhomocysteinemia, as well as the effect of NF-κB/DNMT1 in the pathogenesis.
Materials and Methods
Animal model and separation of mononuclear cells
To establish an animal model with atherosclerosis, 12 male wild-type mice and 36 male apolipoprotein E-deficient (ApoE−/−) mice on a C57BL/6J genetic background were kept in our laboratory for 15 weeks. The animals were purchased from Jackson Laboratory (Bar Harbor, ME) and bred to 5 weeks of age in the Animal Center of Beijing University (Beijing, China). After 1 week of acclimatization, ApoE−/− mice were randomly divided into three groups (n=12 each). Mice of the ApoE−/− control group were fed with regular mouse diet for 15 weeks. The high-methionine group (HM) received the same diet but with a 1.7% methionine supplement. The high-methionine plus folate and vitamin B12 group (HM+FA+VB) received regular mouse diet plus 1.7% methionine, 0.006% folate, and 0.0004% vitamin B12. Wild-type C57BL/6J mice (n=12) were fed with a regular mouse diet as blank control group. After 15-week experimental diets, mice were euthanized with 20% ethylcarbamate (2 mL/100 g) enterocoelia injection, and blood was immediately collected to measure serum Hcy, NF-κB, MCP-1, and DNMT1 levels. The aortas were stored at −80°C until analysis.
Blood used for separation of mononuclear cells should be fresh and free of clots in preservative-free anticoagulant. Histopaque 1083 (Sigma, St. Louis, MO) was added to centrifuge tubes. Whole blood was carefully layered onto the Histopaque 1083 surface and centrifuged at 400 g for exactly 30 min at room temperature. After centrifugation, the upper layer was aspirated carefully with a plastic pipe until only 3 mm of the opaque interface containing the mononuclear cells remained. Isotonic phosphate-buffered saline (PBS) was then added, and this was centrifuged at 250 g for 10 min for three times to remove any remaining Histopaque 1083 from the mononuclear cells. The cells suspended in isotonic PBS were used for the following assays.
Cell culture
The TH-1 monocyte cell line was obtained from Sichuan University (Chengdu, China). THP-1 cells were cultivated in Roswell Park Memorial Institute (RPMI) 1640 with 15% fetal bovine serum (FBS), penicillin (100 U/mL)-streptomycin (100 μg/mL) at 37°C in a 5% CO2 atmosphere, to a density of 107 cells/mL. Then THP-1 cells were differentiated into macrophages by incubation for 24 h with 50 nmol/L phorbol myristate acetate (PMA, Sigma). THP-1 cell-derived macrophages (West China Hospital of Sichuan University) were treated with oxidized low density lipoprotein (50 mg/L, Sigma) and Hcy (0, 100 μmol/L, Sigma) for 24 h at 37°C and stained with oil red O. Foam-cell formation was observed under an inverted microscope (Olympus, Tokyo, Japan). Foam cells were divided into three groups: foam cell, foam cell intervened by Hcy (100 μmol/L), and foam cell intervened by Hcy (100μmol/L) and pyrrolidine dithiocarbamate (PDTC, 50 μmol/L, Sigma).
Determination of serum Hcy, SAM, and SAH levels
After 30 min standing at room temperature, serum was obtained by centrifugation (3000 g for 10 min at 4°C), and serum Hcy concentrations were measured by automatic biochemistry analyzer. Serum SAM and SAH concentrations were measured by high-performance liquid chromatography (HPLC; D-2000 Elite HPLC, Hitachi High Technologies, Tokyo, Japan). The supernatant of each sample was filtered through a 0.22-μm filter (Millipore, Billerica, MA) and then loaded onto a C18 column (250 mm×4.6 mm internal diameter, 5-μm particle) fitted with a matched guard column, run by a Waters HPLC system (D-2000 Elite HPLC, Hitachi High Technologies), and connected to an ultraviolet detector. Absorption of eluted compounds was monitored at λex=254 nm. Chromatograms were recorded by a D-2000 Elite HPLC integrator with its quantification accomplished by automatic peak area integration. SAM and SAH standards (Sigma) were used to identify the elution peaks.
Tissue preparation and atherosclerotic lesion examination
The analysis of paraffin histological sections was used for evaluating atherosclerotic lesions. The aortas were separated carefully and then put into 4% (w/v) formalin for 24 h. The samples subsequently underwent washing, dehydration, clearing, embedding, slicing, coating, grilling, and hematoxylin-eosin (HE) staining. Finally, the sections were observed via microscope (Olympus).
Serum NF-κB, MCP-1, and DNMT1 concentrations and NF-κB and DNMT1 levels in foam cells measured by enzyme-linked immunosorbent assay
Serum NF-κB, MCP-1, and DNMT1 concentrations and NF-κB and DNMT1 levels in foam cells were determined by using commercially available enzyme-linked immunosorbent assay kit (ELISA; R&D Systems, Emeryville, CA) according to the manufacturer's instructions. All reagents were kept at room temperature for at least 30 min before assays began. All standards, controls, and samples were run in duplicate. Finally, absorbance was read on a microplate reader at 450 nm.
Nested methylation-specific polymerase chain reaction for MCP-1
Genomic DNA was isolated from the peripheral blood mononuclear cells using the Wizard® Genomic DNA Purification Kit (Promega, Madison, WI). DNA concentration and purity were detected by spectrophotometry at 260 and 280 nm. The A260/A280 absorbance ratio was consistent at approximately 1.8. It integrated DNA denaturation and bisulfite conversion processes into one-step by EZ DNA Methylation-Gold™ Kit (ZYMO, Irvine, CA). Following bisulfite treatment of genomic DNA, unmethylated cytosine residues are converted into uracil. Their improved sequence specificity facilitates relative quantification of methylated and unmethylated alleles that are simultaneously amplified in a single tube. MCP-1 outer primers were 5′-TTG TTG AAA TGA ATT TTA AGG GTT T-3′ and 5′-CCC AAA TAA CTC CAA CCT AAC TAT C-3′. The process of reaction was as follows: 94°C for 5 min, 94°C for 30 sec, 65°C for 30 sec, and 72°C for 1 min, repeating for 30 cycles, decreasing 0.5°C every cycle; 94°C for 30 sec, 50°C for 30 sec, and 72°C for 1 min, repeating for 20 cycles; and 72°C for 7 min. MCP-1 methyl and unmethyl primer: left M primer, 5′-TTT AAG GGT TTT TAG ATT TTA TCG T-3′; right M primer, 5′-AAC TCT CTA CCC TAT TTC CTT CGT A-3′; left U primer, 5′-TTT AAG GGT TTT TAG ATT TTA TTG T-3′; right U primer, 5′-AAC TCT CTA CCC TAT TTC CTT CAT A-3′. The process of reaction was as follows: 94°C for 5 min, 94°C for 30 sec, 61.6°C for 30 sec, and 72°C for 1 min, repeating for 30 cycles, decreasing 0.5°C every cycle; 94°C for 30 sec, 46.6°C for 30 sec, and 72°C for 1 min, repeating for 20 cycles; and 72°C for 7 min. The PCR products were analyzed by 2% agarose gel electrophoresis.
Statistical analysis
The results were expressed as means plus or minus standard error (means±SEM). Differences among groups were analyzed by one-way analysis of variance and analysis of multiple comparisons within treatment groups was carried out using Student-Newman-Keuls's test; p<0.05 was considered as statistically significant.
Results
Analysis of aortic pathological section with HE staining and levels of serum Hcy, SAM, and SAH in mice
We found that atherosclerotic lesion areas of ApoE−/− control group, HM group, and HM+FA+VB group were increased respectively compared with those of the blank control group (Fig. 1A). In the blank control group, the aorta walls were round, and smooth muscle cells were not seen underneath the endoderm (Fig. 2A). The aorta walls were rough, and the intima revealed hyperplasia and had few foam cells and atherosclerotic substances in the ApoE−/− control group (Fig. 2B). In the HM group, a large amount of atherosclerotic substances, many foam cells, and a few inflammatory cells were seen (Fig. 2C). In the HM+FA+VB group, the thickness of aorta walls was uneven, the thickness ratio of the intima to media was decreased, and there were fewer foam cells under the fiber caps, fewer atheronecrotic substances, and fewer inflammatory cells compared with the HM group (Fig. 2D). In summary, the atherosclerosis mouse model was established successfully.
FIG. 1.

Methionine is converted through SAM and SAH to Hcy, which is subsequently remethylated back to methionine. Many investigations have revealed several potential mechanisms that Hcy induced the formation of atherosclerosis. (A) Serum Hcy concentrations. (B) Statistical analysis of lesion area of atherosclerosis. Values were means±SEM. *p<0.05, **p<0.01 vs. blank control; ■p<0.05, ■■p<0.01 vs. HM, n=10. SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; Hcy, homocysteine; HM, high-methionine.
FIG. 2.

Atherosclerotic lesion (HE, ×400 magnification) on the aortas in mice fed with diets varying in methionine for 15 weeks. The cross-sections were from aortas from each group, and these were chosen from mice whose average lesion area approximated the mean value for that group. The arrows indicate atherosclerotic plaques. (A) blank control group; (B) ApoE−/− control group; (C) HM group; (D) HM+FA+VB group. HE, hematoxylin and eosin; ApoE−/−, apolipoprotein E–deficient; FA, folic acid; VB, vitamin B12.
Hcy is a key intermediate of methionine metabolism. After 15 weeks, serum Hcy concentrations were confirmed in mice fed with different diets. The levels of serum Hcy were elevated 38.9% in the ApoE−/− control group, ∼2.5-fold in the HM group, and 96.0% in the HM+FA+VB group; a remarkable increase was observed in HM group compared with blank control group (p<0.01). Apoliproprotein E (ApoE) plays a central role in lipid metabolism and is polymorphic with three common alleles—e2, e3, and e4. ApoE4 carrier may promote greater binding of Hcy to lipoproteins and a faster clearance of the lipoprotein-bound Hcy fraction.23 Therefore serum Hcy levels were higher in mice of the ApoE−/− control group with the regular mouse diet. In comparison with the HM group, serum Hcy concentrations were slightly lower: ∼1.6-fold in the ApoE−/− control group (p<0.01), and 80.6% in the HM+FA+VB group (p<0.05, Fig. 1B). Taken together, our data suggested that Hcy may induce the formation of atherosclerosis in ApoE−/− mice, and folate and vitamin B12 could reduce the effect of Hcy-induced atherosclerosis.
There are other metabolic intermediates of methionine, including SAM, which provides a methyl group for DNA methylation, and SAH, which can inhibit DNMT1 activation. After 15-week experimental diets, serum SAM concentrations were elevated by 3-, 3.6-, and 2.45-fold in the ApoE−/− control, HM, and HM+FA+VB groups, respectively, compared with the blank control group, and serum SAH concentrations of the HM group were obviously higher in comparison with the blank control group (Fig. 3, Table 1).
FIG. 3.

A representative chromatograms showing elution profiles for SAM and SAH by high-performance liquid chromatography (HPLC) in all groups. SAM and SAH standards were used to identify the elution peaks, chromatograms were recorded by an integrator. (A) blank control group; (B) ApoE−/− control group; (C) HM group; (D) HM+FA+VB group.
Table 1.
Levels of Serum S-Adenosylmethionine and S-Adenosylhomocysteine
| Group | SAM (nmol/L) | SAH (nmol/L) |
|---|---|---|
| Blank control | 4.533±1.56 | 12.42±0.04 |
| ApoE−/− control | 18.22±3.73** | 12.72±0.14 |
| HM | 20.82±0.82** | 13.12±0.17** |
| HM+FA+VB | 15.68±1.14** | 12.97±0.19* |
Values are means±SEM.
p<0.05, **p<0.01 vs. blank control, n=10.
SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; ApoE−/−, apolipoprotein E–deficient; HM, high-methionine; HM+FA+VB, HM plus folate and vitamin B12.
Analysis of MCP-1 promoter DNA methylation and serum MCP-1 concentrations in mice
DNA methylation has emerged as an important epigenetic modification and a means to regulate gene transcription. After 15 weeks, MCP-1 promoter DNA methylation was determined in the blank control, ApoE−/− control, HM, and HM+FA+VB groups (Fig. 4A, B). Our data suggested that in the HM group, the change of MCP-1 promoter DNA hypomethylation was the most obvious, with levels ∼6.2% higher than those in the blank control group (p<0.05). In the HM+FA+VB group, antagonistic effects could be observed on the hypomethylation change (Fig. 4A, B). In general, DNA methylation inhibited transcription and hypomethylation was associated with gene activation. As a result, serum MCP-1 levels were the highest in the HM group (p<0.05; Fig. 4C). MCP-1 played a lead role in leukocyte infiltration into the vessel and activation during the inflammatory process of atherosclerosis. The atherosclerotic lesions in the HM group were the most severe and widespread, and MCP-1 promoter DNA hypomethylation and atherosclerotic area were positively correlated (γ=0.5175, p<0.05; Fig. 4D).
FIG. 4.

The monocyte chemoattractant protein-1 (MCP-1) promoter was amplified from bisulfite-converted DNA and analyzed by nested methylation-specific polymerase chain reaction (nMS-PCR). MCP-1 promoter DNA hypomethylation in mice (A), statistical analysis of MCP-1 promoter DNA hypomethylation (B), serum MCP-1 levels in mice (C), correlation of MCP-1 hypomethylation and atherosclerosis area (D) (r=0.5175, p=0.0006, n=10). M, amplified band by methylation-specific primer; U, amplified band by unmethylation-specific primer. Values were means±SEM. *p<0.05 vs. blank control, n=10.
Serum NF-κB and DNMT1 levels by ELISA analysis in mice and foam cells
Mice were fed with different experimental diets for 15 weeks, after which the amounts of NF-κB and DNMT1 were estimated by ELISA (Table 2). Hcy could induce NF-κB activation in the early stage of atherosclerosis, and serum NF-κB levels were the highest in the HM group (p<0.05). In the HM+FA+VB group, serum NF-κB levels were 2.9-fold lower than in the HM group (p<0.01). Serum DNMT1 concentrations were decreased by 20.8% in the HM group in comparison with the blank group (p<0.01) and slightly higher in the HM+FA+VB group compared with the HM group (p<0.05). Serum NF-κB and DNMT1 concentrations were negatively correlated (γ=0.3394, p<0.05; Fig. 5).
Table 2.
Levels of Serum Nuclear Factor-κB and DNA Methyltransferase 1
| Group | NF-κB (μmol/L) | DNMT1 (U/L) |
|---|---|---|
| Blank control | 190.40±11.17 | 17.42±0.56 |
| ApoE−/− control | 200.40±18.22 | 15.92±0.63 |
| HM | 261.50±6.45* | 14.42±0.36** |
| HM+FA+VB | 67.31±4.22† | 16.37±0.42‡ |
Values are means±SEM.
p<0.05, **p<0.01 vs. blank control.
p<0.05, †p<0.01 vs. HM.
NF-κB, nuclear factor-κB; DNMT1, DNA methyltransferase 1.
FIG. 5.
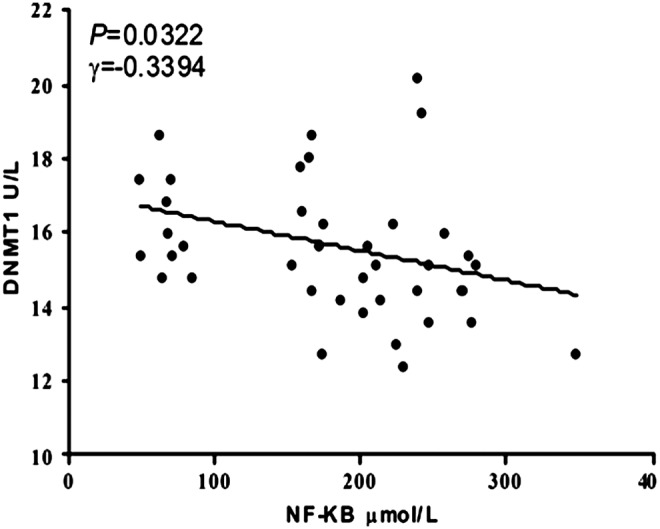
Correlation between serum nuclear factor-κB (NF-κB) and DNA methyltransferase 1 (DNMT1) concentrations in wild-type and ApoE−/− mice (r=−0.3394, p=0.0322, n=10).
Our results might indicate a negative correlation between NF-κB and DNMT1 in wild-type and ApoE−/− mice. In order to further confirm the interaction between NF-κB and DNMT1, we studied foam cells (Fig. 6) to explore the relationship of NF-κB and DNMT1. NF-κB is a transcription factor involved in cell proliferation, adhesion, invasion, and metastasis, and PDTC is an antioxidant and potent NF-κB inhibitor that has been used to suppress the activity of NF-κB in other cell types. DNMTs are also responsible for genomic DNA methylation, which is then maintained by DNMT1. Our results appear to confirm that NF-κB concentration was decreased by PDTC intervention (p<0.01; Fig. 7A) and DNMT1 level was increased in the foam cell+Hcy+PDTC group in comparison with the HM group (p<0.05; Fig. 7B). Taken together, these findings showed that NF-κB might suppress the activity of DNMT1 with the involvement of DNA hypomethylation.
FIG. 6.
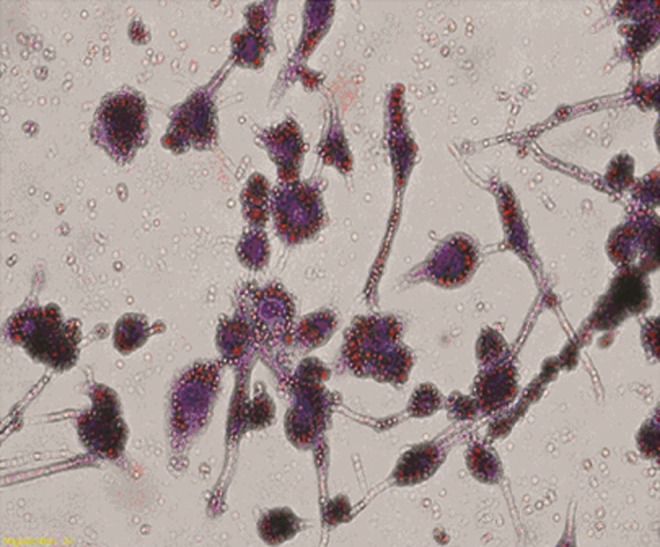
Foam cell formation assay. Phorbol myristate acetate (PMA)-induced THP-1 monocytes to transformate for macrophages with pseudopodium for 48 h and macrophages were incubated with oxidized low density lipoprotein (ox-LDL; 50 μg/mL). Foam cells were irregular shapes and bubble-like change with oil-red O staining (magnification ×400). Red particles in the cytoplasm represent lipid droplets.
FIG. 7.
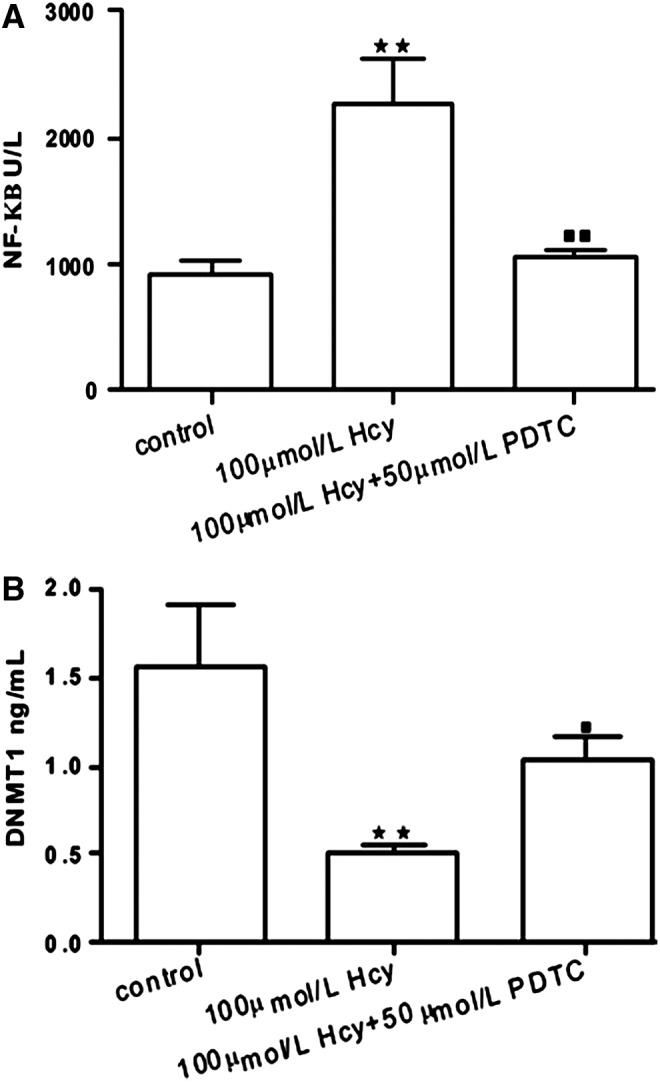
NF-κB and DNMT1 levels in foam cells. Values are expressed as means with their standard errors depicted by vertical bars. **p<0.01 vs. foam cell, ■p<0.05, ■■p<0.01 vs. 100 μmol/L Hcy.
Discussion
Over the past decade, an accumulating body of evidence has indicated that Hcy is an independent risk factor for atherosclerosis, and an obvious association between Hcy and atherosclerosis has been reported in many retrospective and prospective studies.24,25 However, the mechanisms of hyperhomocysteinemia-induced atherosclerosis is not yet well understood. We chose wild-type and ApoE−/− mice fed with a high-methionine diet to confirm the role of MCP-1 in the formation of hyperhomocysteinemia-induced atherosclerosis.Our data provided the first evidence that atherosclerosis models were established successfully by analyzing pathological sections with HE staining and serum Hcy concentrations.
A related study suggested that MCP-1 is a part of inflammation and acts as a basic mediator in a biological system that plays a significant role in the early stages of atherosclerosis formation.26 In another study, ApoE−/− mice with increased MCP-1 secretion were at an increased risk of atherosclerosis.27 The structure of MCP-1/CCL2 [location: 11C-E1; 1146.5 cM; sequence: chromosome: 11; NC-000077.6 (82035577…82037452)] is illustrated in Figure 8. Simultaneously, MCP-1 SNP-2518 may be a valuable genetic marker for assessing the risk of developing distant metastasis after radiation therapy in nasopharyngeal carcinoma patients.28 There is research showing that MCP-1 promoter hypomethylation may be affected by blood glucose and triglyceride.29 However, the interaction of hyperhomocysteinemia and MCP-1 promoter DNA methylation has not been demonstrated by much experimental evidence. In our study, the MCP-1 promoter was hypomethylated and the level of MCP-1 hypomethylation was the highest in the HM group. Recently, small interfering RNAs (siRNAs) have been found to participate in gene regulation together with methylation and can induce the methylation of the promoter, which thus silences the gene.30 As a result of siRNA participation, serum Hcy levels were increased nearly 2.5 times and a concomitant increase of lesion area also occurred in ApoE−/− mice fed with the high methionine diet, but there was only a slight reduction in MCP-1 DNA methylation and a modest increase in serum MCP-1. The current prevailing explanation for Hcy-induced DNA hypomethylation is that DNA methylation modification needs SAM as the methyl donor. SAM is converted into SAH, which potentially inhibits transmethylation reactions.31 The accumulation of SAH can affect the DNA methylation pattern by causing a feedback inhibition of SAM-dependent methyltransferases.32 In this study, our results suggested that SAM and SAH could also explain MCP-1 promoter DNA hypomethylation. After the transmethylation reaction, SAM converts to SAH, undergoing hydrolysis to form Hcy; Hcy can be remethylated to methionine.33 An increased level of Hcy would interfere with this methionine cycle, and an increased level of Hcy is associated with DNA hypomethylation.34 Therefore we hypothesized that Hcy could also take part in MCP-1 promoter DNA hypomethylation by the methionine cycle. The correlation of MCP-1 hypomethylation and atherosclerotic lesion area proved that MCP-1 promoter DNA hypomethylation might promote the formation of atherosclerosis.
FIG. 8.

The epigenomics and structure of MCP-1/CCL2 are suggested by NCBI (A, B).
DNA methylation is maintained by DNMT1 and serves as a key mechanism that controls gene expression in a variety of chronic diseases such as atherosclerosis.35 DNA methylation affects the expression of genes via inhibiting the association of DNA promoter recognition site and special transcription factor like NF-κB. Hcy activates NF-κB in endothelial cells via oxidative stress.36 Consistent with those results, our research also suggested that serum NF-κB concentrations in the HM group were higher than those in the blank control group, and we found that serum NF-κB and DNMT1 levels were negatively correlated in ApoE−/− mice. Hcy up-regulates the expression of Fas, a death receptor, via NF-κB activation and promotes apoptosis in endothelial cells through activation of the extrinsic cell death pathway.37,38 Thus, intracellular substances are released into the blood. Then we tested NF-κB and DNMT1 concentrations in serum as indicators of intracellular levels. So in the in vitro experiment, foam cells were chosen as the object, and PDTC was supplemented to inhibit the effect of NF-κB in the hope of further verifying the relationship between NF-κB and DNMT1. Once NF-κB binds to target gene promoters, the target gene transcription can be activated, which in turn leads to DNMT1 down-regulation and DNA hypomethylation. The results confirmed that the DNMT1 levels were increased concomitant with the decreased concentration of NF-κB in the foam cell+Hcy+PDTC group. In in vivo and in vitro experiments, the results showed that Hcy might play a role in DNA methylation of atherosclerosis by NF-κB/DNMT1.
In conclusion, accumulating evidence suggests MCP-1 promoter DNA hypomethylation may promote the formation of atherosclerotic plaque under hyperhomocysteinemia through NF-κB/DNMT1. This fact may be significant for therapy of atherosclerosis that is associated with gene expression due to hypomethylation of a gene's regulatory regions. The induction of MCP-1 promoter DNA hypomethylation by hyperhomocysteinemia is a new element in their mechanisms. In all, our findings reveal a novel role of Hcy in the pathogenesis of atherosclerosis.
Acknowledgment
This work was supported by the Specialized Research Fund for the National Natural Science Foundation of China (30960124, 81160044) and by the New Century Excellent Talents in University (NCET-10-0916) awarded to Y.J.
Disclosure Statement
The authors declare that no competing financial interests exist.
References
- 1.Kalhan SC. Marczewski SE. Methionine, homocysteine, one carbon metabolism and fetal growth. Rev Endocr Metab Disord. 2012:15. doi: 10.1007/s11154-012-9215-7. (abstract). [DOI] [PubMed] [Google Scholar]
- 2.Woo KS. Sanderson JE. Sun YY, et al. Hyperhomocyst(e)inemia is a risk factor for arterial endothelial dysfunction in humans. Circulation. 2000;101:116. doi: 10.1161/01.cir.101.12.e116. [DOI] [PubMed] [Google Scholar]
- 3.Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. 2012;110:875–888. doi: 10.1161/CIRCRESAHA.111.257535. [DOI] [PubMed] [Google Scholar]
- 4.Catalgol B. Kartal Ozer N. Lipid rafts and redox regulation of cellular signaling in cholesterol induced atherosclerosis. Curr Cardiol Rev. 2010;6:309–324. doi: 10.2174/157340310793566181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuttolomondo A. Di Raimondo D. Pecoraro R, et al. Atherosclerosis as an inflammatory disease. Curr Pharm Des. 2012:29. doi: 10.2174/138161212802481237. (abstract). [DOI] [PubMed] [Google Scholar]
- 6.Poddar R. Sivasubramanian N. DiBello PM, et al. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: implications for vascular disease. Circulation. 2001;103:2717–2723. doi: 10.1161/01.cir.103.22.2717. [DOI] [PubMed] [Google Scholar]
- 7.Satiroglu O. Uydu HA. Demir A, et al. Association between plasma monocyte chemoattractant protein-1 levels and the extent of atherosclerotic peripheral artery disease. Tohoku J Exp Med. 2011;224:301–306. doi: 10.1620/tjem.224.301. [DOI] [PubMed] [Google Scholar]
- 8.Schepers A. Eefting D. Bonta PI, et al. Anti-MCP-1 gene therapy inhibits vascular smooth muscle cells proliferation and attenuates vein graft thickening both in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2006;26:2063–2069. doi: 10.1161/01.ATV.0000235694.69719.e2. [DOI] [PubMed] [Google Scholar]
- 9.Lu Q. Ma D. Zhao S. DNA methylation changes in cervical cancers. Methods Mol Biol. 2012;863:155–176. doi: 10.1007/978-1-61779-612-8_9. [DOI] [PubMed] [Google Scholar]
- 10.Vo AT. Millis RM. Epigenetics and breast cancers. Obstet Gynecol Int. 2012;2012:602720. doi: 10.1155/2012/602720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meehan RR. Stancheva I. DNA methylation and control of gene expression in vertebrate development. Essays Biochem. 2001;37:59–70. doi: 10.1042/bse0370059. [DOI] [PubMed] [Google Scholar]
- 12.Dudziec E. Gogol-Döring A. Cookson V, et al. Integrated epigenome profiling of repressive histone modifications, DNA methylation and gene expression in normal and malignant urothelial cells. PLoS One. 2012;7:e32750. doi: 10.1371/journal.pone.0032750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turunen MP. Aavik E. Ylä-Herttuala S. Epigenetics and atherosclerosis. Biochim Biophys Acta. 2009;1790:886–891. doi: 10.1016/j.bbagen.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 14.Yideng J. Jianzhong Z. Ying H, et al. Homocysteine-mediated expression of SAHH, DNMTs, MBD2, and DNA hypomethylation potential pathogenic mechanism in VSMCs. DNA Cell Biol. 2007;26:603–611. doi: 10.1089/dna.2007.0584. [DOI] [PubMed] [Google Scholar]
- 15.Yideng J. Zhihong L. Jiantuan X, et al. Homocysteine-mediated PPARalpha,gamma DNA methylation and its potential pathogenic mechanism in monocytes. DNA Cell Biol. 2008;27:143–150. doi: 10.1089/dna.2007.0658. [DOI] [PubMed] [Google Scholar]
- 16.Mitra S. Khaidakov M. Lu J, et al. Prior exposure to oxidized low-density lipoprotein limits apoptosis in subsequent generations of endothelial cells by altering promoter methylation. Am J Physiol Heart Circ Physiol. 2011;301:H506–513. doi: 10.1152/ajpheart.00252.2011. [DOI] [PubMed] [Google Scholar]
- 17.Stark GR. Wang Y. Lu T. Lysine methylation of promoter-bound transcription factors and relevance to cancer. Cell Res. 2011;21:375–380. doi: 10.1038/cr.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwang SY. Woo CW. Au-Yeung KK, et al. Homocysteine stimulates monocyte chemoattractant protein-1 expression in the kidney via nuclear factor-κB activation. Am J Physiol Renal Physiol. 2008;294:236–244. doi: 10.1152/ajprenal.00331.2007. [DOI] [PubMed] [Google Scholar]
- 19.Gupta SC. Sundaram C. Reuter S. Aggarwal BB. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010;1799:775–787. doi: 10.1016/j.bbagrm.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y. Mayo MW. Nagji AS, et al. Phosphorylation of RelA/p65 promotes DNMT-1 recruitment to chromatin and represses transcription of the tumor metastasis suppressor gene BRMS1. Oncogene. 2012;31:1143–1154. doi: 10.1038/onc.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S. Liu Z. Xie Z, et al. Bortezomib induces DNA hypomethylation and silenced gene transcription by interfering with Sp1/NF-κB–dependent DNAmethyltransferase activity in acute myeloid leukemia. Blood. 2008;10:2364–2373. doi: 10.1182/blood-2007-08-110171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marumo T. Schini-Kerth VB. Fisslthaler B. Busse R. Platelet-derived growth factor-stimulated superoxide anion production modulates activation of transcription factor NF-κB and expression of monocyte chemoattractant protein 1 in human aortic smooth muscle cells. Circulation. 1997;96:2361–2367. doi: 10.1161/01.cir.96.7.2361. [DOI] [PubMed] [Google Scholar]
- 23.Ravaglia G. Forti P. Maioli F, et al. Apolipoprotein E e4 allele affects risk of hyperhomocysteinemia in the elderly. Am J Clin Nutr. 2006;84:1473–1480. doi: 10.1093/ajcn/84.6.1473. [DOI] [PubMed] [Google Scholar]
- 24.Tehlivets O. Homocysteine as a risk factor for atherosclerosis: is its conversion to s-adenosyl-L-homocysteine the key to deregulated lipid metabolism? J Lipids. 2011;2011:702853. doi: 10.1155/2011/702853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veeranna V. Zalawadiya SK. Niraj A, et al. Homocysteine and reclassification of cardiovascular disease risk. J Am Coll Cardiol. 2011;58:1025–1033. doi: 10.1016/j.jacc.2011.05.028. [DOI] [PubMed] [Google Scholar]
- 26.Sahinarslan A. Kocaman SA. Topal S, et al. The relation of serum monocyte chemoattractant protein-1 level with coronary atherosclerotic burden and collateral degree in stable coronary artery disease. Turk Kardiyol Dern Ars. 2011;39:269–275. doi: 10.5543/tkda.2011.01290. [DOI] [PubMed] [Google Scholar]
- 27.Liu XL. Zhang PF. Ding SF, et al. Local gene silencing of monocyte chemoattractant protein-1 prevents vulnerable plaque disruption in apolipoprotein e-knockout mice. PLoS One. 2012;7:33497. doi: 10.1371/journal.pone.0033497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tse KP. Tsang NM. Chen KD, et al. MCP-1 promoter polymorphism at 2518 is associated with metastasis of nasopharyngeal carcinoma after treatment. Clin Cancer Res. 2007;13:6320–6326. doi: 10.1158/1078-0432.CCR-07-1029. [DOI] [PubMed] [Google Scholar]
- 29.Liu ZH. Chen LL. Deng XL, et al. Source methylation status of CpG sites in the MCP-1 promoter is correlated to serum MCP-1 in type 2 diabetes. J Endocrinol Invest. 2011:3. doi: 10.3275/7981. (abstract). [DOI] [PubMed] [Google Scholar]
- 30.Li XQ. Guo YY. De W. DNA methylation and microRNAs in cancer. World J Gastroenterol. 2012;18:882–888. doi: 10.3748/wjg.v18.i9.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shu S. Mahadeo DC. Liu X, et al. S-adenosylhomocysteine hydrolase is localized at the front of chemotaxing cells, suggesting a role for transmethylation during migration. Proc Natl Acad Sci USA. 2006;103:19788–19793. doi: 10.1073/pnas.0609385103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caudill MA. Wang JC. Melnyk S, et al. Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl-deficient cystathionine beta-synthase heterozygous mice. J Nutr. 2001;131:2811–2818. doi: 10.1093/jn/131.11.2811. [DOI] [PubMed] [Google Scholar]
- 33.Li L. Xie J. Zhang M. Wang S. Homocysteine harasses the imprinting expression of IGF2 and H19 by demethylation of differentially methylated region between IGF2/H19 genes. Acta Biochim Biophys Sin. 2009;41:464–471. doi: 10.1093/abbs/gmp033. [DOI] [PubMed] [Google Scholar]
- 34.Castro R. Rivera I. Martins C, et al. Intracellular S-adenosylhomocysteine increased levels are associated with DNA hypomethylation in HUVEC. J Mol Med. 2005;83:831–836. doi: 10.1007/s00109-005-0679-8. [DOI] [PubMed] [Google Scholar]
- 35.Bressler J. Shimmin LC. Boerwinkle E. Hixson JE. Global DNA methylation and risk of subclinical atherosclerosis in young adults: The Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study. Atherosclerosis. 2011;219:958–962. doi: 10.1016/j.atherosclerosis.2011.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Au-Yeung KK. Woo CW. Sung FL, et al. Hyperhomocysteinemia activates nuclear factor-kappaB in endothelial cells via oxidative stress. Circ Res. 2004;94:28–36. doi: 10.1161/01.RES.0000108264.67601.2C. [DOI] [PubMed] [Google Scholar]
- 37.Skurk C. Walsh K. Death receptor induced apoptosis: a new mechanism of homocysteine-mediated endothelial cell cytotoxicity. Hypertension. 2004;43:1168–1170. doi: 10.1161/01.HYP.0000127811.48554.12. [DOI] [PubMed] [Google Scholar]
- 38.Suhara T. Fukuo K. Yasuda O, et al. Homocysteine enhances endothelial apoptosis via upregulation of Fas-mediated pathways. Hypertension. 2004;43:1208–1213. doi: 10.1161/01.HYP.0000127914.94292.76. [DOI] [PubMed] [Google Scholar]