

Abstract
Many women are unable to quit smoking during pregnancy and therefore are prescribed drugs, including nicotine (NRT) to aid with smoking cessation. However, the consequences to the offspring of pregnant NRT users have not been well studied. The goals of this study were to determine the consequences of fetal and neonatal exposure to nicotine on lung development and function. Female rats were exposed to nicotine for 2 weeks prior to mating until weaning. Lungs were collected from saline and nicotine treated rats from birth to adulthood to assess postnatal lung structure and function. .Although nicotine exposure altered alveolarization at weaning, an effect which resolved by adulthood, it did not affect lung function at any of the ages investigated. However, nicotine exposure significantly decreased lung vascularization. The current study suggests that perinatal exposure to nicotine alters lung development, an effect which may be mediated via decreased VEGF signaling.
Keywords: vascularization, lung function, airway responsiveness, methacholine challenge
Introduction
Maternal cigarette smoking is associated with a higher risk of poor pregnancy outcomes including spontaneous abortion, premature delivery and fetal death (1,2). It is also highly correlated with an increased incidence of sudden infant death syndrome, pulmonary disorders and increased respiratory morbidity in the offspring (2,3). Furthermore, epidemiological studies report abnormal pulmonary function in infants and children exposed to maternal cigarette smoke (4–6). Despite the numerous chemicals present in cigarette smoke, many of the deleterious side effects on the fetus and newborn are thought to arise from the presence of nicotine.
In Canada, nicotine replacement therapy (NRT) is recommended as a safe smoking cessation aid for pregnant women (7). However, the use of NRT during pregnancy continues to be controversial as it can have toxic effects on multiple organ systems in the fetus (8,9). Indeed, nicotine passes readily from the maternal to fetal circulation (10) and binds to nicotinic acetylcholine receptors (nAChR) in target cells. These target cells include airway, vessel and alveolar wall cells in the fetal lung (11).
Lung development is a dynamic process that occurs throughout gestation and infancy. There are five developmental stages: embryonic, pseudoglandular, canalicular, saccular and lastly, alveolar. The alveolarization stage in human lungs begins in the 36th week of gestation and continues for 3 years after birth (12) whereas in rats alveolarization is initialized postnatally concludes around postnatal day 21 (13). Rodent and primate models have demonstrated that fetal and/or neonatal exposure to nicotine can adversely affect postnatal lung structure, primarily involving impaired alveolarization and increased individual alveolar size (11,14–16). In primates these nicotine-induced changes in lung development are associated with impaired lung function, but these assessments were only made immediately post-term (16). If the developmental structural defects seen in rats following nicotine exposure persist postnatally, it would be expected that there would be functional consequences similar to those seen in other emphysemic disorders of the lung, which would include increased compliance, increased resistance and possibly increased non-specific airway responsiveness. However, this remains to be investigated.
To date, the mechanisms by which nicotine can cause impaired alveolarization are not well characterized, but there is some evidence to suggest that it may be related to altered angiogenesis in the lung. The growth of the airways and alveoli are closely coordinated with the growth of their associated vasculature (17). This synchronized development is controlled by growth factors such as vascular endothelial growth factor (VEGF). In mice the expression of angiogenic factors including VEGF and its receptors (VEGF-R1 and VEGF-R2) increase postnatally concomitant with alveolarization (18). Furthermore, neonatal rats treated with either anti-angiogenic drugs or a VEGF-R2 antagonist have decreased alveolar number in association with reduced lung vessel density (19). Although nicotine is generally considered to be pro-angiogenic (20), we have previously shown that fetal and neonatal exposure to nicotine in rats resulted in reduced vessel density and decreased expression of VEGF and its receptor (VEGF-R2) in the adult ovary postnatally (21). Since decreased angiogenesis is associated with impaired alveolarization (19), alterations in constituents of the signalling pathway might at least partly explain the mechanism by which developmental exposure to nicotine can negatively affect lung development in rodents. The goals of the current study were to 1) investigate the effect of fetal and neonatal exposure to nicotine on lung development; 2) assess the functional consequences of developmental changes in lung structure into adulthood and 3) examine whether expression of key constituents of the VEGF signalling pathway in the lung are affected by nicotine exposure.
Materials and Methods
Maintenance and treatment of animals
All animal experiments were approved by the Animal Research Ethics Board at McMaster University, in accordance with the guidelines of the Canadian Council for Animal Care. Nulliparous 200–250g female Wistar rats (Harlan, Indianapolis, IN) were maintained under controlled lighting (12:12 L:D) and temperature (22°C) with ad libitum access to food and water. Dams were randomly assigned (n=12 per group) to receive saline (vehicle) or nicotine bitartrate (1mg3kg−13d−1, Sigma-Aldrich, St. Louis MO) via subcutaneous injection daily from 2 weeks prior to mating until weaning at postnatal day 21 (PND21). With this dose of nicotine, the concentration of active nicotine (nicotine free base concentration) is consistent with daily nicotine exposure in a typical smoker (22). The maternal steady state serum cotinine (major metabolite of nicotine) levels resulting from this exposure is 135.9 ± 7.86 ng/ml (23) and is within the range of cotinine concentrations reported in pregnant smokers during both early pregnancy and late pregnancy (24). At postnatal day 1 (PND1) litters were culled to eight to assure uniformity of litter size between treated and control litters. To eliminate any confounding effects of the female reproductive cycle, only male offspring were used in this study. After weaning, male offspring were selected randomly for the experiments described below.
Lung morphometry
Lung tissue was collected from a subset of pups at birth (PND1), 3 (weaning), and 12 weeks of age (n=6 per group at each age). To avoid litter effects, no more than one animal from a single litter was tested at each age. Animals at PND1 were euthanized by decapitation and at 3 and 12 weeks of age, animals were euthanized by CO2 inhalation. The left lung from each animal was inflation-fixed with 10% neutral buffered formalin (EM Science, Gibbstown, NJ) at a pressure of 20 cmH2O for 24 hours. The right lung was weighed and lung volume was measured by water volume displacement (25). The inflation-fixed lungs were cut into blocks, processed then paraffin embedded as previously described (26). Briefly, the left lung was sectioned into 4 planes prior to tissue processing. The bottom half of the left lung was sectioned into 2 transverse slices while the top half of the left lung was section into 2 sagittal segments. The segments were processed in a tissue processor (Leica Microsystems GmbH, Wetzlar, Germany). The tissues were dehydrated by sequential 1-hour incubations in sequential 70%, 95% and ethanol baths followed by 3 1-hour incubations in xylenes. The tissues were then paraffinized by 3 sequential 1-hour incubations in paraplast (Fisher Scientific, Pittsburg, PA) at 60°C. The tissues were embedded in paraffin such that the indicated planes would be accessible for sectioning at a thickness of 5 μm. Two of the four planes were selected at random for analysis.
Tissue sections (5μm) were stained with hematoxylin and eosin. For each section (8 sections per animal), 5 randomly distributed images (20x magnification) were captured using an Olympus BX-61 inverted microscope (Olympus Canada Inc., Markham, ON), avoiding blood vessels, airways, and terminal bronchioles. Mean linear intercept (Lm), an index of alveolar size, was determined by point and intersection counting (27) using the ImagePro Plus 5.1 software package (MediaCybernetics Inc., Bethesda, MD). Briefly, a grid composed of 5 evenly spaced vertical lines (336.9 μm each) and 5 evenly spaced horizontal lines (451.2 μm each) was superimposed on each frame to give a total line length of 3940.5 μm per captured image. Lm was measured as follows:
| ( 27) | Eqn. 3 |
where L is the total length of the lines in the grid superimposed on each image and n is the number of times an alveolar septum intersects the grid. The Lm for each animal was averaged over all the sections from both planes. This technique for assessing alveolar size is in agreement with recommendations made by the American Thoracic Society (28). All analyses were performed by a single investigator blinded to the treatment groups.
Lung function
Lung mechanics were measured in saline- and nicotine-exposed animals at 3 and 12 weeks of age using the computer-controlled flexiVent small animal ventilator (SCIREQ, Montreal, QC) using previously established methods (29). Briefly, rats (n=5–6/group) were anesthetized withxylazine (10 mg/kg i.p.) and pentobarbital (30 mg/kg i.p., tracheostomized with a blunted 19-gauge needle and then connected to the flexiVent system. Pancuronium bromide (20 mg/kg i.v.) was administered to achieve paralysis and prevent respiratory effort during measurement. The animals were ventilated quasi-sinusoidally (90 breaths/min, tidal volume 10 ml/kg and a maximum pressure limit of 30 cmH2O) with a positive end-expiratory pressure (PEEP) of 2 cmH2O. Quasi-static pressure-volume (PV) relationships were then determined by increasing the lung volume from functional residual capacity (FRC) to total lung capacity (TLC) in 7 equal steps and then decreasing the lung volume back to FRC in 7 steps. Each volume change was maintained for 1.14 s before proceeding to the next increment. The data on the deflation arm of the PV curve were fitted to the Salazar-Knowles equation. Static compliance (Cst), an index of lung elasticity, was calculated as the slope of the deflation curve between PEEP and 5 cmH2O.
Airway responsiveness
Airway responsiveness was determined by infusing 20 μL of methacholine through a jugular vein catheter at concentrations of 0 (saline vehicle), 10, 100, 330, 1000 and 3300 μg/kg. The peak resistance achieved with each dose was measured. Baseline airway resistance was determined by the response to the saline vehicle. Airway responsiveness to methacholine was assessed by the slope of the linear regression between the peak respiratory resistance and the log10 of the methacholine dose between the 100 and 3300 μg/kg doses. Heart rate and oxygen saturation were monitored throughout the procedure using a Biox 3700 infrared pulse oximeter (Ohmeda, Boulder, CO) with the probe placed on the rat’s hind limb.
Lung blood vessel density
Immunohistochemical detection of vascularization was performed on 5μm sections of formalin fixed, paraffin embedded lung tissue (n=6 per group). Sections were of uniform size and randomly oriented on glass slides. Tissue sections were deparaffinized in xylene, rehydrated and washed in PBS. Endogenous peroxidase activity was quenched by incubating tissue sections in 3% hydrogen peroxide (in methanol) for 10 minutes. The sections were next incubated with 10% normal goat serum and 1% BSA for 10 minutes at room temperature, and then with the primary antibody (anti-CD31; 1:500; BD Pharmingen, Franklin Lakes, NJ) overnight at 4 °C. Sections were then washed in PBS, and immunostaining was performed by the avidin-biotin-peroxidase technique. Tissues were incubated with biotin-labeled anti-mouse secondary antibody (1:100 dilution, Sigma) for 2 hours at room temperature, washed and exposed to horseradish peroxidase (Extravidin; 1:50 dilution; Sigma) for 1 hour at room temperature, with diaminobenzadine as the chromogen. Tissue sections were then counterstained with Carazzi’s hematoxylin, dehydrated and mounted with Permount (Fisher Scientific, Fair Lawn, NJ). To ensure staining specificity, primary and secondary antibody omission controls were used. In all cases, omission of either the primary or secondary antibody abolished immunostaining. Blood vessel density was quantified using integrated morphometry software (MetaMorph, CA) in which the number of blood vessels per field of view were counted (n=6 animals per group, 5 fields of view per animal). The five fields of view were selected at random from a random selection of two of the four sections obtained from the left lung. Quantification was performed on tissue slides viewed at 40x magnification. Measurements were made by a single observer blinded to the treatment groups.
VEGF, VEGF-R1 and VEGF-R2 protein expression
Protein expression was measured by western blotting in whole lung homogenates from nicotine and saline-exposed offspring at PND1, 3 and 12 weeks of age. 20 μg of total protein was extracted from the lung (n=4 per group) using RIPA lysis buffer (15 mM Tris–HCl, 1% (v/v) Triton X-100, 0.1% (w/v) SDS, 167 mM NaCl, 0.5% (w/v) sodium deoxycholatic acid), with Complete Mini EDTA-free protease inhibitors (Roche Applied Science, Laval QC, Canada) and subjected to SDS-PAGE using 8 or 12% separating gels. The separated proteins were electro-transferred to PVDF blotting membrane (BioRad Laboratories, CA). Membranes were blocked overnight with 5 % (w/v) skim milk in TTBS (TBS, 0.5% (v/v) Tween 20) at 4°C and then incubated for 1 h at room temperature in primary antibody on a rocking platform. For detection of VEGF, the membrane was cut horizontally at approximately 40kDa and the upper molecular weight portion was incubated in rabbit polyclonal anti-alpha tubulin as a loading control (50 kDa running weight; 1:5000 dilution, AbCam, MA). The lower molecular weight portion of the blot was incubated with the primary antibody for VEGF (rabbit polyclonal, 1:2000, 21kDa, Santa Cruz Biotechnology, CA). For detection of the VEGF receptors, the blots were cut at approximately 100kDa and the lower molecular weight portion was incubated in rabbit polyclonal anti-alpha tubulin and the upper molecular weight portion of the blots were incubated with the primary antibodies for VEGF-R2 (rabbit polyclonal, 1:2000, Santa Cruz Biotechnology, CA) and VEGF-R1 (rabbit polyclonal, 1:2000, Santa Cruz Biotechnology, CA).
Statistical analysis
All statistical analyses were performed using SigmaStat (v.3.1, SPSS, Chicago, IL). The results are expressed as mean ± SEM. Data were checked for normality and equal variance and were tested using unpaired Student’s t-tests (α = 0.05). Where data failed normality or equal variance tests, data was reanalyzed using Mann-Whitney rank sum test.
Results
Lung morphometry
Nicotine exposure during fetal development did not affect lung weight at birth (Figure 1A). However, the nicotine-exposed offspring had a significant reduction in the lung to body weight ratio at 3 weeks of age, which disappeared in adulthood (i.e. 12 weeks of age, Figure 1C). There was no effect of nicotine exposure on lung volume at any age examined (Figure 1B). To determine whether fetal and neonatal exposure to nicotine affected the structure of the airspaces, Lm was measured as an index of airspace size. At PND1 (saccular stage of lung development), there was a significant reduction in the airspace size in the nicotine-exposed animals (p=0.047; Figure 2). At 3 weeks of age following lung alveolarization, the airspace size in the nicotine-exposed animals was significantly larger than in the saline controls (p=0.007; Figure 2). In addition the nicotine group displayed short secondary septal buds whereas the saline group had fully-formed secondary septa at 3 weeks of age (Figure 2). At 12 weeks of age, the nicotine-induced changes in airspace size were no longer evident (p>0.05; Figure 2)
Figure 1.

Total lung weight (A), left lung volume (B) and total lung weight normalized to body weight (C) of male offspring exposed to saline (closed circles) or nicotine (open circles) during fetal and neonatal development. Data are presented as mean ± SEM. Values marked with an asterisk (*) are significantly different (p<0.05) from the saline controls at the same age.
Figure 2.
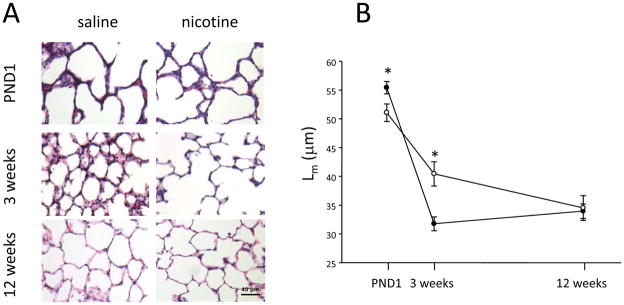
(A) Representative H&E-stained lung sections from saline and nicotine-exposed animals at postnatal day 1 (PND1), 3 and 12 weeks of age. (B) Mean linear intercept in the lungs of male rats exposed to saline (closed circles) or nicotine (open circles) during fetal and neonatal life. Data are presented as mean ± SEM. Values marked with an asterisk (*) are significantly different (p<0.05) from the saline controls at the same age.
Lung function and airway responsiveness
There was no effect of fetal and neonatal exposure to nicotine on lung compliance or baseline airway resistance at 3 or 12 weeks of age (Figure 3). Moreover, nicotine treatment did not significantly change the response of the airways to methacholine (Figure 4).
Figure 3.

Lung compliance (A) and baseline airway resistance (B) of male saline- (closed bars) and nicotine-exposed (open bars) animals at 3 and 12 weeks of age.
Figure 4.

Dose-response curve showing the airway response to methacholine challenge in 3week (A) and 12 week (C) old male rats exposed to saline (closed circles) or nicotine (open circles) during fetal and neonatal development. Slope of the concentration effect curves in saline- (black bars) and nicotine-exposed (open bars) offspring at 3 (B) and 12 (D) weeks of age. All data are presented as mean ± SEM.
Lung vessel density and VEGF expression
Nicotine-exposed offspring exhibited a significant reduction in lung vessel density at all ages studied (Figure 5). Protein expression of VEGF and VEGF-R1 were not significantly altered by nicotine exposure (data not shown). In contrast, VEGF-R2 expression in the lung was significantly lower (p<0.05) at weaning (3 weeks of age) in the nicotine-exposed animals, an effect which was not observed at either PND1 or 12 weeks of age (Figure 6).
Figure 5.
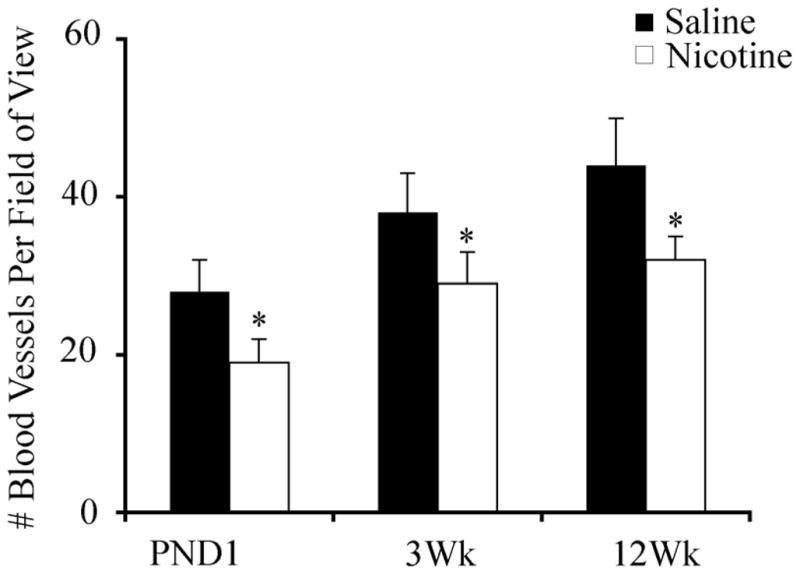
Lung vessel density at PND1, 3 and 12 weeks of age in saline (closed bars) and nicotine (open bars) exposed offspring (n=6 per group). Data are presented as mean ± SEM. Values marked with an asterisk (*) are significantly different (p<0.05) from the saline controls at the same age.
Figure 6.

(A) Representative western blots of VEGF-R1, VEGF-R2, and the loading control α-tubulin in lungs of rats exposed to saline or nicotine during fetal and neonatal development. Quantification of VEGF-R1 (B) and VEGF-R2 (C) protein expression at PND1, 3 and 12 weeks of age for saline (closed bars) and nicotine (open bars) exposed offspring. Data are presented as relative optical density ± SEM normalized to the α-tubulin control. Values marked with an asterisk (*) are significantly different (p<0.05) from the saline controls at the same age.
Discussion
Previous epidemiological studies indicate that maternal cigarette smoking is associated with an increased incidence of pulmonary disorders in the offspring including lower respiratory tract infections as well as wheezing and asthma (2,3). Because animal models have indicated that nicotine itself may adversely affect lung development (30), we hypothesized that maternal nicotine exposure during gestation and lactation would adversely impact the development of lung structures in the offspring and that these changes would result in altered pulmonary function. Indeed, the current study demonstrates that fetal nicotine exposure alone (PND1) and in combination with neonatal nicotine exposure altered airspace size. At birth (i.e., PND1) saccule size was significantly reduced in the nicotine-exposed offspring, an effect which has not previously been reported in rats. However, following alveolarization the nicotine exposed animals had larger airspace size. During alveolarization secondary alveolar septa bud and elongate from the primary saccular wall, resulting in a rapid division of the original saccular airspaces and the establishment of smaller airspaces, namely the alveoli (17). In this study, the saline-exposed animals showed normal alveolarization, with smaller airspaces at 3 weeks relative to those at PND1. In the nicotine group, this decrease was considerably more modest and resulted in significantly larger airspaces by 3 weeks of age, suggesting nicotine exposure during lung development adversely impacted the alveolarization process. This finding is consistent with other studies in rats which have reported larger mean alveolar volume as well as decreases in alveolar number and airspace wall surface area per unit volume by 3 weeks of age following nicotine exposure (31). Similarly, in monkeys, gestational nicotine exposure resulted in a significant increase in the mean linear intercept at birth, when the alveolar stage is nearly complete (11). Although other studies have reported that differences in airspace size in animals exposed to nicotine during pregnancy and lactation persisted after the exposure had been terminated (15), in this study airspace size was not significantly different in the saline- and nicotine-treated animals at 12 weeks of age. These results therefore suggest that fetal and neonatal nicotine exposure may delay but not prevent alveolarization. Although the inhibitory effect of nicotine on postnatal alveolarization was transient, there is significant clinical and experimental evidence to suggest that early abnormal lung development may be a risk factor for respiratory disease in later life such as COPD and asthma (32) which may explain, in part, the adverse respiratory outcomes in children born to mothers who smoke during pregnancy (33–36).
Although nicotine exposure did not affect lung compliance under baseline (i.e., non-stressed) conditions, fetal nicotine exposure has been shown to alter lung mechanics in response to hypoxia (37). Therefore it is possible that lung function in nicotine-exposed animals may be different under conditions of stress such as hypoxia, exercise and/or inflammation. Although lung function was not affected by nicotine exposure, our results suggest that nicotine, by altering lung structure, may affect other aspects of lung function, namely gas exchange. Decreased alveolar complexity as observed in our model is equivalent to a decrease in the surface area of the lung available for taking up O2 and eliminating excess CO2. Moreover, vessel density in the lungs of nicotine-exposed animals was significantly reduced which might also adversely affect gas exchange. Indeed neonates born to women who smoked during pregnancy have reduced tissue oxygenation relative to those born to non-smoking mothers (38). The consequences of this decreased vessel density in our animal model is unknown, but it does not appear to affect peripheral oxygenation at weaning or in adulthood as there was no difference in oxygen saturation levels in nicotine-exposed offspring at either 3 (saline 93 ± 1.0% vs nicotine 93 ± 2.2%, p>0.05) or 12 (saline 96 ± 0.8% vs nicotine 94 ± 1.2%, p>0.05) weeks of age. However it is plausible that this decreased surface area and reduced vessel density as a result of nicotine exposure might result in a decreased capacity for gas exchange and therefore contribute to an increased risk of exercise intolerance and poor tolerance of acute respiratory infections as has been shown in survivors of bronchopulmonary dysplasia; a disease characterized by reduced lung vascularization and impaired alveolar development (39).
We hypothesized that the ability of nicotine to alter alveolarization and decrease lung vessel density may be mediated through a decrease in VEGF signaling in the lung. Indeed, the biologic basis for impaired VEGF signaling leading to decreased vascular growth and impaired alveolarization is well established (40). We did not find any changes in the expression of VEGF or VEGF-R1, however, the expression of the VEGF-R2 receptor in the lung was significantly lower at 3 weeks of age in the nicotine-exposed animals. As VEGF-R2 is responsible for mediating VEGF-induced proliferation, differentiation and survival of endothelial cells, the concomitant changes observed in VEGF-R2 protein expression and alveolarization at 3 weeks of age suggest that decreased VEGF signaling may play an important role in mediating nicotine’s adverse effects on lung structure and the observed reduction in lung vessel density. This interpretation is supported by previous studies showing that the VEGF signaling system is crucial in both the development and maintenance of the alveoli. For instance, treatment of adult rats with SU-5416, a non-selective VEGF receptor inhibitor, resulted in a decrease in the arterial density of the lung accompanied by a 20% enlargement of the airspaces (19). Similarly, treatment of neonatal rats with a single dose of SU-5416 resulted in impaired development of alveoli (i.e., decreased alveolar number and increased airspace size) by 3 weeks of age; an effect that persisted for 3–4 months (41). Although in our study the decrease in VEGF-R2 was temporally associated with impaired alveolarization, the effects of nicotine exposure on lung structure were transient and did not persist into adulthood. However, the decreased lung vessel density in nicotine-exposed animal persisted and could have significant implications for respiratory health as the animal ages, including increased susceptibility to infection and progressive loss of normal lung function. VEGF has been shown to enhance the immune response to respiratory syncytial virus infection in the neonatal ovine lung (42, 43). The impaired VEGF signaling induced by nicotine exposure may reduce resistance to respiratory infection and provide a mechanistic link between maternal nicotine use and the increased predisposition to respiratory infections observed in children born to smoking mothers (44).
Nicotine replacement therapy has been widely developed as a pharmacotherapy of smoking cessation, and is considered to be of benefit for pregnant women who are highly dependent and have been unable to quit smoking by other means (7,44,45). However, results from this study suggest that nicotine alone may adversely affect lung development and provides further support to the recent concerns about the safety of nicotine replacement therapy during pregnancy and lactation (9).
Acknowledgments
We thank Michelle Ross and Jennifer Wattie for technical assistance and the staff of the McMaster University Central Animal Facility for their help with the animal work.
Funding
Funding for this project was provided by Operating grants from the Canadian Institutes of Health Research to ACH. (MOP 86474). MAP was funded by an Ashley Studentship for Research in Tobacco Control. NRL has a career award from the Father Sean O’Sullivan Research Centre, St. Joseph’s Healthcare Hamilton ON. JJP has a career award from the Canadian Institutes of Health Research.
Footnotes
Conflict of interest
The authors have no conflict of interest to declare.
References
- 1.Einarson A, Riordan S. Smoking in pregnancy and lactation: a review of risks and cessation strategies. Eur J Clin Pharmacol. 2009;65(4):325–330. doi: 10.1007/s00228-008-0609-0. [DOI] [PubMed] [Google Scholar]
- 2.Rogers JM. Tobacco and pregnancy. Reprod Toxicol. 2009;28(2):152–160. doi: 10.1016/j.reprotox.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 3.Stocks J, Dezateux C. The effect of parental smoking on lung function and development during infancy. Respirology. 2003;8(3):266–285. doi: 10.1046/j.1440-1843.2003.00478.x. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham J, Dockery DW, Speizer FE. Maternal smoking during pregnancy as a predictor of lung function in children. Am J Epidemiol. 1994;139(12):1139–52. doi: 10.1093/oxfordjournals.aje.a116961. [DOI] [PubMed] [Google Scholar]
- 5.Moshammer H, Hoek G, Luttmann-Gibson H, et al. Parental smoking and lung function in children: an international study. Am J Respir Crit Care Med. 2006;173(11):1255–1263. doi: 10.1164/rccm.200510-1552OC. [DOI] [PubMed] [Google Scholar]
- 6.Tager IB, Ngo L, Hanrahan JP. Maternal smoking during pregnancy. Effects on lung function during the first 18 months of life. Am J Respir Crit Care Med. 1995;152(3):977–983. doi: 10.1164/ajrccm.152.3.7663813. [DOI] [PubMed] [Google Scholar]
- 7.Ontario Medical Association. Rethinking stop-smoking medications: myths and facts. 1999. [Google Scholar]
- 8.Bruin JE, Gerstein HC, Holloway AC. Long-term consequences of fetal and neonatal nicotine exposure: a critical review. Toxicol Sci. 2010;116(2):364–374. doi: 10.1093/toxsci/kfq103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ginzel KH, Maritz GS, Marks DF, et al. Critical review: nicotine for the fetus, the infant and the adolescent? J Health Psychol. 2007;12(2):215–224. doi: 10.1177/1359105307074240. [DOI] [PubMed] [Google Scholar]
- 10.Lambers DS, Clark KE. The maternal and fetal physiologic effects of nicotine. Semin Perinatol. 1996;20(2):115–126. doi: 10.1016/s0146-0005(96)80079-6. [DOI] [PubMed] [Google Scholar]
- 11.Sekhon HS, Jia Y, Raab R, et al. Prenatal nicotine increases pulmonary alpha7 nicotinic receptor expression and alters fetal lung development in monkeys. J Clin Invest. 1999;103(5):637–647. doi: 10.1172/JCI5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burri PH. Structural aspects of postnatal lung development – Alveolar formation and growth. Neonatology. 2006;89(4):313–22. doi: 10.1159/000092868. [DOI] [PubMed] [Google Scholar]
- 13.Bolle I, Eder G, Takenaka S, et al. Postnatal lung function in the developing rat. J Appl Physiol. 2006;104(4):1167–76. doi: 10.1152/japplphysiol.00587.2007. [DOI] [PubMed] [Google Scholar]
- 14.Maritz GS, Dennis H. Maternal nicotine exposure during gestation and lactation interferes with alveolar development in the neonatal lung. Reprod Fertil Dev. 1998;10(3):255–261. doi: 10.1071/r98036. [DOI] [PubMed] [Google Scholar]
- 15.Maritz GS, Windvogel S. Chronic maternal nicotine exposure during gestation and lactation and the development of the lung parenchyma in the offspring. Response to nicotine withdrawal. Pathophysiology. 2003;10(1):69–75. doi: 10.1016/j.pathophys.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 16.Sekhon HS, Keller JA, Benowitz NL, et al. Prenatal nicotine exposure alters pulmonary function in newborn rhesus monkeys. Am J Respir Crit Care Med. 2001;164(6):989–994. doi: 10.1164/ajrccm.164.6.2011097. [DOI] [PubMed] [Google Scholar]
- 17.Burri PH. Fetal and postnatal development of the lung. Annu Rev Physiol. 1984;6:617–628. doi: 10.1146/annurev.ph.46.030184.003153. [DOI] [PubMed] [Google Scholar]
- 18.Tsao PN, Li H, Wei SC, et al. Expression of angiogenic factors and their receptors in postnatal mouse developing lung. J Formos Med Assoc. 2004;103(2):137–43. [PubMed] [Google Scholar]
- 19.Jakkula M, Le Cras TD, Gebb S, et al. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol. 2000;279(3):L600–607. doi: 10.1152/ajplung.2000.279.3.L600. [DOI] [PubMed] [Google Scholar]
- 20.Costa F, Soares R. Nicotine: a pro-angiogenic factor. Life Sci. 2009;84(23–24):785–790. doi: 10.1016/j.lfs.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Petrik JJ, Gerstein HC, Cesta CE, et al. Effects of rosiglitazone on ovarian function and fertility in animals with reduced fertility following fetal and neonatal exposure to nicotine. Endocrine. 2009;36(2):281–290. doi: 10.1007/s12020-009-9229-4. [DOI] [PubMed] [Google Scholar]
- 22.Matta SG, Balfour DJ, Benowitz NL, et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 2007;190(3):269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- 23.Holloway AC, Kellenberger LD, Petrik JJ. Fetal and neonatal exposure to nicotine disrupts ovarian function and fertility in adult female rats. Endocrine. 2006;30(2):213–216. doi: 10.1385/ENDO:30:2:213. [DOI] [PubMed] [Google Scholar]
- 24.George L, Granath F, Johansson AL, et al. Self-reported nicotine exposure and plasma levels of cotinine in early and late pregnancy. Acta Obstet Gynecol Scand. 2006;85(11):1331–1337. doi: 10.1080/00016340600935433. [DOI] [PubMed] [Google Scholar]
- 25.Garber SJ, Zhang H, Foley JP, et al. Hormonal regulation of alveolarization: structure-function correlation. Respir Res. 2006 Mar 27;7:47. doi: 10.1186/1465-9921-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ellis R, Leigh R, Southam D, et al. Morphometric analysis of mouse airways after chronic allergen challenge. Lab Invest. 2003;83(9):1285–1291. doi: 10.1097/01.lab.0000087586.25982.b5. [DOI] [PubMed] [Google Scholar]
- 27.Massaro D, Teich N, Maxwell S, et al. Postnatal development of alveoli. Regulation and evidence for a critical period in rats. J Clin Invest. 1985;76(4):1297–1305. doi: 10.1172/JCI112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsai CCW, Hyde DM, Ochs M, et al. An Official Research Policy Statement of the American Thoracic Society/European Respiratory Society: Standards for Quantitative Assessment of Lung Structure. Am J Respir Crit Care Med. 2010;181:394–418. doi: 10.1164/rccm.200809-1522ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ask K, Labiris R, Farkas L, et al. Comparison between conventional and “clinical” assessment of experimental lung fibrosis. J Transl Med. 2008 Apr 10;6:16. doi: 10.1186/1479-5876-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maritz GS. Are nicotine replacement therapy, varenicline or bupropion options for pregnant mothers to quit smoking? Effects on the respiratory system of the offspring. Ther Adv Respir Dis. 2009;3(4):193–210. doi: 10.1177/1753465809343712. [DOI] [PubMed] [Google Scholar]
- 31.Maritz GS. Maternal nicotine exposure during gestation and lactation of rats induce microscopic emphysema in the offspring. Exp Lung Res. 2002;28(5):391–403. doi: 10.1080/01902140290092010. [DOI] [PubMed] [Google Scholar]
- 32.Shi W, Bellusci S, Warburton D. Lung development and adult lung diseases. Chest. 2007;132:651–656. doi: 10.1378/chest.06-2663. [DOI] [PubMed] [Google Scholar]
- 33.Gilliland FD, Berhane K, McConnell R, et al. Maternal smoking during pregnancy, environmental tobacco smoke exposure and childhood lung function. Thorax. 2000;55(4):271–276. doi: 10.1136/thorax.55.4.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilliland FD, Li YF, Peters JM. Effects of maternal smoking during pregnancy and environmental tobacco smoke on asthma and wheezing in children. Am J Respir Crit Care Med. 2001;163(2):429–436. doi: 10.1164/ajrccm.163.2.2006009. [DOI] [PubMed] [Google Scholar]
- 35.Lannero E, Wickman M, Pershagen G, et al. Maternal smoking during pregnancy increases the risk of recurrent wheezing during the first years of life (BAMSE) Respir Res. 2006 Jan 5;7:3. doi: 10.1186/1465-9921-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Midodzi WK, Rowe BH, Majaesic CM, et al. Early life factors associated with incidence of physician-diagnosed asthma in preschool children: results from the Canadian Early Childhood Development cohort study. J Asthma. 2010;47(1):7–13. doi: 10.3109/02770900903380996. [DOI] [PubMed] [Google Scholar]
- 37.Sandberg KL, Poole SD, Hamdan A, et al. Prenatal nicotine exposure transiently alters the lung mechanical response to hypoxia in young lambs. Respir Physiol Neurobiol. 2007;156:283–292. doi: 10.1016/j.resp.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 38.Pichler G, Heinzinger J, Klaritsch P, et al. Impact of smoking during pregnancy on peripheral tissue oxygenation in term neonates. Neonatology. 2008;93(2):132–137. doi: 10.1159/000108408. [DOI] [PubMed] [Google Scholar]
- 39.Stenmark KR, Abman SH. Lung vascular development: implications for the pathogenesis of bronchopulmonary dysplasia. Annu Rev Physiol. 2005;67:623–61. doi: 10.1146/annurev.physiol.67.040403.102229. [DOI] [PubMed] [Google Scholar]
- 40.Voelkel NF, Vandivier RW, Tuder RM. Vascular endothelial growth factor in the lung. Am J Physiol Lung Cell Mol Physiol. 2006;290(2):L209–21. doi: 10.1152/ajplung.00185.2005. [DOI] [PubMed] [Google Scholar]
- 41.Le Cras TD, Markham NE, Tuder RM, et al. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Physiol. 2002;283(3):L555–L562. doi: 10.1152/ajplung.00408.2001. [DOI] [PubMed] [Google Scholar]
- 42.Meyerholz DK, Gallup JM, Lazic T, et al. Pretreatment with recombinant human vascular endothelial growth factor reduces virus replication and inflammation in a perinatal lamb model of respiratory syncytial virus infection. Viral Immunol. 2007;20(1):188–96. doi: 10.1089/vim.2006.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sow FB, Gallup JM, Meyerholz DK, Ackermann MR. Gene profiling studies in the neonatal ovine lung show enhancing effects of VEGF on the immune response. Dev Comp Immunol. 2009;33(6):761–71. doi: 10.1016/j.dci.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prescott SL. Effects of early cigarette smoke exposure on early immune development and respiratory disease. Paediatr Respir Rev. 2008;9(1):3–9. doi: 10.1016/j.prrv.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 45.Okuyemi KS, Ahluwalia JS, Harris KJ. Pharmacotherapy of smoking cessation. Arch Fam Med. 2000;9(3):270–281. doi: 10.1001/archfami.9.3.270. [DOI] [PubMed] [Google Scholar]
- 46.Oncken CA, Kranzler HR. Pharmacotherapies to enhance smoking cessation during pregnancy. Drug Alcohol Rev. 2003;22(2):191–202. doi: 10.1080/09595230100100633. [DOI] [PubMed] [Google Scholar]